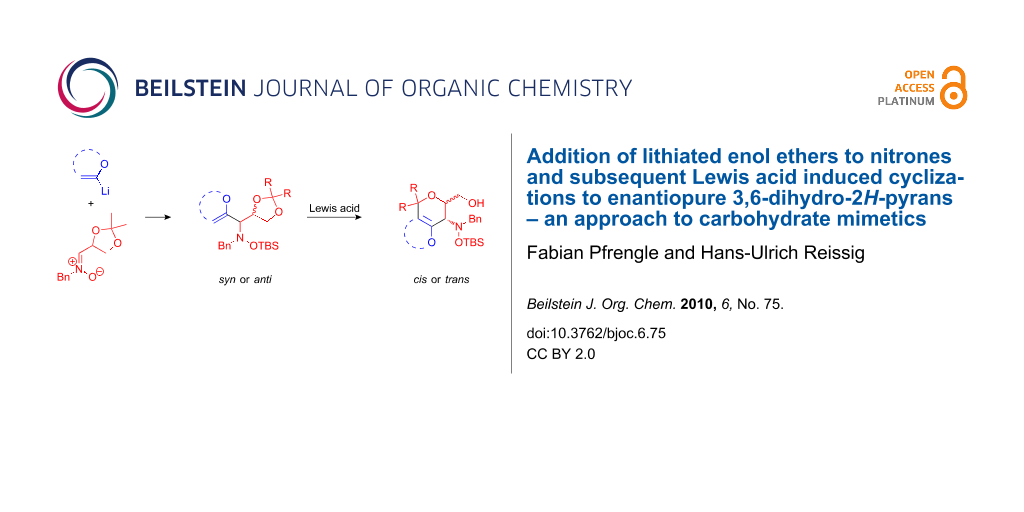
Abstract
A stereodivergent synthesis of enantiopure 3,6-dihydro-2H-pyrans is presented. The addition of lithiated enol ethers to carbohydrate-derived nitrones afforded syn- or anti-configured hydroxylamine derivatives 4a–d that were cyclized under Lewis acidic conditions to yield functionalized dihydropyrans cis- or trans-5a–d containing an enol ether moiety. This functional group was employed for a variety of subsequent reactions such as dihydroxylation or bromination. Bicyclic enol ether 19 was oxidatively cleaved to provide the highly functionalized ten-membered ring lactone 20. The synthesized enantiopure aminopyrans 24, 26, 28 and 30 can be regarded as carbohydrate mimetics. Trimeric versions of 24 and 28 were constructed via their attachment to a tricarboxylic acid core.

Graphical Abstract
Introduction
The pyran structural motif can be found in numerous bioactive natural products. Possible strategies towards their efficient preparation include Prins cyclizations, intramolecular substitutions, ring closure metathesis, and hetero Diels–Alder reactions [1,2]. Our group recently reported the synthesis of enantiopure aminopyrans employing as the key step a Lewis acid induced rearrangement of 1,2-oxazines to bicyclic ketones. This strategy allowed a simple access to unusual carbohydrates with C2-branched 4-amino sugar units and related carbohydrate mimetics [3-7]. Moreover, pursuing a similar strategy, we described the stereocontrolled preparation of 3,6-dihydro-2H-pyrans D as precursors for the synthesis of enantiopure aminopyrans E (Scheme 1). The sequence of nucleophilic addition of lithiated enol ethers A to nitrones B and Lewis acid promoted cyclization of the resulting 1,3-dioxolanyl-substituted hydroxylamine derivatives C, delivered the enantiopure dihydropyrans D in a highly stereodivergent fashion [8]. In this report we disclose full details about this new method, including expansion of the reaction scope and further evaluation of the synthetic potential of the easily accessible functionalized dihydropyrans D.
![[1860-5397-6-75-i1]](/bjoc/content/inline/1860-5397-6-75-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 3,6-dihydro-2H-pyrans D.
Scheme 1: Synthesis of 3,6-dihydro-2H-pyrans D.
Results and Discussion
The addition of organometallic species to nitrones has been extensively studied in the past. Highly stereoselective reactions of enantiopure nitrones allow rapid access to functionalized intermediates that have been elegantly used for the preparation of a wide range of natural products [9]. It is known that pre-complexation of carbohydrate-derived nitrones by Lewis acids can reverse the stereochemical outcome of these reactions [10] allowing, for example, a stereodivergent access to enantiopure 1,2-oxazines [11].
