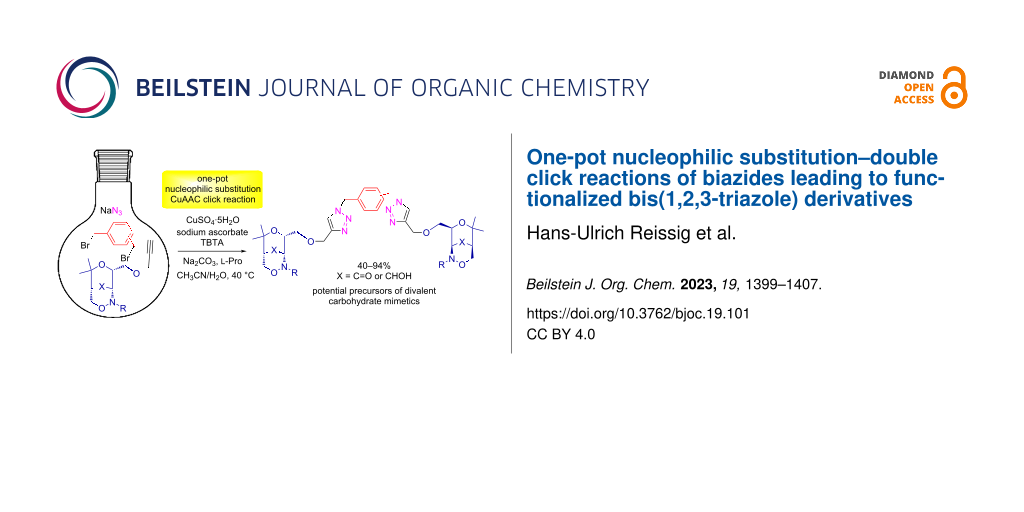
Abstract
The nucleophilic substitution of benzylic bromides with sodium azide was combined with a subsequent copper-catalyzed (3 + 2) cycloaddition with terminal alkynes. This one-pot process was developed with a simple model alkyne, but then applied to more complex alkynes bearing enantiopure 1,2-oxazinyl substituents. Hence, the precursor compounds 1,2-, 1,3- or 1,4-bis(bromomethyl)benzene furnished geometrically differing bis(1,2,3-triazole) derivatives. The use of tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) as ligand for the click step turned out to be very advantageous. The compounds with 1,2-oxazinyl end groups can potentially serve as precursors of divalent carbohydrate mimetics, but the reductive cleavage of the 1,2-oxazine rings to aminopyran moieties did not proceed cleanly with these compounds.

Graphical Abstract
Introduction
The concept of click reactions [1,2], in particular, the discovery of the copper-catalyzed alkyne azide (3 + 2) cycloaddition (CuAAC) [3,4], has dramatically changed the approaches to many problems in chemistry, supramolecular chemistry, materials science, biological chemistry and related fields (selected reviews: [5-15]). Mechanistic aspects of the CuAAC have been studied in detail [16,17]. Whereas the traditional 1,3-dipolar cycloaddition (Huisgen reaction) [18-20] of azides and alkynes requires often – but not always – relatively harsh conditions and proceeds with moderate regioselectivity only [21], the copper-catalyzed version can generally be executed at room temperature and it affords exclusively 1,4-disubstituted 1,2,3-triazole derivatives, thus allowing a controlled and highly efficient connection of a variety of molecular building blocks. This “Lego-approach” found countless applications and the bestowal of the Nobel Prize in 2022 to M. Meldal, K. B. Sharpless and C. R. Bertozzi did not come as a surprise.
In most cases the (3 + 2) cycloadditions were performed with isolated (and purified) organic azides, but it was early found that one-pot processes generating the potentially hazardous azides [22] in situ are possible [23]. Later, examples were published showing that these methods are also compatible with the conditions of CuAAC. The earliest case was probably published by Fokin et al. [24,25], one of the inventors of the original copper-catalyzed (3 + 2) cycloaddition. Many examples of nucleophilic substitutions employing sodium azide and organic substrates with potential leaving groups have been reported. The resulting organic azides were trapped in situ by a suitable alkyne to give the 1,2,3-triazoles [26-36]. Fairly recent review articles summarize these results [37,38].
For several years, our group was interested in preparing multivalent carbohydrate mimetics [39-43] on the basis of efficient coupling reactions of aminopyran and aminooxepane derivatives with suitable linker elements. Hence, the aminopyran derivatives A could be converted into several multivalent compounds B by amine or amide bond formations [44-47]. The transformation of the corresponding azidopyrans and azidooxepanes C or E into multivalent 1,2,3-triazole derivatives D and F by Meldal–Sharpless cycloadditions with suitable alkynes proceeded generally in good yields and furnished another set of multivalent carbohydrate mimetics [48-50]. In the current report, we want to disclose our experience with an “inverted” approach to multivalent systems [51]: bicyclic 1,2-oxazine derivatives of type G [52,53], which can be regarded as internally protected aminopyrans [54], should be converted into divalent compounds via coupling of the terminal propynyl group with benzylic biazides. Since biazides are potentially explosive [22] it was very desirable to avoid their isolation and to generate these reactive species in situ from the corresponding benzylic halides. In this study we investigated the compatibility of the nucleophilic substitution of 1,2-, 1,3- or 1,4-bis(bromomethyl)benzene H with sodium azide and the copper-catalyzed alkyne–azide cycloadditions with compounds of type G to provide divalent compounds I (Scheme 1). These may serve as precursors of divalent carbohydrate mimetics with aminopyran end groups.
