Abstract
A straightforward convergent synthesis has been carried out for the tetrasaccharide repeating unit of the O-specific cell wall lipopolysaccharide of the strain Sp7 of Azospirillum brasilense. The target tetrasaccharide has been synthesized from suitably protected monosaccharide intermediates in 42% overall yield in seven steps by using a [2 + 2] block glycosylation approach.

Graphical Abstract
Introduction
The intensive use of chemicals for the treatment of plant diseases led to environmental pollution, pathogen resistance, an increase in production costs, and serious risks to human health. Among several alternative approaches for the protection of crops against pathogens, biological control based on plant growth-promoting bacteria (PGPB) seems to be among the most promising ones [1]. Azospirillum brasilense (A. brasilense) is a plant growth-promoting bacterium, found abundant in the rizosphere of several leguminous plants and regular crops such as wheat, rice, corn etc. Azospirillum is a Gram-negative rizobacterium, which exerts a variety of beneficial effects to the plant by fixing nitrogen in the soil [2-4]. Besides their nitrogen-fixing capability, Azospirillum also produce several vitamins and phytohormones, which promote the overall crop production [5-7]. There are also reports on the moderate biocontrolling capabilities of Azospirillum against crown gall disease in plants [8,9], leaf and/or vascular diseases of tomato [10,11] as well as inhibitory properties against the development of bacterial diseases or promoting disease resistance on rice crops [12]. It has been demonstrated that Azospirilla produce a diverse range of macromolecules such as exopolysaccharides (EPS), lipopolysaccharides (LPS) and capsular polysaccharides (CPS), which influence their interactions with plants [13-15]. There are only a limited number of reports on structural studies of the LPS present in the cell wall of Azospirilla species despite their well-known role in interactions between plants and bacteria [16-21]. Recently, Sigida et al. reported the structure of the repeating unit of the LPS present in the cell wall of Azospirillum brasilense strain Sp7: a tetrasaccharide consisting of D-galactose, D-glucosamine, L-rhamnose and L-fucose [22].
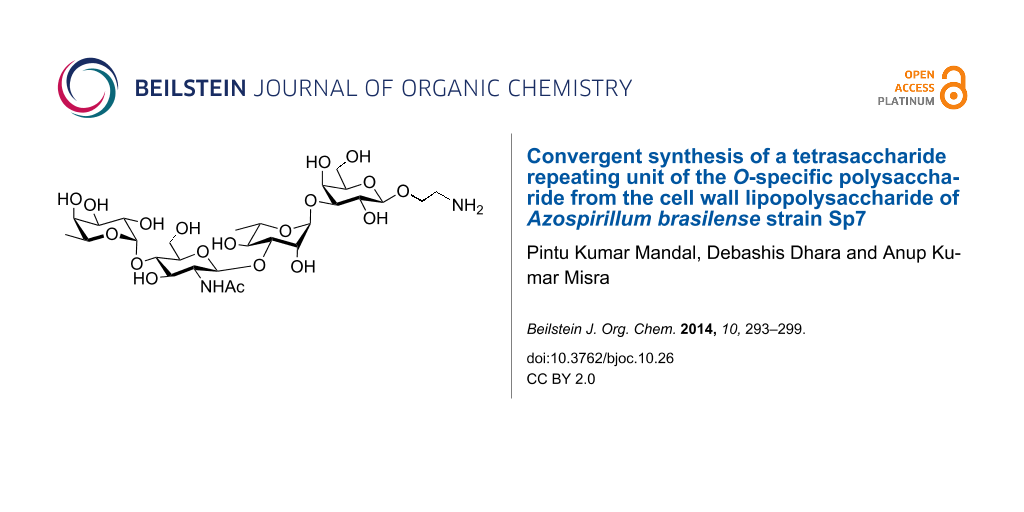
Biological plant growth-promoting agents are becoming attractive as a means for the enhancement of the crop production while minimizing the hazards associated with the use of chemical fertilizers [23,24]. For a detailed understanding of the association of the LPS present in the cell wall of Azospirillum in the initial stage of the interactions with plant roots large quantities of the LPS or its repeating unit are required. The dependence on natural sources for a sufficient quantity of the desired tetrasaccharide is inconvenient. Therefore, the development of chemical syntheses facilitate the access to the required tetrasaccharide. A limited number of reports on synthetic studies of the LPS repeating units from the cell wall of Azospirillum are available to date [25-29]. A convenient chemical synthesis of the tetrasaccharide repeating unit corresponding to the O-specific polysaccharide of the LPS of A. brasilense Sp7 strain as its 2-aminoethyl glycoside (Figure 1) is presented herein.
![[1860-5397-10-26-1]](/bjoc/content/figures/1860-5397-10-26-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the synthesized tetrasaccharide and its precursor intermediates. PMB: p-methoxybenzyl.
Figure 1: Structure of the synthesized tetrasaccharide and its precursor intermediates. PMB: p-methoxybenzyl.
Results and Discussion
The target tetrasaccharide 1 has been synthesized as its 2-aminoethyl glycoside (Figure 1) to facilitate its conjugation with a suitable substrate (e.g. a protein, a lipid or another aglycone) for biochemical applications. A stereoselective [2 + 2] block glycosylation of the disaccharide derivative 8 with the disaccharide thioglycoside 9 led to the formation of tetrasaccharide 10, which was finally deprotected to give the desired tetrasaccharide 1. A number of suitably functionalized monosaccharide intermediates 2, 3 [30], 4 [31], and 5 [32] were prepared from the reducing sugars by using literature reports. The application of a one-pot reaction sequence for the stereoselective glycosylation and the removal of the p-methoxybenzyl (PMB) group [33] as well as the utilization of the “armed–disarmed” glycosylation technique [34,35] are notable points of this synthesis.
The transformation of 2-azidoethyl β-D-galactopyranoside (6) [36] into 2-azidoethyl 2,6-di-O-acetyl-β-D-galactopyranoside (7) was carried out in 72% yield by using the reaction sequence consisting of 3,4-O-isopropylidenation with 2,2-dimethoxypropane in the presence of p-toluenesulfonic acid [37], conventional acetylation of the free hydroxy groups followed by acidic hydrolysis of the acetonide functionality. Selective acetylation of compound 7 by the formation of an orthoester intermediate [38] resulted in the formation of 2-azidoethyl 2,4,6-tri-O-acetyl-β-D-galactopyranoside (2) in 74% yield (Scheme 1).