When lithiated enol ethers 1 were used as the organometallic species in the addition to nitrones 2 [12-14], either syn- or anti-configured hydroxylamine derivatives 3 were obtained, depending strongly on the reaction conditions (Table 1) [15,16]. Subsequent treatment under basic conditions with tert-butyldimethylsilyl triflate afforded TBS-protected compounds syn- and anti-4 in good overall yields [17]. The stereochemical outcome of the reaction is in line with the addition of other nucleophiles to nitrone 2a [9-11]. The configurations of compounds 4 could be unambiguously assigned by X-ray crystallographic analysis of an aminopyran derivative derived from anti-4b (compound 29, Scheme 12) [8]. The enol ethers used, i.e., ethyl vinyl ether, dihydropyran, and dihydrofuran, all added smoothly to glyceraldehyde-derived nitrones 2a or 2b after lithiation with tert-butyllithium. Whereas in the reaction with uncomplexed nitrones, 1.5 equivalents of the respective lithiated enol ether were sufficient to obtain the desired syn-products with good yields and stereoselectivities, 5 equivalents of the nucleophile were necessary to achieve reasonable yields in the addition to the nitrone/chlorodiethylaluminium complexes. Despite this, diastereomeric ratios in favour of the anti-products were generally excellent.
Table 1: Stereodivergent additions of lithiated enol ethers 1 to glyceraldehyde-derived nitrones 2.
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-75-i15.svg?max-width=637&scale=1.0)
|
||||
| Lithiated enol ether | Nitrone | Conditionsa | Product | Yield and diastereomeric ratio |
|---|---|---|---|---|
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-75-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-6-75-i17.svg?max-width=637&scale=1.0)
|
a) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-6-75-i18.svg?max-width=637&scale=1.0)
|
syn: 61%
anti: 9% (d.r. = 88:12) |
| 1a | 2a | b) |
![[Graphic 5]](/bjoc/content/inline/1860-5397-6-75-i19.svg?max-width=637&scale=1.0)
|
syn: 4%
anti: 48% (d.r. = 93:7) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-6-75-i20.svg?max-width=637&scale=1.0)
|
2a | a) |
![[Graphic 7]](/bjoc/content/inline/1860-5397-6-75-i21.svg?max-width=637&scale=1.0)
|
syn: 44%
anti: 18% (d.r. = 72:28) |
| 1b | 2a | b) |
![[Graphic 8]](/bjoc/content/inline/1860-5397-6-75-i22.svg?max-width=637&scale=1.0)
|
syn: 8%
anti: 56% (d.r. = 88:12) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-6-75-i23.svg?max-width=637&scale=1.0)
|
2a | a) |
![[Graphic 10]](/bjoc/content/inline/1860-5397-6-75-i24.svg?max-width=637&scale=1.0)
|
syn: 58%
anti: 10% (d.r. = 86:14) |
| 1c | 2a | b) |
![[Graphic 11]](/bjoc/content/inline/1860-5397-6-75-i25.svg?max-width=637&scale=1.0)
|
syn: 9%
anti: 54% (d.r. = 86:14) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-6-75-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-6-75-i27.svg?max-width=637&scale=1.0)
|
a) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-6-75-i28.svg?max-width=637&scale=1.0)
|
syn: 70%
(d.r. > 95:5) |
aCondition a): 1. THF, −78 °C, 1 h (1.5 equiv of enol ether); 2. TBSOTf, 2,6-lutidine, 30 min; Condition b): 1. 2 + Et2AlCl, THF, 5 min, then addition to 1, THF, −78 °C, 15 min (5 equiv of enol ether); 2. TBSOTf, 2,6-lutidine, 30 min.
Lithiated ethyl vinyl ether was also added to pre-complexed D-arabinose-derived nitrone 2c (Scheme 2). After a subsequent TBS-protection step, the desired hydroxylamine derivative anti-4e was obtained in low yield, but with excellent diastereoselectivity.
![[1860-5397-6-75-i2]](/bjoc/content/inline/1860-5397-6-75-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Addition of lithiated enol ether 1a to nitrone 2c/Et2AlCl.
Scheme 2: Addition of lithiated enol ether 1a to nitrone 2c/Et2AlCl.