![[1860-5397-19-101-i1]](/bjoc/content/inline/1860-5397-19-101-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Earlier approaches to multivalent carbohydrate mimetics B, D or F based on enantiopure aminopyran and aminooxepane derivatives and goal of this study employing alkyne component G and biazides in situ-generated from H.
Scheme 1: Earlier approaches to multivalent carbohydrate mimetics B, D or F based on enantiopure aminopyran a...
Results and Discussion
We started our investigations by preparing the [2-(trimethylsilyl)ethoxy]methyl-substituted 1,2,3-triazole derivative 3 as simple model compound applying different conditions (Scheme 2). The highest yield for 3 was recorded when benzyl azide (1) and the simple alkyne 2 were combined in the presence of 0.2 equiv of copper(I) iodide in triethylamine as solvent (Scheme 2, reaction 1). After 16 hours at room temperature and chromatographic purification compound 3 was isolated in 79% yield as colorless liquid. Interestingly, under similar conditions, but employing two equivalents of copper(I) iodide in the presence of Hünig’s base [55] in acetonitrile at 40 °C provided 4,4'-bis(1,2,3-triazole) 4 in low yield (Scheme 2, reaction 2). Performing this reaction at room temperature gave a mixture of 3 and 4. The formation of oxidative dimers in minor amounts has already been observed by Sharpless and co-workers in their original report on CuAAC reactions [4] and was systematically investigated by Burgess et al. [56] who found that the base plays a crucial role during the formation of this type of bistriazoles. This dimerization was also discussed in a review article [57] dealing with the various types of bis(1,2,3-triazoles). Since we were not interested in compounds such as 4 we did not further investigate details in order to optimize this process. Instead, we looked at the one-pot nucleophilic substitution to generate benzyl azide 3 in situ from benzyl bromide (5) and sodium azide and to directly trap the intermediate with alkyne 2. Under conditions summarized in reaction 3 of Scheme 2 we obtained the desired 1,2,3-triazole derivative 3 in 82% yield. Copper(II) sulfate pentahydrate (0.07 equivalents based on 2) in the presence of sodium ascorbate as reducing agent and sodium carbonate as base as well as ʟ-proline as ligand in a DMF/water mixture at 60 °C provided this promising result. These conditions applied are similar to those described by Fokin et al. [24], which had also been employed by other groups [37,38].
![[1860-5397-19-101-i2]](/bjoc/content/inline/1860-5397-19-101-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of model compound 3 under conventional conditions and as a one-pot process employing benzyl bromide (5), alkyne 2 and sodium azide as precursors for the click reaction.
Scheme 2: Synthesis of model compound 3 under conventional conditions and as a one-pot process employing benz...
The conditions found were studied with a second model reaction in order to examine its suitability for the synthesis of potential carbohydrate mimetic precursors (Scheme 3). Sodium azide, benzyl bromide (5) and enantiopure bicyclic 1,2-oxazin-4-one derivative 6 [53] which bears a propargylic ether moiety as substituent undergo a one-pot reaction with reasonable efficiency and furnished the expected 1,2,3-triazole derivative 7 in 61% yield under the approved conditions.
![[1860-5397-19-101-i3]](/bjoc/content/inline/1860-5397-19-101-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: One-pot reaction employing enantiopure alkynyl-substituted 1,2-oxazin-4-one derivative 6 leading to 1,2,3-triazole 7.
Scheme 3: One-pot reaction employing enantiopure alkynyl-substituted 1,2-oxazin-4-one derivative 6 leading to...
Next, we turned our attention to the generation of divalent systems, starting with the reactions of 1,3- (8) and 1,2-bis(bromomethyl)benzene (11), respectively, in the presence of sodium azide and alkyne 2 (Scheme 4). The conditions employed above converted the meta-substituted dihalide into the expected symmetric bis(1,2,3-triazole) derivative 9 in very good yield, but we also isolated azide 10 in small quantities, where the intermediate biazide has undergone only one cycloaddition (Scheme 4, reaction 1). Under the same conditions the ortho-dihalide 11 delivered a very similar result, furnishing the expected product 12 in 83% yield. But again, the corresponding azide 13 was obtained in low yield (Scheme 4, reaction 2). In order to improve the process we examined the influence of the ligand tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) which has been identified by Sharpless et al. [58] as a very beneficial component in CuAAC reactions. After comprehensive optimization, we found that the addition of 0.2 equiv of this ligand not only allowed to lower the reaction temperature from 60 °C to 40 °C, but it also induced full consumption of the intermediate biazide derived from dihalide 11 (Scheme 4, reaction 3); 0.2 equiv of copper(II) sulfate pentahydrate, 0.4 equiv of sodium ascorbate and 0.4 equiv of ʟ-proline in very little of acetonitrile/water as solvent furnished the exclusively isolated bis(1,2,3-triazole) derivative 12 in excellent 94% yield. These conditions of the one-pot nucleophilic substitution double-click reaction became the standard reaction conditions and were applied in most of the following experiments. When the reaction of 2 with 11 in the presence of sodium azide was performed under similar conditions, but without TBTA (not shown), 58% of 12 and 12% of 13 were isolated, clearly emphasizing the rate enhancing effect of this ligand on the (3 + 2) cycloaddition step.