![[1860-5397-10-26-i1]](/bjoc/content/inline/1860-5397-10-26-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (a) 2,2-dimethoxypropane, p-TsOH, DMF, room temperature, 12 h; (b) acetic anhydride, pyridine, room temperature, 6 h; (c) 80% aq AcOH, 80 °C, 1.5 h, 72% overall yield; (d) triethyl orthoacetate, p-TsOH, DMF, 2 h then 80% aq AcOH, room temperature, 1 h, 74%.
Scheme 1: Reagents and conditions: (a) 2,2-dimethoxypropane, p-TsOH, DMF, room temperature, 12 h; (b) acetic ...
D-Galactosyl acceptor 2 and 3-O-PMB-protected L-rhamnosyl donor 3 were coupled with a stereoselective glycosylation promoted by iodonium ions in the presence of a combination of N-iodosuccinimide (NIS) and trifluoromethanesulfonic acid (TfOH) [39,40]. The participating 2-O-acetyl group in donor 3 ensured the α-selectivity. By raising the temperature after completion of the coupling, the 3-O-PMB group was removed in the same pot [33] furnishing disaccharide acceptor 8 in 77% yield. The formation of compound 8 was confirmed by NMR analysis (signals at δ 4.80 (br s, H-1B), 4.44 (d, J = 8.0 Hz, H-1A) and at δ 101.0 (C-1A), 99.4 (C-1B) in the 1H and 13C NMR spectra respectively) (Scheme 2).
![[1860-5397-10-26-i2]](/bjoc/content/inline/1860-5397-10-26-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents: (a) N-iodosuccinimide (NIS), TfOH, CH2Cl2, MS 4 Å, −30 °C, 1 h, then 0 °C, 1 h, 77%; (b) NIS, TfOH, CH2Cl2/Et2O 1:1, MS 4 Å, −25 °C, 30 min, 75%; (c) NIS, TfOH, CH2Cl2, MS 4 Å, −10 °C, 1 h, 72%; (d) NH2NH2·H2O, CH3CH2OH, 80 °C, 8 h; (e) acetic anhydride, pyridine, room temperature, 1 h; (f) H2, 20% Pd(OH)2/C, CH3OH, room temperature, 24 h; (g) 0.1 M CH3ONa, CH3OH, room temperature, 3 h, 69% overall yield.
Scheme 2: Reagents: (a) N-iodosuccinimide (NIS), TfOH, CH2Cl2, MS 4 Å, −30 °C, 1 h, then 0 °C, 1 h, 77%; (b) ...
The 1,2-cis-glycosylation of thioglycoside 4 with thioglycoside 5 in the presence of a combination of NIS–TfOH [39,40] in the mixed solvent CH2Cl2/Et2O 1:1 furnished disaccharide thioglycoside derivative 9 in 75% yield. A minor quantity (~8%) of the 1,2-trans-glycosylated product was also formed under the reaction conditions but could be removed by column chromatography. The use of Et2O in the reaction solvent facilitated the formation of 1,2-cis glycoside as the major product. The concept of the Fraser–Reid’s “armed–disarmed” effect was successfully exploited in this glycosylation reaction [34,35]. Compound 5 acted as an activated or armed glycosyl donor because of the presence of an electron-donating benzyl group at the C-2 position. The presence of an electron-withdrawing N-phthaloyl group at the C-2 position of compound 4 ensured its functionality as a deactivated or disarmed glycosyl acceptor. The formation of compound 9 was confirmed by its spectroscopic analysis (signals at δ 5.50 (d, J = 10.0 Hz, H-1c), 4.97 (d, J = 3.5 Hz, H-1D), in the 1H NMR and δ 100.5 (C-1D), 80.6 (C-1C) in the 13C NMR spectra).
Finally, the 1,2-trans-glycosylation of compound 8 with the thioglycoside donor 9 in the presence of NIS–TfOH [39,40] furnished tetrasaccharide derivative 10 in 72% yield. The formation of compound 10 was confirmed by NMR analysis (signals at δ 5.53 (d, J = 8.5 Hz, H-1C), 4.94 (d, J = 3.0 Hz, H-1D), 4.85 (br s, H-1B), 4.47 (d, J = 8.0 Hz, H-1A) and at δ 101.1 (C-1A), 100.0 (C-1D), 98.7 (2 C, C-1C, C-1B) in the 1H and 13C NMR spectra respectively). Compound 10 was transformed to the target compound 1 in an overall 69% yield following a sequence of reactions consisting of (a) the conversion of the N-phthaloyl group to an acetamido group [41], (b) the hydrogenolysis with hydrogen over Pearlman’s catalyst [42], and (c) the saponification with sodium methoxide. A NMR analysis of compound 1 unambiguously confirmed its formation (signals at δ 4.92 (br s, H-1B), 4.85 (d, J = 3.5 Hz, H-1D), 4.61 (d, J = 8.5 Hz, H-1C), 4.40 (d, J = 7.5 Hz, H-1A), and 102.5 (C-1C), 102.4 (C-1A), 101.9 (C-1B), 99.6 (C-1D) in the 1H and 13C NMR spectra, respectively) (Scheme 2).
Conclusion
In summary, a straightforward and convergent synthesis of the tetrasaccharide 1 as its 2-aminoethyl glycoside corresponding to the O-specific polysaccharide of the LPS of A. brasilense strain Sp7 has been presented. The use of thioglycosides both as glycosyl donor and acceptor according to the concept of the “armed–disarmed” effect as well as the removal of PMB ether in a one-pot reaction following the glycosylation reduced the overall number of steps.
Experimental
General methods
All reactions were monitored by thin-layer chromatography over silica gel-coated TLC plates. TLC spots were visualized by warming plates sprayed with ceric sulfate (2% Ce(SO4)2 in 5% H2SO4 in EtOH) on a hot plate. Silica gel 230–400 mesh was used for column chromatography. 1H NMR and 13C NMR, DEPT 135, 2D COSY, and HSQC spectra were recorded on a Bruker Avance DRX 500 MHz spectrometer by using CDCl3 + CCl4 and D2O as solvents and TMS as an internal reference unless stated otherwise. Chemical shift values δ are expressed in ppm. ESIMS spectra were recorded on a JEOL spectrometer. Elementary analysis was carried out on Carlo ERBA analyzer. IR spectra were recorded on Shimadzu spectrophotometers. Optical rotations were determined on an Autopol III polarimeter. Commercially available grades of organic solvents of adequate purity were used in all reactions.