Next, the protected hydroxylamine derivatives syn-4 and anti-4 were treated with Lewis acid to provide enantiopure 3,6-dihydro-2H-pyrans cis- and trans-5 (Table 2). The cyclization proceeded in moderate to excellent yields when two equivalents of trimethylsilyl triflate were used as Lewis acid. Catalytic amounts of Lewis acid were not sufficient to obtain the dihydropyrans, even after prolonged reaction times. The reactions with stoichiometric amounts of TMSOTf gave monocyclic, bicyclic, and spirocyclic dihydropyrans depending on the 1,3-dioxolanyl-substituted enol ether employed (Table 2). Unfortunately, attempts to generate an eight-membered (or six-membered) ring by cyclization of anti-4e were unsuccessful, only decomposition was observed.
![[Graphic 15]](/bjoc/content/inline/1860-5397-6-75-i29.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-6-75-i30.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-6-75-i31.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-6-75-i32.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-6-75-i33.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-6-75-i34.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-6-75-i35.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-6-75-i36.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-6-75-i37.svg?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-6-75-i38.svg?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-6-75-i39.svg?max-width=637&scale=1.0)
![[Graphic 26]](/bjoc/content/inline/1860-5397-6-75-i40.svg?max-width=637&scale=1.0)
![[Graphic 27]](/bjoc/content/inline/1860-5397-6-75-i41.svg?max-width=637&scale=1.0)
![[Graphic 28]](/bjoc/content/inline/1860-5397-6-75-i42.svg?max-width=637&scale=1.0)
![[Graphic 29]](/bjoc/content/inline/1860-5397-6-75-i43.svg?max-width=637&scale=1.0)
![[Graphic 30]](/bjoc/content/inline/1860-5397-6-75-i44.svg?max-width=637&scale=1.0)
![[Graphic 31]](/bjoc/content/inline/1860-5397-6-75-i45.svg?max-width=637&scale=1.0)
As a mechanistic rationale, shown below for the transformation of syn-4a into cis-5a, we propose that the Lewis acid coordinates to the distal oxygen atom of the dioxolane moiety in 4 leading to a ring opening and formation of a stabilized carbenium ion 6 (Scheme 3). Subsequent attack of the oxocarbenium ion on the enol ether moiety leads to ring closure affording the cationic pyran intermediate 7. Subsequent proton transfer to the (moderately basic) hydroxylamine nitrogen re-establishes the enol ether moiety. During aqueous work-up the OTMS-group is apparently removed due to the fairly acidic conditions to produce cis-5a. Overall, the described cyclization can be classified as an aldol-type reaction of an enol ether to an acetal, or as a Prins-type reaction.
![[1860-5397-6-75-i3]](/bjoc/content/inline/1860-5397-6-75-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanistic proposal for the transformation of syn-4a into cis-5a in the presence of TMSOTf.
Scheme 3: Mechanistic proposal for the transformation of syn-4a into cis-5a in the presence of TMSOTf.
Encouraged by these results, we decided to try a related Friedel–Crafts-type cyclization. Thus, 2-lithiofuran was added to nitrone 2a [18] and then protected with TBSOTf to give hydroxylamine derivative syn-4f in 55% (Scheme 4). Remarkably, subsequent treatment with 2 equiv of TMSOTf did not lead to the desired bicyclic product. Since no conversion was observed, the lower reactivity of syn-4f, caused by the considerably weaker nucleophilicity of the furan compared to an enol ether [19], might in the future be overcome with more forcing reaction conditions.
![[1860-5397-6-75-i4]](/bjoc/content/inline/1860-5397-6-75-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Unsuccessful attempt to cyclize hydroxylamine derivative syn-4f.
Scheme 4: Unsuccessful attempt to cyclize hydroxylamine derivative syn-4f.
The enol ether moiety in dihydropyrans 5 represents a very versatile handle for further synthetic modifications (Scheme 5 and Scheme 6). Starting from cis-5a, acidic hydrolysis with concurrent loss of the TBS-group by treatment with saturated methanolic HCl followed by the addition of a saturated solution of NaHCO3 in water gave ketone 8 in good yield (Scheme 5 and Scheme 6). Dihydroxylation using the K2OsO4/N-methylmorpholinoxide-system afforded α-hydroxyketone 9 via in situ hydrolysis of an initially formed hemiacetal. Bromination with NBS in MeCN/H2O led to α-bromoketone 10 and its desilylated derivative 11 as single diastereomers. We presume that the NBS attacks the enol ether moiety, as is also the case with the dihydroxylation reagent OsO4, from the sterically less hindered side, leading to incorporation of the bromo substituent trans to the hydroxymethyl and the bulky hydroxylamine moiety. The partial removal of the TBS-group is undoubtedly caused by HBr formed in the course of the reaction.