![[1860-5397-19-101-i4]](/bjoc/content/inline/1860-5397-19-101-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: One-pot reactions of dihalides 8 and 11 with sodium azide and alkyne 2 leading to symmetric divalent bis(1,2,3-triazoles) 9 and 12 as major products.
Scheme 4: One-pot reactions of dihalides 8 and 11 with sodium azide and alkyne 2 leading to symmetric divalen...
With enantiopure alkynyl-substituted 1,2-oxazin-4-one derivative 6, a potential aminopyran precursor, as alkyne component similar experience was gathered (Scheme 5). This sterically more demanding component reacted slower under the initially examined conditions and provided the desired bis(1,2,3-triazole) 14 and the mono-cycloadduct 15 in almost equal quantities (Scheme 5, reaction 1). Again, the addition of TBTA as ligand strongly improved the performance of the CuAAC reaction: the symmetric divalent compound 14 with two 1,2-oxazinyl end groups was isolated in very satisfying 87% yield (Scheme 5, reaction 2). When 1,4-bis(bromomethyl)benzene (16) was employed as starting material, these conditions afforded the para-substituted bis(1,2,3-triazole) 17 in very good yield although the azidomethyl-substituted compound 18 was also isolated in 6% yield in this case (Scheme 5, reaction 3).
![[1860-5397-19-101-i5]](/bjoc/content/inline/1860-5397-19-101-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: One-pot reactions employing enantiopure alkynyl-substituted 1,2-oxazin-4-one derivative 6 leading to bis(1,2,3-triazoles) 14 and 17 as major products.
Scheme 5: One-pot reactions employing enantiopure alkynyl-substituted 1,2-oxazin-4-one derivative 6 leading t...
Finally, the one-pot click reactions were examined with the alkynyl-substituted 1,2-oxazin-4-ol derivative 19 which is smoothly available from the corresponding ketone 6 by highly stereoselective reduction with sodium borohydride [53]. Starting from 1,3- (8) or 1,4-bis(bromomethyl)benzene (16), respectively, the approved conditions in the presence of TBTA led to the expected bis(1,2,3-triazoles) 20 or 21 in moderate or very good yield (Scheme 6). We cannot decide whether the lower yields in this series are caused by the unprotected hydroxy group of precursor 19 or the corresponding products. Although we did not isolate the conceivable mono-adducts we cannot rigorously exclude their formation.
![[1860-5397-19-101-i6]](/bjoc/content/inline/1860-5397-19-101-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: One-pot reaction employing enantiopure alkynyl-substituted 1,2-oxazin-4-ol derivative 19 leading to bis(1,2,3-triazoles) 20 and 21.
Scheme 6: One-pot reaction employing enantiopure alkynyl-substituted 1,2-oxazin-4-ol derivative 19 leading to...
Compared to bis(1,2,3-triazoles) 14 and 17, compounds 20 and 21 are one step closer to the desired divalent aminopyran-substituted carbohydrate mimetics, since they already contain a free hydroxy group instead of the carbonyl group. However, their reductive transformation into divalent carbohydrate mimetics turned out to be a difficult task. We started the experiments with the hydrogenolysis of bicyclic 1,2-oxazin-4-ol 19 as simple model compound (Scheme 7). A methanol solution of 19 under an atmosphere of hydrogen was stirred for 17 h in the presence of palladium on carbon and provided the expected (propyloxy)methyl-substituted aminopyran 22 in 81% yield (Scheme 7, reaction 1). The reductive removal of the N-benzyl group and the cleavage of the N–O bond occurred apparently without problems. With the second model compound, 1,2,3-triazole 23, which is almost quantitatively available by sodium borohydride reduction of 7, we encountered the first problems. Under similar conditions of the hydrogenolysis the expected product 24 could be isolated as major component and characterized (Scheme 7, reaction 2), but the obtained sample contained unknown byproducts. It is possible, that the N-benzyl group attached to the 1,2,3-triazole moiety is partially removed under these conditions and/or that even the C–O bond connecting the 1,2,3-triazole part with the aminopyran part is reductively cleaved since this bond also has benzylic character. In earlier investigations with other triazolyl-substituted aminopyran derivatives we encountered similar selectivity problems due to the sensitivity of benzylic bonds to the applied hydrogenolysis conditions [59].
![[1860-5397-19-101-i7]](/bjoc/content/inline/1860-5397-19-101-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reductive ring-openings of 1,2-oxazine derivatives 19 and 23 as simple model compounds by hydrogenolysis in the presence of palladium on carbon leading to aminopyran derivatives 22 and 24.
Scheme 7: Reductive ring-openings of 1,2-oxazine derivatives 19 and 23 as simple model compounds by hydrogeno...