2-Azidoethyl 2,6-di-O-acetyl-β-D-galactopyranoside (7): To a solution of compound 6 (3 g, 12.05 mmol) in dry DMF (10 mL) was added 2,2-dimethoxypropane (1.9 mL, 24.09 mmol) followed by p-TsOH (150 mg), and the reaction mixture was allowed to stir at room temperature for 12 h. The reaction mixture was neutralized with Et3N (1.5 mL) and concentrated under reduced pressure. A solution of the crude product in acetic anhydride and pyridine (10 mL, 1:1; v/v) was kept at room temperature for 6 h, and the solvents were removed under reduced pressure. A solution of the crude product in 80% aq AcOH (100 mL) was allowed to stir at 80 °C for 1.5 h. The solvents were removed under reduced pressure, and the crude product was purified over SiO2 by using hexane/EtOAc 4:1 as an eluent to give pure compound 7 (2.9 g, 72%). Colourless oil; [α]D25 −16 (c 1.0, CHCl3); IR (neat): 3418, 2917, 2857, 1702, 1623, 1585, 1443, 1374, 1316, 1275, 1115, 1069, 1028, 786, 712 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.96 (dd, J = 10.0, 8.0 Hz, 1H, H-2), 4.43 (d, J = 8.0 Hz, 1H, H-1), 4.36–4.32 (m, 1H, H-6a), 4.27 (dd, J = 7.0, 6.5 Hz, 1H, H-3), 4.03–3.99 (m, 1H, H-6b), 3.90 (br s, 1H, H-5), 3.68–3.62 (m, 2H, H-4, OCH2), 3.50–3.45 (m, 2H, OCH2, CH2N3), 3.27–3.23 (m, 1H, CH2N3), 2.12 (s, 3H, COCH3), 2.07 (s, 3H, COCH3); 13C NMR (125 MHz, CDCl3) δ 171.6 (COCH3), 170.9 (COCH3), 100.7 (C-1), 72.7 (C-5), 72.5 (C-2), 72.3(C-4), 68.7 (C-3), 68.1 (OCH2), 62.7 (C-6), 50.6 (CH2N3), 21.0 (COCH3), 20.8 (COCH3); ESIMS (m/z): 356.1 [M + Na]+; Anal. calcd for C12H19N3O8: C, 43.24; H, 5.75; found: C, 43.13; H, 5.87.
2-Azidoethyl 2,4,6-tri-O-acetyl-β-D-galactopyranoside (2): To a solution of compound 7 (2.5 g, 7.51 mmol) in dry DMF (10 mL) was added triethyl orthoacetate (5.5 mL, 30.03 mmol) followed by p-TsOH (100 mg), and the reaction mixture was allowed to stir at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure, and a solution of the crude product in 80% aq AcOH (80 mL) was stirred at room temperature for 1 h. The solvents were removed under reduced pressure, and the crude product was purified over SiO2 by using hexane/EtOAc 4:1 as an eluent to give pure compound 2 (2.1 g, 74%). Yellow oil; [α]D25 −19 (c 1.0, CHCl3); IR (neat): 3459, 3021, 2927, 2847, 1734, 1555, 1323, 1238, 1133, 1102, 1079, 782, 698 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.25 (d, J = 3.0 Hz, 1H, H-4A), 4.92 (dd, J = 10.0, 8.0 Hz, 1H, H-2A), 4.43 (d, J = 8.0 Hz, 1H, H-1A), 4.09–4.02 (m, 2H, H-6abA), 3.97 (ddd, J = 10.6, 4.7, 3.5 Hz, 1H, OCH2), 3.81–3.74 (m, 2H, H-5A, H-3A), 3.62 (ddd, J = 10.6, 8.4, 3.4 Hz, 1H, OCH2), 3.43 (ddd, J = 13.3, 8.4, 3.5 Hz, 1H, CH2N3), 3.24 (ddd, J = 13.3, 4.7, 3.4 Hz, 1H, CH2N3), 2.11 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 1.99 (s, 3H, COCH3); 13C NMR (125 MHz, CDCl3) δ 170.9 (COCH3), 170.8 (COCH3), 170.4 (COCH3), 100.8 (C-1A), 72.1 (C-5A), 71.1 (C-2A), 70.9 (C-4A), 69.7 (C-3A), 68.2 (OCH2), 61.9 (C-6A), 50.5 (CH2N3), 20.9 (COCH3), 20.7 (COCH3), 20.6 (COCH3); ESIMS (m/z): 398.1 [M + Na]+; Anal. calcd for C14H21N3O9: C, 44.80; H, 5.64; found: C, 44.62; H, 5.83.
2-Azidoethyl (2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1→3)-2,4,6-tri-O-acetyl-β-D-galactopyranoside (8): To a solution of compound 2 (1.5 g, 4.00 mmol) and compound 3 (2.2 g, 4.80 mmol) in anhydrous CH2Cl2 (10 mL) was added MS 4 Å (2 g), and the reaction mixture was stirred under argon at room temperature for 30 min and cooled to −30 °C. To the cooled mixture were added NIS (1.3 g, 5.76 mmol) and TfOH (30 μL), and the mixture was stirred at the same temperature for 1 h. The temperature of reaction mixture was raised to 0 °C and it was stirred at 0 °C for an additional 1 h. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (100 mL). The combined organic layer was successively washed with 5% Na2S2O3, satd. NaHCO3, and water, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 by using hexane/EtOAc 4:1 as an eluant to give pure compound 8 (2 g, 77%). Yellow oil; [α]D25 +35 (c 1.0, CHCl3); IR (neat): 3418, 2911, 2769, 2211, 1664, 1532, 1498, 1411, 1379,1255, 1198, 1063, 982, 661 cm −1; 1H NMR (500 MHz, CDCl3) δ 7.32–7.25 (m, 5H, Ar-H), 5.28 (d, J = 3.0 Hz, 1H, H-4A), 5.17 (dd, J = 10.0, 8.0 Hz, 1H, H-2A), 4.85–4.83 (m, 1H, H-2B), 4.80 (br s, 1H, H-1B), 4.72 (d, J = 11.5 Hz, 1H, PhCH2), 4.67 (d, J = 11.5 Hz, 1H, PhCH2), 4.44 (d, J = 8.0 Hz, 1H, H-1A), 4.11–4.05 (m, 2H, H-6abA), 4.00–3.96 (m, 1H, OCH2), 3.86–3.74 (m, 4H, H-5B, H-3B, H-5A, H-3A), 3.64–3.61 (m, 1H, OCH2), 3.49–3.42 (m, 1H, CH2N3), 3.32–3.34 (m, 2H, H-4B, CH2N3), 2.11 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 1.27 (d, J = 6.2 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3) δ 170.2 (2 C, COCH3), 169.9 (COCH3), 169.7 (COCH3), 138.3-127.8 (Ar-C), 101.0 (C-1A), 99.4 (C-1B), 80.9 (C-4B), 76.7 (C-3A), 74.3 (PhCH2), 72.7 (C-3B), 71.4 (C-5A), 70.3 (C-2A), 68.9 (2 C, C-2B, C-4A), 68.0 (C-5B), 67.9 (OCH2), 61.9 (C-6A), 50.5 (CH2N3), 20.9 (COCH3), 20.7 (COCH3), 20.6 (2C, COCH3), 17.9 (CCH3); ESIMS (m/z): 676.1 [M + Na]+; Anal. calcd for C29H39N3O14: C, 53.29; H, 6.01; found: C, 53.10; H, 6.22.