![[1860-5397-6-75-i5]](/bjoc/content/inline/1860-5397-6-75-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Functionalizations of the enol ether moiety of cis-5a leading to compounds 8–11.
Scheme 5: Functionalizations of the enol ether moiety of cis-5a leading to compounds 8–11.
When these reaction conditions were applied to the diastereomeric dihydropyran trans-5a, different behaviour was noted (Scheme 6). A smooth hydrolysis of the enol ether moiety leading to ketone 12 was neither feasible with methanolic HCl nor with aqueous HCl. After dihydroxylation, a partial hydrolysis of the hemiacetal initially formed in the dihydroxylation was observed leading to an inseparable mixture of compounds 13 and 14. The TBS-protection group was completely removed during bromination of the enol ether moiety with the formation of compound 15 in good yield [20].
![[1860-5397-6-75-i6]](/bjoc/content/inline/1860-5397-6-75-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Transformations of the enol ether moiety of trans-5a leading to products 13–15.
Scheme 6: Transformations of the enol ether moiety of trans-5a leading to products 13–15.
The internal enol ether moiety of the bicyclic compound trans-5b could also be converted into the corresponding α-bromoketone with the bromination reagent NBS to give the ring opened product 16 (Scheme 7). The two possible diastereomers were formed in a ratio of 92:8, as indicated by 1H NMR spectroscopy and have not been separated.
![[1860-5397-6-75-i7]](/bjoc/content/inline/1860-5397-6-75-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Bromination of bicyclic dihydropyran trans-5b affording 16.
Scheme 7: Bromination of bicyclic dihydropyran trans-5b affording 16.
Bromination of the cyclohexylidene-substituted dihydropyran cis-5d afforded α-bromo ketone 17 which, upon treatment with NaBH4, produced epoxypyran 18 (Scheme 8). Epoxide 18 was obtained as a single diastereomer and should be a promising substrate for subsequent nucleophilic addition reactions leading to another series of enantiopure aminopyrans.
![[1860-5397-6-75-i8]](/bjoc/content/inline/1860-5397-6-75-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of epoxypyran 18 by bromination of cis-5d.
Scheme 8: Synthesis of epoxypyran 18 by bromination of cis-5d.
An additional option for the use of the enol ether moiety is oxidative cleavage [21]. Thus, benzyl protection of compound cis-5b followed by treatment of the resulting 19 with RuCl3 and NaIO4 gave the ten-membered ring lactone 20 by selective cleavage of the internal double bond (Scheme 9).
![[1860-5397-6-75-i9]](/bjoc/content/inline/1860-5397-6-75-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Oxidative cleavage of dihydropyran 19 to lactone 20.
Scheme 9: Oxidative cleavage of dihydropyran 19 to lactone 20.
With the objective of preparing a diaminopyran, we attempted to substitute the bromide atom in compound 15 with the azido group. Surprisingly, instead of the desired α-azido ketone, the formation of nitrone 21 was observed, which apparently involves an unusual internal redox reaction (Scheme 10). Sodium azide presumably acts as base to initiate the reaction by deprotonation of the hydroxylamine moiety. A hydride shift from the benzylic position to the 3-position of the pyran ring produces the nitrone moiety of 21 and simultaneously displaces the axially positioned bromo substituent by an intramolecular substitution. This mechanistic hypothesis was supported by the fact that heating 15 in DMF in the absence of NaN3 did not lead to conversion of the substrate.
![[1860-5397-6-75-i10]](/bjoc/content/inline/1860-5397-6-75-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Transformation of α-bromoketone 15 into nitrone 21 by an internal redox reaction.
Scheme 10: Transformation of α-bromoketone 15 into nitrone 21 by an internal redox reaction.
When the diastereomeric α-bromoketone 11 was reacted with sodium azide, the identical nitrone 21 was obtained in moderate yield (Scheme 11). We presume that an initially formed nitrone 22 probably undergoes base induced epimerization to generate the thermodynamically more stable trans-substituted nitrone 21. As shown below, the nitrone moiety of 21 can be regarded as a masked hydroxylamine, however, it should also be very useful for further diversification of our pyran derivatives, e.g., by 1,3-dipolar cycloaddition leading to isoxazole derivatives [22] or, by addition of nucleophiles to this electrophilic unit [9].
![[1860-5397-6-75-i11]](/bjoc/content/inline/1860-5397-6-75-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Transformation of α-bromoketone 11 into nitrone 21.