Despite of the discouraging results with model compound 23 we nevertheless examined the reductive cleavage of bis(1,2,3-triazole) 21. It turned out that the hydrogenolysis of this compound was very capricious and (in part) depended on the batch of palladium on carbon used. In most cases, incomplete consumption of starting material was observed, even after long reaction time. The best result is depicted in Scheme 8 (reaction 1), when after five days of hydrogenolysis and 0.8 equivalents of palladium at least 40% of impure divalent aminopyran derivative 25 was isolated. The major component of the isolated material is certainly fitting to the proposed structure according to the NMR data and their comparison with related compounds, but the sample again contains unknown impurities, which are probably due to additional bond cleavage events. Compound 25 contains four bonds of heteroatoms to carbon atoms which have benzylic character and are possibly attacked under the reaction conditions, in particular, considering the long reaction time and the fairly high amount of catalyst employed.
![[1860-5397-19-101-i8]](/bjoc/content/inline/1860-5397-19-101-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Attempted reductive ring-openings of compound 21 by hydrogenolysis or by samarium diiodide leading to impure samples of divalent aminopyran derivatives 25 or 26.
Scheme 8: Attempted reductive ring-openings of compound 21 by hydrogenolysis or by samarium diiodide leading ...
As an alternative method, which should be more chemoselective, we examined the reduction with samarium diiodide [60]. This versatile one-electron transfer reagent is known to cleave N–O bonds with high selectivity [61,62] and was applied several times by our group with good success for reductive ring-openings of 1,2-oxazine derivatives [63,64]. A slight excess of samarium diiodide in tetrahydrofuran completely consumed compound 21 within five hours. Compound 26 was isolated in 50% yield, but again the sample was not clean. The NMR data revealed that the N-benzyl groups were still intact, however, we cannot exclude that partial reductions had occurred at other positions of this relatively complex molecule. Thus, better conditions for the clean transformation of compounds such as 21 into carbohydrate mimetics have still to be developed. We abstained from the conversion of compounds 24, 25 or 26 into their sulfated form due to the presence of the impurities in these samples. However, only the sulfated forms of this type of multivalent carbohydrate mimetics had earlier shown respectable biological activity as ligands of P- and L-selectins [44-50].
Conclusion
We found very good conditions for a one-pot nucleophilic substitution of benzylic bromides with sodium azide and direct subsequent CuAAC reactions with alkynes. The conditions were tested with the simple alkyne model compound 2, but could be applied to more complex alkynes such as 1,2-oxazin-4-one derivative 6 or 1,2-oxazin-4-ol derivative 19. With 1,2-, 1,3- or 1,4-bis(bromomethyl)benzene as precursors bis(1,2,3-triazole) derivatives were isolated in good to excellent yields. The use of TBTA as ligand for the click step turned out to be very advantageous. The reductive ring openings of bis(1,2,3-triazole) derivative 21 to divalent carbohydrate mimetics with hydrogen under palladium catalysis or with samarium diiodide did not proceed cleanly and need further optimization.
Supporting Information
| Supporting Information File 1: Experimental procedures, spectroscopic and analytical characterization data of new compounds as well as copies of the NMR spectra. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5
Return to citation in text: [1] -
Rutjes, F. P. J. T. Click Chemistry; Science of Synthesis; Thieme: Stuttgart, Germany, 2022. doi:10.1055/b000000077
Return to citation in text: [1] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4
Return to citation in text: [1] [2] -
Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479
Return to citation in text: [1] -
Hänni, K. D.; Leigh, D. A. Chem. Soc. Rev. 2010, 39, 1240–1251. doi:10.1039/b901974j
