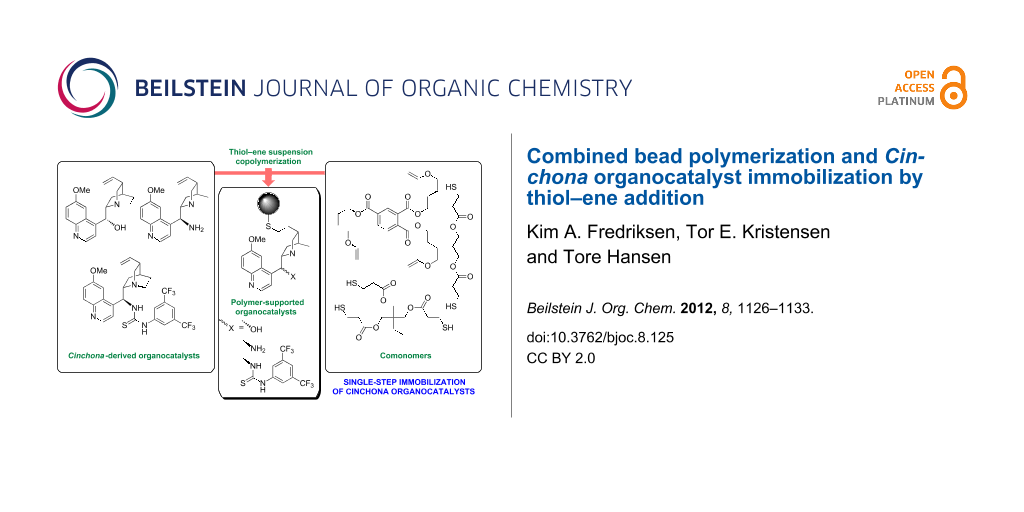
Abstract
In this work, we report an unusually concise immobilization of Cinchona organocatalysts using thiol–ene chemistry, in which catalyst immobilization and bead polymerization is combined in a single step. A solution of azo initiator, polyfunctional thiol, polyfunctional alkene and an unmodified Cinchona-derived organocatalyst in a solvent is suspended in water and copolymerized on heating by thiol–ene additions. The resultant spherical and gel-type polymer beads have been evaluated as organocatalysts in catalytic asymmetric transformations.

Graphical Abstract
Introduction
Polymer-supported chiral organocatalysts have emerged as a rapidly expanding field of research in recent years [1], in part due to the traditionally emphasized advantages of polymeric immobilization (facilitated separation and recovery procedures, recycling etc.), but perhaps even more due to the enhanced activity and selectivity sometimes exhibited by such organocatalysts, especially under aqueous conditions [2]. Recently, the use of polymer-supported organocatalysts in continuous-flow systems has also surfaced in the literature, and their development is quickly gaining momentum [3-5]. Regrettably, cost issues linger over the field as a whole, due to the lengthy and laborious syntheses involved in the preparation of these polymer-supported entities, hampering the more widespread utilization of polymer-supported reagents or catalysts as part of conventional chemical synthesis.
Consequently, we have been engaged in the development of scalable and expedient syntheses of polymer-supported organocatalysts for some time now [6,7]. In our bottom-up approach for the preparation of polymer-supported organocatalysts, the catalyst immobilization and the preparation of the polymer scaffold are closely connected, to facilitate the synthesis of larger quantities of supported catalyst [6,7]. Acrylic derivatives of established organocatalysts are prepared on a gram scale by using nonchromatographic procedures and copolymerized with suitable comonomers to give cross-linked and microporous beads. Such polymer beads have provided good to excellent results as organocatalysts in various asymmetric transformations [6,7].