Scheme 11: Transformation of α-bromoketone 11 into nitrone 21.
Compounds 8 and 21 proved to be excellent precursors for the stereoselective preparation of enantiopure aminopyrans with high structural similarities to carbohydrates. Reduction of the carbonyl group of 8 by NaBH4 followed by hydrogenolysis of the N,O- and N-benzyl-bonds furnished aminopyran 24 (Scheme 12). Similarly, nitrone 21 was smoothly converted to the diastereomeric aminopyran 26 by analogous reductive transformations.
![[1860-5397-6-75-i12]](/bjoc/content/inline/1860-5397-6-75-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of diastereomeric aminopyrans 24 and 26.
Scheme 12: Synthesis of diastereomeric aminopyrans 24 and 26.
α-Hydroxyketones 9 and 13 were used as precursors for the preparation of compounds with an additional hydroxyl group, such as aminopyrans 28 and 30 (Scheme 13). Reduction of 9 with NaBH4 in the presence of CeCl3 and subsequent hydrogenolysis furnished compound 28 in excellent yield. The presence of CeCl3 during the reduction of the carbonyl group proved to be essential to achieve excellent diastereoselectivity. The mixture of compounds 13 and 14, which was formed during dihydroxylation of dihydropyran cis-5a (Scheme 6), was also treated with NaBH4 to afford the diastereomeric compound 29 [23]. A final hydrogenation step gave aminopyran 30 in high overall yield. Aminopyrans 24, 26, 28 and 30 can be regarded as mimetics of differently configured (2-deoxy) 4-amino sugars [24-26].
![[1860-5397-6-75-i13]](/bjoc/content/inline/1860-5397-6-75-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of diastereomeric aminopyrans 28 and 30.
Scheme 13: Synthesis of diastereomeric aminopyrans 28 and 30.
Aminopyrans 24 and 28 have already been linked via amide bonds to the thiol shell of gold nanoparticles (particle diameter 6 nm). After sulfation of the hydroxyl groups the multivalent conjugates obtained displayed strong binding to P- and L-selectins, thus demonstrating that compounds such as 24 and 28 are of interest for the development of new anti-inflammatory agents [27,28]. Inspired by these first findings, we decided to prepare also smaller conjugates for evaluation as selectin ligands. Thus, the free hydroxyl groups of aminopyrans 24 and 28 were temporarily protected as TMS ethers in order to achieve smooth reaction with 1,3,5-benzenetricarboxylic acid chloride (Scheme 14) [29]. After successful conversion to the triamide, the TMS-groups were removed by treatment with trifluoroacetic acid to give the desired compounds 31 and 32. The low yield of 32 was due to difficulties encountered in the purification of the compound which was carried out by crystallization.
![[1860-5397-6-75-i14]](/bjoc/content/inline/1860-5397-6-75-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis of triamides 31 and 32.
Scheme 14: Synthesis of triamides 31 and 32.
Conclusion
We have presented an approach to novel 3,6-dihydro-2H-pyrans that can be transformed into a series of new carbohydrate mimetics and into other enantiopure heterocycles. The stereodivergent addition of lithiated enol ethers to carbohydrate-derived nitrones produced 1,3-dioxolanyl-substituted hydroxylamine derivatives as suitable substrates for Lewis-acid induced cyclizations furnishing mono-, di-, or spirocyclic dihydropyrans. Subsequent reactions such as bromination, acidic hydrolysis or dihydroxylation provided differently substituted and configured pyranone derivatives as precursors for the stereoselective synthesis of aminopyran derivatives that can be regarded as carbohydrate mimetics. Trimeric versions of these carbohydrate mimetics were constructed via their attachment to a tricarboxylic acid core to produce novel polyhydroxylated compounds that will be evaluated as potential selectin binders at a future date.