Return to citation in text: [1] -
Mamidyala, S. K.; Finn, M. G. Chem. Soc. Rev. 2010, 39, 1252–1261. doi:10.1039/b901969n
Return to citation in text: [1] -
Pedersen, D. S.; Abell, A. Eur. J. Org. Chem. 2011, 2399–2411. doi:10.1002/ejoc.201100157
Return to citation in text: [1] -
Fokin, V. V.; Matyjaszewski, K. CuAAC: The Quintessential Click Reaction. In Organic Chemistry – Breakthroughs and Highlights; Ding, K.; Dai, L.-X., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp 247–277. doi:10.1002/9783527664801.ch7
Return to citation in text: [1] -
Totobenazara, J.; Burke, A. J. Tetrahedron Lett. 2015, 56, 2853–2859. doi:10.1016/j.tetlet.2015.03.136
Return to citation in text: [1] -
Singh, M. S.; Chowdhury, S.; Koley, S. Tetrahedron 2016, 72, 5257–5283. doi:10.1016/j.tet.2016.07.044
Return to citation in text: [1] -
Kacprzak, K.; Skiera, I.; Piasecka, M.; Paryzek, Z. Chem. Rev. 2016, 116, 5689–5743. doi:10.1021/acs.chemrev.5b00302
Return to citation in text: [1] -
Döhler, D.; Michael, P.; Binder, W. H. Acc. Chem. Res. 2017, 50, 2610–2620. doi:10.1021/acs.accounts.7b00371
Return to citation in text: [1] -
Tiwari, V. K.; Mishra, B. B.; Mishra, K. B.; Mishra, N.; Singh, A. S.; Chen, X. Chem. Rev. 2016, 116, 3086–3240. doi:10.1021/acs.chemrev.5b00408
Return to citation in text: [1] -
Santos, C. S.; de Oliveira, R. J.; de Oliveira, R. N.; Freitas, J. C. R. ARKIVOC 2020, No. i, 219–271. doi:10.24820/ark.5550190.p011.293
Return to citation in text: [1] -
Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457–460. doi:10.1126/science.1229506
Return to citation in text: [1] -
Berg, R.; Straub, B. F. Beilstein J. Org. Chem. 2013, 9, 2715–2750. doi:10.3762/bjoc.9.308
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 633–645. doi:10.1002/anie.196306331
Return to citation in text: [1] -
Breugst, M.; Reissig, H.-U. Angew. Chem., Int. Ed. 2020, 59, 12293–12307. doi:10.1002/anie.202003115
Return to citation in text: [1] -
Kirmse, W.; Horner, L. Justus Liebigs Ann. Chem. 1958, 614, 1–3. doi:10.1002/jlac.19586140102
Return to citation in text: [1] -
Bräse, S.; Banert, K., Eds. Organic Azides – Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010. doi:10.1002/9780470682517
Return to citation in text: [1] [2] -
Maksikova, A. V.; Serebryakova, E. S.; Tikhonova, L. G.; Vereshchagin, L. I. Chem. Heterocycl. Compd. 1980, 16, 1284–1285. doi:10.1007/bf00501836
Return to citation in text: [1] -
Feldman, A. K.; Colasson, B.; Fokin, V. V. Org. Lett. 2004, 6, 3897–3899. doi:10.1021/ol048859z
Return to citation in text: [1] [2] -
Appukkuttan, P.; Dehaen, W.; Fokin, V. V.; Van der Eycken, E. Org. Lett. 2004, 6, 4223–4225. doi:10.1021/ol048341v
Return to citation in text: [1] -
Kacprzak, K. Synlett 2005, 943–946. doi:10.1055/s-2005-864809
Return to citation in text: [1] -
Wang, Z.-X.; Zhao, Z.-G. J. Heterocycl. Chem. 2007, 44, 89–92. doi:10.1002/jhet.5570440115
Return to citation in text: [1] -
Odlo, K.; Høydahl, E. A.; Hansen, T. V. Tetrahedron Lett. 2007, 48, 2097–2099. doi:10.1016/j.tetlet.2007.01.130
Return to citation in text: [1] -
Bonnamour, J.; Legros, J.; Crousse, B.; Bonnet-Delpon, D. Tetrahedron Lett. 2007, 48, 8360–8362. doi:10.1016/j.tetlet.2007.09.118
Return to citation in text: [1] -
Fukuzawa, S.-i.; Shimizu, E.; Kikuchi, S. Synlett 2007, 2436–2438. doi:10.1055/s-2007-986638
Return to citation in text: [1] -
Kumar, D.; Patel, G.; Reddy, V. B. Synlett 2009, 399–402. doi:10.1055/s-0028-1087556
Return to citation in text: [1] -
Huang, Y.; Gard, G. L.; Shreeve, J. M. Tetrahedron Lett. 2010, 51, 6951–6954. doi:10.1016/j.tetlet.2010.10.149
Return to citation in text: [1] -
Stefani, H. A.; Canduzini, H. A.; Manarin, F. Tetrahedron Lett. 2011, 52, 6086–6090. doi:10.1016/j.tetlet.2011.09.004
Return to citation in text: [1] -
Johansson, J. R.; Lincoln, P.; Nordén, B.; Kann, N. J. Org. Chem. 2011, 76, 2355–2359. doi:10.1021/jo200134a
Return to citation in text: [1] -
Zhang, J.; Wu, J.; Shen, L.; Jin, G.; Cao, S. Adv. Synth. Catal. 2011, 353, 580–584. doi:10.1002/adsc.201000791
Return to citation in text: [1] -