Ethyl (2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→4)-3-O-acetyl-6-O-benzyl-2-deoxy-2-N-phthalimido-1-thio-β-D-glucopyranoside (9): To a solution of compound 4 (1 g, 2.06 mmol) and compound 5 (1.03, 2.16 mmol) in anhydrous CH2Cl2/Et2O (15 mL, 1:1, v/v) was added MS 4 Å (1 g), and the reaction mixture was allowed to stir at room temperature for 30 min under argon and cooled to −25 °C. To the cooled reaction mixture were added NIS (510 mg, 2.27 mmol) and TfOH (10 μL), and it was stirred at the same temperature for 30 min. The reaction mixture was diluted with CH2Cl2 (50 mL), filtered through a Celite® bed, and washed with CH2Cl2. The combined organic layer was successively washed with 5% Na2S2O3, satd. NaHCO3, and water, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 by using hexane/EtOAc 5:1 as an eluant to give pure compound 9 (1.4 g, 75%). Yellow oil; [α]D25 +17 (c 1.0, CHCl3); IR (neat): 3087, 2856, 1732, 1719, 1625, 1520, 1375, 1239, 1173, 1097, 1072, 989, 911, 823, 755, 698 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.85–7.24 (m, 24H, Ar-H), 5.70 (dd, J = 8.5, 1.5 Hz, 1H, H-3C), 5.50 (d, J = 10.5 Hz, 1H, H-1C), 4.97 (d, J = 3.5 Hz, 1H, H-1D), 4.94 (d, J = 12.5 Hz, 1H, PhCH2), 4.79 (dd, J = 12.0 Hz, 2H, PhCH2), 4.69 (d, J = 12.5 Hz, 1H, PhCH2), 4.63 (dd, J = 11.5 Hz, 2H, PhCH2), 4.39 (dd, J = 12.0 Hz, 2H, PhCH2), 4.27 (t, J = 10.0 Hz, 1H, H-2C), 4.05 (d, J = 11.0 Hz, 1H, H-6aC), 3.99–3.97 (m, 1H, H-5D), 3.88–3.84 (m, 2H, H-5C, H-2D), 3.79–3.71 (m, 3H, H-4C, H-3D, H-6bC), 3.60 (br s, 1H, H-4D), 2.79–2.57 (m, 2H, SCH2CH3), 1.87 (s, 3H, COCH3), 1.25 (t, J = 7.5 Hz, 3H, SCH2CH3), 1.02 (d, J = 6.5 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3) δ 170.6 (COCH3), 167.8, 167.4 (Phth), 138.7–123.5 (Ar-C), 100.5 (C-1D), 80.6 (C-1C), 79.3 (C-3D), 79.0 (C-2D), 78.2 (C-4C), 77.9 (C-4D), 76.7 (C-5D), 75.0 (PhCH2), 73.9 (2C, C-3C, PhCH2), 73.2 (PhCH2), 72.9 (PhCH2), 69.6 (C-6C), 67.8 (C-5C), 54.2 (C-2C), 24.2 (SCH2CH3), 20.9 (COCH3), 16.4 (CCH3), 15.2 (SCH2CH3); ESIMS (m/z): 924.2 [M + Na]+; Anal. calcd for C52H55NO11S: C, 69.24; H, 6.15; found: C, 69.06; H, 6.30.