Experimental
Addition of lithiated enol ethers to glyceraldehyde-derived nitrones without precomplexation of the nitrone
The respective enol ether (1.5 equiv) was dissolved in THF (2 mL/mmol of enol ether) and cooled to −78 °C. tBuLi (1.6 M in pentane, 1.5 equiv) was added and the reaction mixture stirred for 1 h during which time it was allowed to warm to 0 °C. After further stirring for 1 h at this temperature, it was cooled once more to −78 °C. A solution of the respective nitrone (1 equiv) in THF (0.7 mL/mmol nitrone) was added dropwise over a 15 min period. The mixture was stirred at this temperature for 1 h and the reaction quenched by the addition of H2O. After the mixture reached room temperature it was extracted three times with Et2O. The combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo. The crude product (1 equiv) was dissolved in CH2Cl2 (2.5 mL/mmol), and 2,6-lutidine (2 equiv) and TBSOTf (1.5 equiv) were added slowly at 0 °C. The mixture was stirred at room temperature for 30 min and then the reaction was quenched by the addition of a sat. NH4Cl solution. The layers were separated and the aqueous phase was extracted three times with CH2Cl2. The combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo. Purification by column chromatography (silica gel, hexane/EtOAc = 20:1) yielded the products as colourless oils.
Addition of lithiated enol ethers to glyceraldehyde-derived nitrones precomplexated by Et2AlCl
The respective enol ether (5 equiv) was dissolved in THF (2 mL/mmol of enol ether) and cooled to −78 °C. tBuLi (1.6 M in pentane, 5 equiv) was added and the reaction mixture stirred for 1 h during which time it was allowed to warm to 0 °C. After further stirring for 3 h at this temperature, it was cooled once more to −78 °C. In a separate flask, a solution of nitrone 2 (1 equiv) in THF (2.5 mL) was treated with Et2AlCl (1 M in hexane, 1 equiv) for 5 min. The prepared solution was added dropwise to the solution of the lithiated enol ether over a 15 min period. The mixture was stirred at this temperature for a further 15 min and the reaction quenched by addition of 2 M NaOH solution. After the mixture reached room temperature it was extracted three times with Et2O. The combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo. The crude product (1 equiv) was dissolved in CH2Cl2 (2.5 mL/mmol), and 2,6-lutidine (2 equiv) and TBSOTf (1.5 equiv) were added slowly at 0 °C. The mixture was stirred at room temperature for 30 min and then the reaction was quenched by the addition of a sat. NH4Cl solution. The layers were separated and the aqueous phase was extracted three times with CH2Cl2. The combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo. Purification by column chromatography (silica gel, hexane/EtOAc = 20:1) yielded the products as colourless oils.
Lewis acid-induced cyclization of 1,3-dioxolanyl-substituted enol ethers
To a solution of the respective 1,3-dioxolanyl-substituted enol ether (1 equiv) in CH2Cl2 (6 mL/mmol) at −30 °C, was added TMSOTf (2 equiv) and the resulting solution stirred until it slowly reached room temperature (6 h). The reaction was quenched by the addition of water. After separation of the layers, the aqueous phase was extracted three times with CH2Cl2. The combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo. Purification by column chromatography (silica gel, hexane/EtOAc = 6:1) yielded the products as colourless oils.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, 1H NMR and 13C NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 3.1 MB | Download |
Acknowledgements
Generous support of this work by the Deutsche Forschungsgemeinschaft (SFB 765), the Fonds der Chemischen Industrie (PhD fellowship to FP) and the Bayer-Schering Pharma AG is most gratefully acknowledged. We thank O. Reimann for his experimental contributions and Dr. R. Zimmer for help during the preparation of the manuscript.
References
-
Larrosa, I.; Romea, P.; Urpí, F. Tetrahedron 2008, 64, 2683–2723. doi:10.1016/j.tet.2007.11.092
Return to citation in text: [1] -
Clarke, P. A.; Santos, S. Eur. J. Org. Chem. 2006, 2045–2053. doi:10.1002/ejoc.200500964
Return to citation in text: [1] -
Pfrengle, F.; Lentz, D.; Reissig, H.-U. Angew. Chem. 2009, 121, 3211–3215. doi:10.1002/ange.200805724
Angew. Chem., Int. Ed. 2009, 48, 3165–3169.
Return to citation in text: [1] -
Pfrengle, F.; Reissig, H.-U. Chem.–Eur. J., in press.
Return to citation in text: [1] -
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227–6231.
Return to citation in text: [1] -
Al-Harrasi, A.; Pfrengle, F.; Prisyashnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996
Return to citation in text: [1] -
Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d
Return to citation in text: [1] -
Pfrengle, F.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 5534–5537. doi:10.1021/ol902354f
Return to citation in text: [1] [2] -
Lombardo, M.; Trombini, C. Synthesis 2000, 759–774. doi:10.1055/s-2000-62
Return to citation in text: [1] [2] [3] -
Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem.–Eur. J. 1995, 1, 505–520. doi:10.1002/chem.19950010804
Return to citation in text: [1] [2] -
Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627
Return to citation in text: [1] [2] -
Nitrones 2a [13] and 2b [14] have been prepared from D-mannitol.