Gonzalez-Olvera, R.; Espinoza-Vázquez, A.; Negrón-Silva, G. E.; Palomar-Pardavé, M. E.; Romero-Romo, M. A.; Santillan, R. Molecules 2013, 18, 15064–15079. doi:10.3390/molecules181215064
Return to citation in text: [1] -
Hassan, S.; Müller, T. J. J. Adv. Synth. Catal. 2015, 357, 617–666. doi:10.1002/adsc.201400904
Return to citation in text: [1] [2] -
Wang, Z.-Y.; Li, J.; Wang, N.; Liu, H.; Wang, K.-K. Asian J. Org. Chem. 2023, 12, 202300105. doi:10.1002/ajoc.202300105
Return to citation in text: [1] [2] -
Sears, P.; Wong, C.-H. Angew. Chem., Int. Ed. 1999, 38, 2300–2324. doi:10.1002/(sici)1521-3773(19990816)38:16<2300::aid-anie2300>3.0.co;2-6
Return to citation in text: [1] -
Koester, D. C.; Holkenbrink, A.; Werz, D. B. Synthesis 2010, 3217–3242. doi:10.1055/s-0030-1258228
Return to citation in text: [1] -
Yang, Y.; Yu, B. Chem. Rev. 2017, 117, 12281–12356. doi:10.1021/acs.chemrev.7b00234
Return to citation in text: [1] -
Hazelard, D.; Compain, P. Org. Biomol. Chem. 2017, 15, 3806–3827. doi:10.1039/c7ob00386b
Return to citation in text: [1] -
Leusmann, S.; Ménová, P.; Shanin, E.; Titz, A.; Rademacher, C. Chem. Soc. Rev. 2023, 52, 3663–3740. doi:10.1039/d2cs00954d
Return to citation in text: [1] -
Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a
Return to citation in text: [1] [2] -
Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/c1ob05583f
Return to citation in text: [1] [2] -
Salta, J.; Dernedde, J.; Reissig, H.-U. Beilstein J. Org. Chem. 2015, 11, 638–646. doi:10.3762/bjoc.11.72
Return to citation in text: [1] [2] -
Salta, J.; Reissig, H.-U. Synthesis 2015, 47, 1893–1898. doi:10.1055/s-0034-1380571
Return to citation in text: [1] [2] -
Bouché, L.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 3697–3703. doi:10.1002/ejoc.201402191
Return to citation in text: [1] [2] -
Salta, J.; Reissig, H.-U. Eur. J. Org. Chem. 2020, 4361–4370. doi:10.1002/ejoc.202000618
Return to citation in text: [1] [2] -
Salta, J.; Arp, F. F.; Kühne, C.; Reissig, H.-U. Eur. J. Org. Chem. 2020, 7333–7342. doi:10.1002/ejoc.202001389
Return to citation in text: [1] [2] -
Fasting, C.; Schalley, C. A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Angew. Chem., Int. Ed. 2012, 51, 10472–10498. doi:10.1002/anie.201201114
Return to citation in text: [1] -
Al-Harrasi, A.; Reißig, H.-U. Angew. Chem., Int. Ed. 2005, 44, 6227–6231. doi:10.1002/anie.200501127
Return to citation in text: [1] -
Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996
Return to citation in text: [1] [2] [3] -
Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d
Return to citation in text: [1] -
Reissig, H.-U. Angew. Chem., Int. Ed. 2021, 60, 9180–9191. doi:10.1002/anie.202101550
Return to citation in text: [1] -
Angell, Y.; Burgess, K. Angew. Chem., Int. Ed. 2007, 46, 3649–3651. doi:10.1002/anie.200700399
Return to citation in text: [1] -
Zheng, Z.-J.; Wang, D.; Xu, Z.; Xu, L.-W. Beilstein J. Org. Chem. 2015, 11, 2557–2576. doi:10.3762/bjoc.11.276
Return to citation in text: [1] -
Chan, T. R.; Hilgraf, R.; Sharpless, K. B.; Fokin, V. V. Org. Lett. 2004, 6, 2853–2855. doi:10.1021/ol0493094
Return to citation in text: [1] -
Yekta, S.; Koóš, P.; Reissig, H.-U. unpublished results.
Return to citation in text: [1] -
Szostak, M.; Spain, M.; Procter, D. J. Chem. Soc. Rev. 2013, 42, 9155–9183. doi:10.1039/c3cs60223k
Return to citation in text: [1] -
Chiara, J. L.; Destabel, C.; Gallego, P.; Marco-Contelles, J. J. Org. Chem. 1996, 61, 359–360. doi:10.1021/jo951571q
Return to citation in text: [1] -
Keck, G. E.; Wager, T. T.; McHardy, S. F. Tetrahedron 1999, 55, 11755–11772. doi:10.1016/s0040-4020(99)00486-x
Return to citation in text: [1] -
Pulz, R.; Al-Harrasi, A.; Reissig, H.-U. Org. Lett. 2002, 4, 2353–2355. doi:10.1021/ol0260573
Return to citation in text: [1] -
Bressel, B.; Reissig, H.-U. Org. Lett. 2009, 11, 527–530. doi:10.1021/ol802514m
Return to citation in text: [1]
| 60. | Szostak, M.; Spain, M.; Procter, D. J. Chem. Soc. Rev. 2013, 42, 9155–9183. doi:10.1039/c3cs60223k |
| 61. | Chiara, J. L.; Destabel, C.; Gallego, P.; Marco-Contelles, J. J. Org. Chem. 1996, 61, 359–360. doi:10.1021/jo951571q |