2-Azidoethyl (2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-(1→4)-(3-O-acetyl-6-O-benzyl-2-deoxy-2-N-phthalimido-β-D-glucopyranoside)-(1→3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1→3)-2,4,6-tri-O-acetyl-β-D-galactopyranoside (10): To a solution of compound 8 (800 mg, 1.23 mmol) and compound 9 (1.3 g, 1.47 mmol) in anhydrous CH2Cl2 (10 mL) was added MS 4 Å (500 mg), and the reaction mixture was stirred under argon at room temperature for 30 min and cooled to −10 °C. To the cooled reaction mixture were added NIS (396 mg, 1.76 mmol) and TfOH (6 μL), and it was stirred at same temperature for 1 h. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (50 mL). The combined organic layer was successively washed with 5% Na2S2O3, satd. NaHCO3 and water, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 by using hexane/EtOAc 5:1 as an eluant to give pure compound 10 (1.3 g, 72%). Colorless oil; [α]D25 +38 (c 1.0, CHCl3); IR (neat): 3029, 2210, 1849, 1628, 1415, 1322, 1042, 988, 756, 667 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.63–6.86 (m, 29H, Ar-H), 5.59 (dd, J = 8.5, 1.5 Hz, 1H, H-3C), 5.53 (d, J = 8.5 Hz, 1H, H-1C), 5.29–5.22 (m, 2H, H-4A, H-2A), 5.04–5.03 (m, 1H, H-2B), 4.94 (d, J = 3.0 Hz, 1H, 1D), 4.90 (d, J = 11.5 Hz, 1H, PhCH2), 4.85 (br s, 1H, H-1B), 4.78 (d, J = 12.0 Hz, 1H, PhCH2), 4.70 (dd, J = 12.0 Hz, 2H, PhCH2), 4.61 (d, J = 12.0 Hz, 2H, PhCH2), 4.57 (d, J = 11.5 Hz, 1H, PhCH2), 4.47 (d, J = 8.0 Hz, 1H, H-1A), 4.46 (d, J = 12.0 Hz, 1H, PhCH2), 4.35 (dd, J = 12.5 Hz, 2H, PhCH2), 4.24–4.18 (m, 2H, H-2C, H-5A), 4.17–4.11 (m, 2H, H-6abA), 4.10–3.99 (m, 3H, H-4C, H-6aC, OCH2), 3.93 (dd, J = 10.0, 3.5 Hz, 1H, H-5D), 3.87–3.78 (m, 4H, H-2D, H-3A, H-3B, H-5C), 3.75–3.69 (m, 4H, H-3D, H-5B, H-6bC, OCH2), 3.57 (br s, 1H, H-4D), 3.55–3.43 (m, 1H, CH2N3), 3.35–3.24 (m, 2H, H-4B, CH2N3), 2.29 (s, 3H, COCH3), 2.18 (s, 3H, COCH3), 2.05 (s, 3H, COCH3), 2.00 (s, 3H, COCH3), 1.79 (s, 3H, COCH3), 1.03 (d, J = 6.0 Hz, 3H, CCH3), 0.98 (d, J = 6.5 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3) δ 170.5 (COCH3), 170.2 (3C, COCH3), 169.7 (COCH3), 167.7, 167.2 (Phth), 101.1 (C-1A), 100.0 (C-1D), 98.7 (2C, C-1C, C-1B), 79.4 (C-3A), 79.2 (C-3D), 78.2 (C-4B), 77.9 (C-4D), 76.4 (C-2D), 76.1 (C-4C), 75.8 (C-5D), 75.1 (C-3B), 74.9 (PhCH2), 73.9 (PhCH2), 73.6 (PhCH2), 73.3 (C-3C), 73.2 (PhCH2), 72.9, (PhCH2), 71.5 (C-5A), 71.4 (C-2B), 70.6 (C-2A), 68.8 (C-4A), 68.4 (OCH2), 68.2 (C-5B), 67.9 (C-6C), 67.5 (C-5C), 61.9 (C-6A), 55.2 (C-2C), 50.6 (CH2N3), 21.0 (COCH3), 20.9 (COCH3), 20.8 (COCH3), 20.7 (2 C, COCH3), 17.7 (CCH3), 16.4 (CCH3); ESIMS (m/z):1515.2 [M + Na]+; Anal. calcd for C79H88N4O25: C, 63.53; H, 5.94; found: C, 63.40; H, 6.12.
2-Aminoethyl (α-L-fucopyranosyl)-(1→4)-(2-acetamido-2-deoxy-β-D-glucopyranoside)-(1→3)-(α-L-rhamnopyranosyl)-(1→3)-β-D-galactopyranoside (1): To a solution of compound 10 (1 g, 0.67 mmol) in EtOH (30 mL) was added hydrazine hydrate (4 mL) and the mixture was allowed to stir at 80 °C for 8 h. The solvents were removed under reduced pressure, and a solution of the crude mass in pyridine (5 mL) and acetic anhydride (5 mL) was kept at room temperature for 1 h. The solvents were evaporated and co-evaporated with toluene under reduced pressure. To a solution of the acetylated product in CH3OH (20 mL) was added 20% Pd(OH)2/C (200 mg) and the reaction mixture was stirred at room temperature under a positive pressure of H2 for 24 h. The reaction mixture was filtered through a Celite® bed, the filtering bed was washed with CH3OH (20 mL), and the combined filtrate was concentrated under reduced pressure. A solution of the hydrogenated product in 0.1 M CH3ONa in CH3OH (10 mL) was allowed to stir at room temperature for 3 h. The reaction mixture was neutralized with Dowex 50W-X8 (H+) resin, filtered and concentrated. The crude product was passed through a Sephadex® LH-20 column by using CH3OH/H2O 3:1 as an eluant to give pure compound 1 (330 mg, 69%). White powder; [α]D25 +10 (c 1.0, H2O); IR (KBr): 3456, 2935, 1622, 1356, 1236, 1165, 1067, 697 cm−1; 1H NMR (500 MHz, D2O) δ 4.92 (br s, 1H, H-1B), 4.85 (d, J = 3.5 Hz, 1H, H-1D), 4.61 (d, J = 8.5 Hz, 1H, H-1C), 4.40 (d, J = 7.5 Hz, 1H, H-1A), 4.27–4.24 (m, 1H, H-5D), 4.19 (br s, 1H, H-2B), 4.04–4.01 (m, 1H, OCH2), 3.90–3.86 (m, 3H, H-2D, H-6aC, OCH2), 3.81–3.78 (m, 2H, H-3B, H-6bC), 3.77–3.62 (m, 9H, H-2C, H-3C, H-3D, H-4A, H-4D, H-4C, H-5B, H-6abA), 3.59–3.53 (m, 2H, H-3A, H-4B), 3.49–3.38 (m, 3H, H-2A, H-5C, H-5A), 3.19–3.16 (m, 2H, CH2N3), 1.93 (s, 3H, COCH3), 1.16 (d, J = 6.5 Hz, 3H, CCH3), 1.09 (d, J = 6.5 Hz, 3H, CCH3); 13C NMR (125 MHz, D2O) δ 175.9 (COCH3), 102.5 (C-1C), 102.4 (C-1A), 101.9 (C-1B), 99.6 (C-1D), 80.2 (C-3A), 79.9 (C-3B), 77.2 (C-5C), 75.2 (C-5A), 75.0 (C-3C), 72.4 (C-2B), 71.9 (C-4B), 70.8 (C-2A), 70.2 (C-4D), 69.7 (C-3D), 69.4 (2C, C-4A, C-4C), 68.3 (C-2D), 68.1 (C-5B), 67.0 (C-5D), 65.8 (OCH2), 60.9 (C-6A), 59.9 (C-6C), 56.2 (C-2C), 39.5 (CH2N3), 22.2 (COCH3), 16.7 (CCH3), 15.2 (CCH3); ESIMS (m/z): 719.2 [M + H]+; Anal. calcd for C28H50N2O19: C, 46.79; H, 7.01; found: C, 46.62; H, 7.20.