Return to citation in text: [1] -
Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T. Synth. Commun. 1994, 24, 2537–2550. doi:10.1080/00397919408010565
Return to citation in text: [1] [2] -
Merino, P.; Franco, S.; Merchán, F. L.; Revuelta, J.; Tejero, T. Tetrahedron Lett. 2002, 43, 459–462. doi:10.1016/S0040-4039(01)02191-8
Return to citation in text: [1] [2] -
Friesen, R. W.; Sturino, C. F. Sci. Synth. 2005, 8, 841–862.
Lithiation of the enol ether was carried out under standard conditions using t-BuLi in THF:.
Return to citation in text: [1] -
Braun, M. Angew. Chem. 1998, 110, 444–465. doi:10.1002/(SICI)1521-3757(19980216)110:4<444::AID-ANGE444>3.0.CO;2-J
Angew. Chem., Int. Ed. 1998, 37, 430–451.
Review on 1-alkenyllithium compounds bearing an α-heteroatom.
Return to citation in text: [1] -
Merino, P.; Tejero, T.; Mannucci, V.; Prestat, G.; Madec, D.; Poli, G. Synlett 2007, 944–948. doi:10.1055/s-2007-973868
Return to citation in text: [1] -
Dondoni, A.; Franco, S.; Merchán, F. L.; Merino, P.; Tejero, T. Tetrahedron Lett. 1993, 34, 5479–5482. doi:10.1016/S0040-4039(00)73939-6
Return to citation in text: [1] -
Mayr, H.; Kempf, B.; Ofial, A. R. Acc. Chem. Res. 2003, 36, 66–77. doi:10.1021/ar020094c
Return to citation in text: [1] -
We assume that NBS and OsO4 attack the enol ether moiety from the sterically less hindered side leading to compounds 13–15. The configuration of compound 15 was unambiguously proven at a later stage (see [23]).
Return to citation in text: [1] -
Silva, L. F., Jr.; Vasconcelos, R. S.; Nogueira, M. A. Org. Lett. 2008, 10, 1017–1020. doi:10.1021/ol800048f
Return to citation in text: [1] -
Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem.–Eur. J. 2009, 15, 7808–7821. doi:10.1002/chem.200900707
Return to citation in text: [1] -
The configuration of compound 29 was proven by X-ray crystallographic analysis (see [8]).
Return to citation in text: [1] [2] -
Chapleur, Y. Carbohydrate Mimics – Concepts and Methods; WILEY-VCH: Weinheim, 1998. doi:10.1002/3527603239
Return to citation in text: [1] -
Sears, P.; Wong, C.-H. Angew. Chem. 1999, 111, 2446–2471. doi:10.1002/(SICI)1521-3757(19990816)111:16<2446::AID-ANGE2446>3.0.CO;2-4
Angew. Chem., Int. Ed. 1999, 38, 2301–2324.
Return to citation in text: [1] -
Blanco, J. L. J.; Ortega-Caballero, F.; Mellet, C. O.; Fernández, J. M. G. Beilstein J. Org. Chem. 2010, 6, No. 20. doi:10.3762/bjoc.6.20
Return to citation in text: [1] -
Roskamp, M. Ph.D. Thesis, Freie Universität Berlin, Germany, 2010.
Return to citation in text: [1] -
Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a
for previous results.