| 62. | Keck, G. E.; Wager, T. T.; McHardy, S. F. Tetrahedron 1999, 55, 11755–11772. doi:10.1016/s0040-4020(99)00486-x |
| 1. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5 |
| 2. | Rutjes, F. P. J. T. Click Chemistry; Science of Synthesis; Thieme: Stuttgart, Germany, 2022. doi:10.1055/b000000077 |
| 18. | Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651 |
| 19. | Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 633–645. doi:10.1002/anie.196306331 |
| 20. | Breugst, M.; Reissig, H.-U. Angew. Chem., Int. Ed. 2020, 59, 12293–12307. doi:10.1002/anie.202003115 |
| 51. | Fasting, C.; Schalley, C. A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Angew. Chem., Int. Ed. 2012, 51, 10472–10498. doi:10.1002/anie.201201114 |
| 16. | Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457–460. doi:10.1126/science.1229506 |
| 17. | Berg, R.; Straub, B. F. Beilstein J. Org. Chem. 2013, 9, 2715–2750. doi:10.3762/bjoc.9.308 |
| 52. | Al-Harrasi, A.; Reißig, H.-U. Angew. Chem., Int. Ed. 2005, 44, 6227–6231. doi:10.1002/anie.200501127 |
| 53. | Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996 |
| 5. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 6. | Hänni, K. D.; Leigh, D. A. Chem. Soc. Rev. 2010, 39, 1240–1251. doi:10.1039/b901974j |
| 7. | Mamidyala, S. K.; Finn, M. G. Chem. Soc. Rev. 2010, 39, 1252–1261. doi:10.1039/b901969n |
| 8. | Pedersen, D. S.; Abell, A. Eur. J. Org. Chem. 2011, 2399–2411. doi:10.1002/ejoc.201100157 |
| 9. | Fokin, V. V.; Matyjaszewski, K. CuAAC: The Quintessential Click Reaction. In Organic Chemistry – Breakthroughs and Highlights; Ding, K.; Dai, L.-X., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp 247–277. doi:10.1002/9783527664801.ch7 |
| 10. | Totobenazara, J.; Burke, A. J. Tetrahedron Lett. 2015, 56, 2853–2859. doi:10.1016/j.tetlet.2015.03.136 |
| 11. | Singh, M. S.; Chowdhury, S.; Koley, S. Tetrahedron 2016, 72, 5257–5283. doi:10.1016/j.tet.2016.07.044 |
| 12. | Kacprzak, K.; Skiera, I.; Piasecka, M.; Paryzek, Z. Chem. Rev. 2016, 116, 5689–5743. doi:10.1021/acs.chemrev.5b00302 |
| 13. | Döhler, D.; Michael, P.; Binder, W. H. Acc. Chem. Res. 2017, 50, 2610–2620. doi:10.1021/acs.accounts.7b00371 |
| 14. | Tiwari, V. K.; Mishra, B. B.; Mishra, K. B.; Mishra, N.; Singh, A. S.; Chen, X. Chem. Rev. 2016, 116, 3086–3240. doi:10.1021/acs.chemrev.5b00408 |
| 15. | Santos, C. S.; de Oliveira, R. J.; de Oliveira, R. N.; Freitas, J. C. R. ARKIVOC 2020, No. i, 219–271. doi:10.24820/ark.5550190.p011.293 |
| 44. | Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a |
| 45. | Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/c1ob05583f |
| 46. | Salta, J.; Dernedde, J.; Reissig, H.-U. Beilstein J. Org. Chem. 2015, 11, 638–646. doi:10.3762/bjoc.11.72 |
| 47. | Salta, J.; Reissig, H.-U. Synthesis 2015, 47, 1893–1898. doi:10.1055/s-0034-1380571 |
| 3. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 4. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 48. | Bouché, L.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 3697–3703. doi:10.1002/ejoc.201402191 |
| 49. | Salta, J.; Reissig, H.-U. Eur. J. Org. Chem. 2020, 4361–4370. doi:10.1002/ejoc.202000618 |
| 50. | Salta, J.; Arp, F. F.; Kühne, C.; Reissig, H.-U. Eur. J. Org. Chem. 2020, 7333–7342. doi:10.1002/ejoc.202001389 |
| 24. | Feldman, A. K.; Colasson, B.; Fokin, V. V. Org. Lett. 2004, 6, 3897–3899. doi:10.1021/ol048859z |
| 25. | Appukkuttan, P.; Dehaen, W.; Fokin, V. V.; Van der Eycken, E. Org. Lett. 2004, 6, 4223–4225. doi:10.1021/ol048341v |
| 37. | Hassan, S.; Müller, T. J. J. Adv. Synth. Catal. 2015, 357, 617–666. doi:10.1002/adsc.201400904 |
| 38. | Wang, Z.-Y.; Li, J.; Wang, N.; Liu, H.; Wang, K.-K. Asian J. Org. Chem. 2023, 12, 202300105. doi:10.1002/ajoc.202300105 |
| 23. | Maksikova, A. V.; Serebryakova, E. S.; Tikhonova, L. G.; Vereshchagin, L. I. Chem. Heterocycl. Compd. 1980, 16, 1284–1285. doi:10.1007/bf00501836 |
| 39. | Sears, P.; Wong, C.-H. Angew. Chem., Int. Ed. 1999, 38, 2300–2324. doi:10.1002/(sici)1521-3773(19990816)38:16<2300::aid-anie2300>3.0.co;2-6 |
| 40. | Koester, D. C.; Holkenbrink, A.; Werz, D. B. Synthesis 2010, 3217–3242. doi:10.1055/s-0030-1258228 |