Supporting Information
| Supporting Information File 1: 1D and 2D NMR spectra of compounds 1, 2, 7, 8, 9, 10. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Compant, S.; Duffy, B.; Nowak, J.; Clément, C.; Barka, E. A. Appl. Environ. Microbiol. 2005, 71, 4951–4959. doi:10.1128/AEM.71.9.4951-4959.2005
Return to citation in text: [1] -
Tripathi, J.; Singh, A. K.; Tiwari, P. Afr. J. Agric. Res. 2013, 8, 3436–3443.
Return to citation in text: [1] -
Bekri, M. A.; Desair, J.; Proost, P.; Searle-van Leeuwen, M.; Vanderleyden, J.; Vande Broek, A. J. Bacteriol. 1999, 181, 2440–2447.
Return to citation in text: [1] -
Okon, Y. Trends Biotechnol. 1985, 3, 223–228. doi:10.1016/0167-7799(85)90012-5
Return to citation in text: [1] -
Bashan, Y.; Holguin, G. Can. J. Microbiol. 1997, 43, 103–121. doi:10.1139/m97-015
Return to citation in text: [1] -
Somers, E.; Ptacek, D.; Gysegom, P.; Srinivasan, M.; Vanderleyden, J. Appl. Environ. Microbiol. 2005, 71, 1803–1810. doi:10.1128/AEM.71.4.1803-1810.2005
Return to citation in text: [1] -
Zakharova, E. A.; Iosipenko, A. D.; Ignatov, V. V. Microbiol. Res. 2000, 155, 209–214. doi:10.1016/S0944-5013(00)80034-8
Return to citation in text: [1] -
Hao, Y.; Charles, T. C.; Glick, B. R. Can. J. Microbiol. 2007, 53, 1291–1299. doi:10.1139/W07-099
Return to citation in text: [1] -
Weller, D. M. Annu. Rev. Phytopathol. 1988, 26, 379–407. doi:10.1146/annurev.py.26.090188.002115
Return to citation in text: [1] -
Romero, A. M.; Correa, O. S.; Moccia, S.; Rivas, J. G. J. Appl. Microbiol. 2003, 95, 832–838. doi:10.1046/j.1365-2672.2003.02053.x
Return to citation in text: [1] -
Bashan, Y.; de-Bashan, L. E. Eur. J. Plant Pathol. 2002, 108, 821–829. doi:10.1023/A:1021274419518
Return to citation in text: [1] -
Holguin, G.; Bashan, Y. Soil Biol. Biochem. 1996, 28, 1651–1660. doi:10.1016/S0038-0717(96)00251-9
Return to citation in text: [1] -
Bogatyrev, V. A.; Dykman, L. A.; Matora, L. Yu.; Schwartsburd, B. I. FEMS Microbiol. Lett. 1992, 96, 115–118. doi:10.1111/j.1574-6968.1992.tb05402.x
Return to citation in text: [1] -
Burdman, S.; Okon, Y.; Jurkevitch, E. Crit. Rev. Microbiol. 2000, 26, 91–110. doi:10.1080/10408410091154200
Return to citation in text: [1] -
Jofré, E.; Lagares, A.; Mori, G. FEMS Microbiol. Lett. 2004, 231, 267–275. doi:10.1016/S0378-1097(04)00003-5
Return to citation in text: [1] -
Fedonenko, Y. P.; Konnova, O. N.; Zatonsky, G. V.; Konnova, S. A.; Kocharova, N. A.; Zdorovenko, E. L.; Ignatov, V. V. Carbohydr. Res. 2005, 340, 1259–1263. doi:10.1016/j.carres.2005.01.042
Return to citation in text: [1] -
Fedonenko, Y. P.; Konnova, O. N.; Zatonsky, G. V.; Shashkov, A. S.; Konnova, S. A.; Zdorovenko, E. L.; Ignatov, V. V.; Knirel, Y. A. Carbohydr. Res. 2004, 339, 1813–1816. doi:10.1016/j.carres.2004.05.013
Return to citation in text: [1] -
Fedonenko, Y. P.; Zatonsky, G. V.; Konnova, S. A.; Zdorovenko, E. L.; Ignatov, V. V. Carbohydr. Res. 2002, 337, 869–872. doi:10.1016/S0008-6215(02)00061-7
Return to citation in text: [1] -
Fedonenko, Y. P.; Konnova, O. N.; Zdorovenko, E. L.; Konnova, S. A.; Zatonsky, G. V.; Shashkov, A. S.; Ignatova, V. V.; Knirel, Y. A. Carbohydr. Res. 2008, 343, 810–816. doi:10.1016/j.carres.2007.12.013
Return to citation in text: [1] -
Boyko, A. S.; Dmitrenok, A. S.; Fedonenko, Y. P.; Zdorovenko, E. L.; Konnova, S. A.; Knirel, Y. A.; Ignatov, V. V. Carbohydr. Res. 2012, 355, 92–95. doi:10.1016/j.carres.2012.04.006
Return to citation in text: [1] -
Fedonenko, Y. P.; Zdorovenko, E. L.; Konnova, S. A.; Kachala, V. V.; Ignatov, V. V. Carbohydr. Res. 2008, 343, 2841–2844. doi:10.1016/j.carres.2008.05.022
Return to citation in text: [1] -
Sigida, E. N.; Fedonenko, Y. P.; Shashkov, A. S.; Zdorovenko, E. L.; Konnova, S. A.; Ignatov, V. V.; Knirel, Y. A. Carbohydr. Res. 2013, 380, 76–80. doi:10.1016/j.carres.2013.07.013
Return to citation in text: [1] -
Pal, K. K.; McSpadden Gardener, B. Plant Health Instruc. 2006. doi:10.1094/PHI-A-2006-1117-02
Return to citation in text: [1] -
Figueiredo, M. V. B.; Seldin, L.; de Araujo, F. F.; Mariano, R. L. R. In Plant Growth and Health Promoting Bacteria; Maheshwari, D. K., Ed.; Springer-Verlag: Berlin, 2011; pp 21–43.