Return to citation in text: [1] -
Irmak, M.; Groschner, A.; Boysen, M. M. K. Chem. Commun. 2007, 177–179. doi:10.1039/b612986b
Return to citation in text: [1]
| 1. | Larrosa, I.; Romea, P.; Urpí, F. Tetrahedron 2008, 64, 2683–2723. doi:10.1016/j.tet.2007.11.092 |
| 2. | Clarke, P. A.; Santos, S. Eur. J. Org. Chem. 2006, 2045–2053. doi:10.1002/ejoc.200500964 |
| 10. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem.–Eur. J. 1995, 1, 505–520. doi:10.1002/chem.19950010804 |
| 21. | Silva, L. F., Jr.; Vasconcelos, R. S.; Nogueira, M. A. Org. Lett. 2008, 10, 1017–1020. doi:10.1021/ol800048f |
| 22. | Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem.–Eur. J. 2009, 15, 7808–7821. doi:10.1002/chem.200900707 |
| 8. | Pfrengle, F.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 5534–5537. doi:10.1021/ol902354f |
| 19. | Mayr, H.; Kempf, B.; Ofial, A. R. Acc. Chem. Res. 2003, 36, 66–77. doi:10.1021/ar020094c |
| 3. |
Pfrengle, F.; Lentz, D.; Reissig, H.-U. Angew. Chem. 2009, 121, 3211–3215. doi:10.1002/ange.200805724
Angew. Chem., Int. Ed. 2009, 48, 3165–3169. |
| 4. | Pfrengle, F.; Reissig, H.-U. Chem.–Eur. J., in press. |
| 5. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227–6231. |
| 6. | Al-Harrasi, A.; Pfrengle, F.; Prisyashnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996 |
| 7. | Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d |
| 20. | We assume that NBS and OsO4 attack the enol ether moiety from the sterically less hindered side leading to compounds 13–15. The configuration of compound 15 was unambiguously proven at a later stage (see [23]). |
| 17. | Merino, P.; Tejero, T.; Mannucci, V.; Prestat, G.; Madec, D.; Poli, G. Synlett 2007, 944–948. doi:10.1055/s-2007-973868 |
| 8. | Pfrengle, F.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 5534–5537. doi:10.1021/ol902354f |
| 15. |
Friesen, R. W.; Sturino, C. F. Sci. Synth. 2005, 8, 841–862.
Lithiation of the enol ether was carried out under standard conditions using t-BuLi in THF:. |
| 16. |
Braun, M. Angew. Chem. 1998, 110, 444–465. doi:10.1002/(SICI)1521-3757(19980216)110:4<444::AID-ANGE444>3.0.CO;2-J
Angew. Chem., Int. Ed. 1998, 37, 430–451. Review on 1-alkenyllithium compounds bearing an α-heteroatom. |
| 18. | Dondoni, A.; Franco, S.; Merchán, F. L.; Merino, P.; Tejero, T. Tetrahedron Lett. 1993, 34, 5479–5482. doi:10.1016/S0040-4039(00)73939-6 |
| 12. | Nitrones 2a [13] and 2b [14] have been prepared from D-mannitol. |
| 13. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T. Synth. Commun. 1994, 24, 2537–2550. doi:10.1080/00397919408010565 |
| 14. | Merino, P.; Franco, S.; Merchán, F. L.; Revuelta, J.; Tejero, T. Tetrahedron Lett. 2002, 43, 459–462. doi:10.1016/S0040-4039(01)02191-8 |
| 11. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 9. | Lombardo, M.; Trombini, C. Synthesis 2000, 759–774. doi:10.1055/s-2000-62 |
| 10. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem.–Eur. J. 1995, 1, 505–520. doi:10.1002/chem.19950010804 |
| 11. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 24. | Chapleur, Y. Carbohydrate Mimics – Concepts and Methods; WILEY-VCH: Weinheim, 1998. doi:10.1002/3527603239 |
| 25. |
Sears, P.; Wong, C.-H. Angew. Chem. 1999, 111, 2446–2471. doi:10.1002/(SICI)1521-3757(19990816)111:16<2446::AID-ANGE2446>3.0.CO;2-4
Angew. Chem., Int. Ed. 1999, 38, 2301–2324. |
| 26. | Blanco, J. L. J.; Ortega-Caballero, F.; Mellet, C. O.; Fernández, J. M. G. Beilstein J. Org. Chem. 2010, 6, No. 20. doi:10.3762/bjoc.6.20 |
| 23. | The configuration of compound 29 was proven by X-ray crystallographic analysis (see [8]). |
| 23. | The configuration of compound 29 was proven by X-ray crystallographic analysis (see [8]). |
| 8. | Pfrengle, F.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 5534–5537. doi:10.1021/ol902354f |
| 13. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T. Synth. Commun. 1994, 24, 2537–2550. doi:10.1080/00397919408010565 |
| 14. | Merino, P.; Franco, S.; Merchán, F. L.; Revuelta, J.; Tejero, T. Tetrahedron Lett. 2002, 43, 459–462. doi:10.1016/S0040-4039(01)02191-8 |
| 27. | Roskamp, M. Ph.D. Thesis, Freie Universität Berlin, Germany, 2010. |
| 28. |
Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a
for previous results. |
| 29. | Irmak, M.; Groschner, A.; Boysen, M. M. K. Chem. Commun. 2007, 177–179. doi:10.1039/b612986b |
© 2010 Pfrengle and Reissig; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)