| 41. | Yang, Y.; Yu, B. Chem. Rev. 2017, 117, 12281–12356. doi:10.1021/acs.chemrev.7b00234 |
| 42. | Hazelard, D.; Compain, P. Org. Biomol. Chem. 2017, 15, 3806–3827. doi:10.1039/c7ob00386b |
| 43. | Leusmann, S.; Ménová, P.; Shanin, E.; Titz, A.; Rademacher, C. Chem. Soc. Rev. 2023, 52, 3663–3740. doi:10.1039/d2cs00954d |
| 22. | Bräse, S.; Banert, K., Eds. Organic Azides – Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010. doi:10.1002/9780470682517 |
| 63. | Pulz, R.; Al-Harrasi, A.; Reissig, H.-U. Org. Lett. 2002, 4, 2353–2355. doi:10.1021/ol0260573 |
| 64. | Bressel, B.; Reissig, H.-U. Org. Lett. 2009, 11, 527–530. doi:10.1021/ol802514m |
| 21. | Kirmse, W.; Horner, L. Justus Liebigs Ann. Chem. 1958, 614, 1–3. doi:10.1002/jlac.19586140102 |
| 26. | Kacprzak, K. Synlett 2005, 943–946. doi:10.1055/s-2005-864809 |
| 27. | Wang, Z.-X.; Zhao, Z.-G. J. Heterocycl. Chem. 2007, 44, 89–92. doi:10.1002/jhet.5570440115 |
| 28. | Odlo, K.; Høydahl, E. A.; Hansen, T. V. Tetrahedron Lett. 2007, 48, 2097–2099. doi:10.1016/j.tetlet.2007.01.130 |
| 29. | Bonnamour, J.; Legros, J.; Crousse, B.; Bonnet-Delpon, D. Tetrahedron Lett. 2007, 48, 8360–8362. doi:10.1016/j.tetlet.2007.09.118 |
| 30. | Fukuzawa, S.-i.; Shimizu, E.; Kikuchi, S. Synlett 2007, 2436–2438. doi:10.1055/s-2007-986638 |
| 31. | Kumar, D.; Patel, G.; Reddy, V. B. Synlett 2009, 399–402. doi:10.1055/s-0028-1087556 |
| 32. | Huang, Y.; Gard, G. L.; Shreeve, J. M. Tetrahedron Lett. 2010, 51, 6951–6954. doi:10.1016/j.tetlet.2010.10.149 |
| 33. | Stefani, H. A.; Canduzini, H. A.; Manarin, F. Tetrahedron Lett. 2011, 52, 6086–6090. doi:10.1016/j.tetlet.2011.09.004 |
| 34. | Johansson, J. R.; Lincoln, P.; Nordén, B.; Kann, N. J. Org. Chem. 2011, 76, 2355–2359. doi:10.1021/jo200134a |
| 35. | Zhang, J.; Wu, J.; Shen, L.; Jin, G.; Cao, S. Adv. Synth. Catal. 2011, 353, 580–584. doi:10.1002/adsc.201000791 |
| 36. | Gonzalez-Olvera, R.; Espinoza-Vázquez, A.; Negrón-Silva, G. E.; Palomar-Pardavé, M. E.; Romero-Romo, M. A.; Santillan, R. Molecules 2013, 18, 15064–15079. doi:10.3390/molecules181215064 |
| 44. | Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a |
| 45. | Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/c1ob05583f |
| 46. | Salta, J.; Dernedde, J.; Reissig, H.-U. Beilstein J. Org. Chem. 2015, 11, 638–646. doi:10.3762/bjoc.11.72 |
| 47. | Salta, J.; Reissig, H.-U. Synthesis 2015, 47, 1893–1898. doi:10.1055/s-0034-1380571 |
| 48. | Bouché, L.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 3697–3703. doi:10.1002/ejoc.201402191 |
| 49. | Salta, J.; Reissig, H.-U. Eur. J. Org. Chem. 2020, 4361–4370. doi:10.1002/ejoc.202000618 |
| 50. | Salta, J.; Arp, F. F.; Kühne, C.; Reissig, H.-U. Eur. J. Org. Chem. 2020, 7333–7342. doi:10.1002/ejoc.202001389 |
| 55. | Reissig, H.-U. Angew. Chem., Int. Ed. 2021, 60, 9180–9191. doi:10.1002/anie.202101550 |
| 54. | Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d |
| 22. | Bräse, S.; Banert, K., Eds. Organic Azides – Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010. doi:10.1002/9780470682517 |
| 58. | Chan, T. R.; Hilgraf, R.; Sharpless, K. B.; Fokin, V. V. Org. Lett. 2004, 6, 2853–2855. doi:10.1021/ol0493094 |
| 53. | Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996 |
| 37. | Hassan, S.; Müller, T. J. J. Adv. Synth. Catal. 2015, 357, 617–666. doi:10.1002/adsc.201400904 |
| 38. | Wang, Z.-Y.; Li, J.; Wang, N.; Liu, H.; Wang, K.-K. Asian J. Org. Chem. 2023, 12, 202300105. doi:10.1002/ajoc.202300105 |
| 53. | Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem. – Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996 |
| 57. | Zheng, Z.-J.; Wang, D.; Xu, Z.; Xu, L.-W. Beilstein J. Org. Chem. 2015, 11, 2557–2576. doi:10.3762/bjoc.11.276 |
| 24. | Feldman, A. K.; Colasson, B.; Fokin, V. V. Org. Lett. 2004, 6, 3897–3899. doi:10.1021/ol048859z |
| 4. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 56. | Angell, Y.; Burgess, K. Angew. Chem., Int. Ed. 2007, 46, 3649–3651. doi:10.1002/anie.200700399 |
© 2023 Reissig and Yu; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.