Return to citation in text: [1] -
Manddal, P. K.; Misra, A. K. Synthesis 2009, 1348–1354. doi:10.1055/s-0028-1088032
Return to citation in text: [1] -
Ghosh, S.; Misra, A. K. Tetrahedron: Asymmetry 2010, 21, A725–A730. doi:10.1016/S0957-4166(11)00020-6
Return to citation in text: [1] -
Ghosh, S.; Misra, A. K. Tetrahedron: Asymmetry 2010, 21, 2755–2761. doi:10.1016/j.tetasy.2010.11.008
Return to citation in text: [1] -
Zhao, H.; Jia, H.; Duan, H.; Zhang, J.; Wang, D.; Liang, X. J. Carbohydr. Chem. 2010, 29, 103–117. doi:10.1080/07328301003786789
Return to citation in text: [1] -
Verma, P. R.; Mukhopadhyay, B. Carbohydr. Res. 2010, 345, 432–436. doi:10.1016/j.carres.2009.11.024
Return to citation in text: [1] -
Mukherjee, C.; Misra, A. K. Glycoconjugate J. 2008, 25, 611–624. doi:10.1007/s10719-008-9107-y
Return to citation in text: [1] -
Sun, D.-Q.; Busson, R.; Herdewijn, P. Eur. J. Org. Chem. 2006, 5158–5166. doi:10.1002/ejoc.200600515
Return to citation in text: [1] -
Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4
Return to citation in text: [1] -
Bhattacharyya, S.; Magnusson, B. G.; Wellmar, U.; Nilsson, U. J. J. Chem. Soc., Perkin Trans. 1 2001, 886–890. doi:10.1039/b009448j
Return to citation in text: [1] [2] -
Fraser-Reid, B.; Lopez, J. C. Top. Curr. Chem. 2011, 301, 1–29. doi:10.1007/978-3-642-20914-7
Return to citation in text: [1] [2] -
Mootoo, D. R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. J. Am. Chem. Soc. 1988, 110, 5583–5584. doi:10.1021/ja00224a060
Return to citation in text: [1] [2] -
Cheng, H.; Cao, X.; Xian, M.; Fang, L.; Cai, T. B.; Ji, J. J.; Tunac, J. B.; Sun, D.; Wang, P. G. J. Med. Chem. 2005, 48, 645–652. doi:10.1021/jm049693a
Return to citation in text: [1] -
Lipták, A.; Imre, J.; Nánási, P. Carbohydr. Res. 1981, 92, 154–156. doi:10.1016/S0008-6215(00)85991-1
Return to citation in text: [1] -
Bouchra, M.; Calinaud, P.; Gelas, J. Carbohydr. Res. 1995, 267, 227–237. doi:10.1016/0008-6215(94)00306-Z
Return to citation in text: [1] -
Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334. doi:10.1016/S0040-4039(00)88799-7
Return to citation in text: [1] [2] [3] -
Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313–4316. doi:10.1016/S0040-4039(00)97609-3
Return to citation in text: [1] [2] [3] -
Kanie, O.; Crawley, S. C.; Palcic, M. M.; Hindsgaul, O. Carbohydr. Res. 1993, 243, 139–164. doi:10.1016/0008-6215(93)84087-M
Return to citation in text: [1] -
Pearlman, W. M. Tetrahedron Lett. 1967, 8, 1663–1664. doi:10.1016/S0040-4039(00)70335-2
Return to citation in text: [1]
| 1. | Compant, S.; Duffy, B.; Nowak, J.; Clément, C.; Barka, E. A. Appl. Environ. Microbiol. 2005, 71, 4951–4959. doi:10.1128/AEM.71.9.4951-4959.2005 |
| 10. | Romero, A. M.; Correa, O. S.; Moccia, S.; Rivas, J. G. J. Appl. Microbiol. 2003, 95, 832–838. doi:10.1046/j.1365-2672.2003.02053.x |
| 11. | Bashan, Y.; de-Bashan, L. E. Eur. J. Plant Pathol. 2002, 108, 821–829. doi:10.1023/A:1021274419518 |
| 33. | Bhattacharyya, S.; Magnusson, B. G.; Wellmar, U.; Nilsson, U. J. J. Chem. Soc., Perkin Trans. 1 2001, 886–890. doi:10.1039/b009448j |
| 8. | Hao, Y.; Charles, T. C.; Glick, B. R. Can. J. Microbiol. 2007, 53, 1291–1299. doi:10.1139/W07-099 |
| 9. | Weller, D. M. Annu. Rev. Phytopathol. 1988, 26, 379–407. doi:10.1146/annurev.py.26.090188.002115 |
| 34. | Fraser-Reid, B.; Lopez, J. C. Top. Curr. Chem. 2011, 301, 1–29. doi:10.1007/978-3-642-20914-7 |
| 35. | Mootoo, D. R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. J. Am. Chem. Soc. 1988, 110, 5583–5584. doi:10.1021/ja00224a060 |
| 5. | Bashan, Y.; Holguin, G. Can. J. Microbiol. 1997, 43, 103–121. doi:10.1139/m97-015 |
| 6. | Somers, E.; Ptacek, D.; Gysegom, P.; Srinivasan, M.; Vanderleyden, J. Appl. Environ. Microbiol. 2005, 71, 1803–1810. doi:10.1128/AEM.71.4.1803-1810.2005 |
| 7. | Zakharova, E. A.; Iosipenko, A. D.; Ignatov, V. V. Microbiol. Res. 2000, 155, 209–214. doi:10.1016/S0944-5013(00)80034-8 |
| 31. | Sun, D.-Q.; Busson, R.; Herdewijn, P. Eur. J. Org. Chem. 2006, 5158–5166. doi:10.1002/ejoc.200600515 |
| 2. | Tripathi, J.; Singh, A. K.; Tiwari, P. Afr. J. Agric. Res. 2013, 8, 3436–3443. |
| 3. | Bekri, M. A.; Desair, J.; Proost, P.; Searle-van Leeuwen, M.; Vanderleyden, J.; Vande Broek, A. J. Bacteriol. 1999, 181, 2440–2447. |
| 4. | Okon, Y. Trends Biotechnol. 1985, 3, 223–228. doi:10.1016/0167-7799(85)90012-5 |
| 32. | Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4 |
| 22. | Sigida, E. N.; Fedonenko, Y. P.; Shashkov, A. S.; Zdorovenko, E. L.; Konnova, S. A.; Ignatov, V. V.; Knirel, Y. A. Carbohydr. Res. 2013, 380, 76–80. doi:10.1016/j.carres.2013.07.013 |
| 25. | Manddal, P. K.; Misra, A. K. Synthesis 2009, 1348–1354. doi:10.1055/s-0028-1088032 |
| 26. | Ghosh, S.; Misra, A. K. Tetrahedron: Asymmetry 2010, 21, A725–A730. doi:10.1016/S0957-4166(11)00020-6 |
| 27. | Ghosh, S.; Misra, A. K. Tetrahedron: Asymmetry 2010, 21, 2755–2761. doi:10.1016/j.tetasy.2010.11.008 |
| 28. | Zhao, H.; Jia, H.; Duan, H.; Zhang, J.; Wang, D.; Liang, X. J. Carbohydr. Chem. 2010, 29, 103–117. doi:10.1080/07328301003786789 |
| 29. | Verma, P. R.; Mukhopadhyay, B. Carbohydr. Res. 2010, 345, 432–436. doi:10.1016/j.carres.2009.11.024 |
| 16. | Fedonenko, Y. P.; Konnova, O. N.; Zatonsky, G. V.; Konnova, S. A.; Kocharova, N. A.; Zdorovenko, E. L.; Ignatov, V. V. Carbohydr. Res. 2005, 340, 1259–1263. doi:10.1016/j.carres.2005.01.042 |
| 17. | Fedonenko, Y. P.; Konnova, O. N.; Zatonsky, G. V.; Shashkov, A. S.; Konnova, S. A.; Zdorovenko, E. L.; Ignatov, V. V.; Knirel, Y. A. Carbohydr. Res. 2004, 339, 1813–1816. doi:10.1016/j.carres.2004.05.013 |
| 18. | Fedonenko, Y. P.; Zatonsky, G. V.; Konnova, S. A.; Zdorovenko, E. L.; Ignatov, V. V. Carbohydr. Res. 2002, 337, 869–872. doi:10.1016/S0008-6215(02)00061-7 |
| 19. | Fedonenko, Y. P.; Konnova, O. N.; Zdorovenko, E. L.; Konnova, S. A.; Zatonsky, G. V.; Shashkov, A. S.; Ignatova, V. V.; Knirel, Y. A. Carbohydr. Res. 2008, 343, 810–816. doi:10.1016/j.carres.2007.12.013 |
| 20. | Boyko, A. S.; Dmitrenok, A. S.; Fedonenko, Y. P.; Zdorovenko, E. L.; Konnova, S. A.; Knirel, Y. A.; Ignatov, V. V. Carbohydr. Res. 2012, 355, 92–95. doi:10.1016/j.carres.2012.04.006 |
| 21. | Fedonenko, Y. P.; Zdorovenko, E. L.; Konnova, S. A.; Kachala, V. V.; Ignatov, V. V. Carbohydr. Res. 2008, 343, 2841–2844. doi:10.1016/j.carres.2008.05.022 |
| 30. | Mukherjee, C.; Misra, A. K. Glycoconjugate J. 2008, 25, 611–624. doi:10.1007/s10719-008-9107-y |
| 13. | Bogatyrev, V. A.; Dykman, L. A.; Matora, L. Yu.; Schwartsburd, B. I. FEMS Microbiol. Lett. 1992, 96, 115–118. doi:10.1111/j.1574-6968.1992.tb05402.x |
| 14. | Burdman, S.; Okon, Y.; Jurkevitch, E. Crit. Rev. Microbiol. 2000, 26, 91–110. doi:10.1080/10408410091154200 |
| 15. | Jofré, E.; Lagares, A.; Mori, G. FEMS Microbiol. Lett. 2004, 231, 267–275. doi:10.1016/S0378-1097(04)00003-5 |
| 12. | Holguin, G.; Bashan, Y. Soil Biol. Biochem. 1996, 28, 1651–1660. doi:10.1016/S0038-0717(96)00251-9 |
| 23. | Pal, K. K.; McSpadden Gardener, B. Plant Health Instruc. 2006. doi:10.1094/PHI-A-2006-1117-02 |
| 24. | Figueiredo, M. V. B.; Seldin, L.; de Araujo, F. F.; Mariano, R. L. R. In Plant Growth and Health Promoting Bacteria; Maheshwari, D. K., Ed.; Springer-Verlag: Berlin, 2011; pp 21–43. |
| 38. | Bouchra, M.; Calinaud, P.; Gelas, J. Carbohydr. Res. 1995, 267, 227–237. doi:10.1016/0008-6215(94)00306-Z |
| 36. | Cheng, H.; Cao, X.; Xian, M.; Fang, L.; Cai, T. B.; Ji, J. J.; Tunac, J. B.; Sun, D.; Wang, P. G. J. Med. Chem. 2005, 48, 645–652. doi:10.1021/jm049693a |
| 37. | Lipták, A.; Imre, J.; Nánási, P. Carbohydr. Res. 1981, 92, 154–156. doi:10.1016/S0008-6215(00)85991-1 |
| 42. | Pearlman, W. M. Tetrahedron Lett. 1967, 8, 1663–1664. doi:10.1016/S0040-4039(00)70335-2 |
| 39. | Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334. doi:10.1016/S0040-4039(00)88799-7 |
| 40. | Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313–4316. doi:10.1016/S0040-4039(00)97609-3 |
| 41. | Kanie, O.; Crawley, S. C.; Palcic, M. M.; Hindsgaul, O. Carbohydr. Res. 1993, 243, 139–164. doi:10.1016/0008-6215(93)84087-M |
| 39. | Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334. doi:10.1016/S0040-4039(00)88799-7 |
| 40. | Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313–4316. doi:10.1016/S0040-4039(00)97609-3 |
| 34. | Fraser-Reid, B.; Lopez, J. C. Top. Curr. Chem. 2011, 301, 1–29. doi:10.1007/978-3-642-20914-7 |
| 35. | Mootoo, D. R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. J. Am. Chem. Soc. 1988, 110, 5583–5584. doi:10.1021/ja00224a060 |
| 39. | Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334. doi:10.1016/S0040-4039(00)88799-7 |
| 40. | Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313–4316. doi:10.1016/S0040-4039(00)97609-3 |
| 33. | Bhattacharyya, S.; Magnusson, B. G.; Wellmar, U.; Nilsson, U. J. J. Chem. Soc., Perkin Trans. 1 2001, 886–890. doi:10.1039/b009448j |
© 2014 Mandal et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)