Cinchona derivatives are used in several types of organocatalysts, and they are all equipped with a pendant vinylic functionality susceptible to activation by chemical transformations based on radical intermediates [1]. As a result, polymeric immobilization of Cinchona derivatives by using thiol–ene addition has a substantial history, founded on procedures developed already in the early 1970s [1]. Cinchona derivatives are either copolymerized with certain comonomers, such as acrylonitrile, directly in a bottom-up fashion to give linear copolymers [1,8], or anchored to prefabricated cross-linked and thiol-funtionalized resins in a traditional post-modification approach [1,9]. However, for the preparation of the preferred beaded and cross-linked polymer resins, so easily handled and separated from reaction mixtures by filtration, this necessitates several steps, as the cross-linked resin must be prepared first by copolymerization, then equipped with thiol functionalities, and finally joined with the Cinchona derivative through thiol–ene coupling.
The thiol–ene addition was described by Theodor Posner already in 1905 [10], and it has been in more or less continuous use since then. The thiol–ene addition can readily be adapted for polymerization, by using polyfunctional alkenes in combination with polyfunctional thiols [11-13]. We envisaged that polymerization into a cross-linked and beaded resin could be combined with immobilization of a Cinchona derivative in a single step under suitable conditions. Such a procedure would enable us to prepare polymer-supported Cinchona organocatalysts directly in a single step and on a large scale, using unmodified Cinchona organocatalyst precursors.
Results and Discussion
Building blocks for the preparation of cross-linked thiol–ene resins
Research oriented towards thiol–ene chemistry has experienced near explosive growth in the past few years, perhaps due to its efficiency and functional tolerance, but possibly even more due to its recent conceptualization as a “click” reaction [13]. In order to prepare different polymer beads with varying degrees of swelling characteristics, we assembled a small collection of useful thiol and alkene building blocks (Figure 1).
![[1860-5397-8-125-1]](/bjoc/content/figures/1860-5397-8-125-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Thiol, alkene and organocatalyst building blocks for combined bead polymerization and Cinchona organocatalyst immobilization.
Figure 1: Thiol, alkene and organocatalyst building blocks for combined bead polymerization and Cinchona orga...
Trithiol 4 is a readily available commercial product, whereas dithiol 5 was easily obtained from esterification of 3-mercaptopropionic acid and propane-1,3-diol [14]. Trivinyl ether 6, polyethylene glycol (PEG) dimethacrylate 7, diacrylate 8 and diallyl ether 9 are all commercially available compounds. For thiol–ene additions via the radical pathway, the order of reactivity of unsaturated compounds 6–9 towards the thiols is generally so: vinyl ether 6 > allyl ether 9 > acrylate 8 > methacrylate 7 [11]. Thiol–ene additions according to the anionic (Michael-type) mechanism have not been investigated in this work. As a minimum for obtaining cross-linked polymeric networks, either trithiol 4 has to be copolymerized with dialkenes 7–9, or dithiol 5 has to be copolymerized with trivinyl ether 6. These two main approaches can then be modified or finely tuned to match suitable swelling characteristics by incorporation of smaller amounts of any of the other constituents 4–9, thereby adjusting the degree of cross-linking.
As for the Cinchona organocatalysts, we wanted to incorporate either unmodified quinine (1), the primary amine organocatalyst 2, or thiourea organocatalyst 3 into the thiol–ene network (Figure 1). While quinine is available directly, primary amine organocatalyst 2 was prepared from quinine, via the azide, in a two-step sequence by using the Bose–Mitsunobu reaction followed by Staudinger reduction, as described by others [15]. Thiourea Cinchona organocatalyst 3 was easily obtained from catalyst 2 by reaction with the appropriate aromatic isothiocyanate [15].
Single step thiol–ene polymerization and Cinchona organocatalyst immobilization
With the assortment of building blocks depicted in Figure 1 available, we could now obtain immobilized versions (10–12) of unmodified Cinchona catalysts 1–3 directly by oil-in-water type thiol–ene suspension copolymerization. A solution of polyfunctional thiol, polyfunctional alkene and Cinchona organocatalyst in a water immiscible solvent, such as chlorobenzene or toluene, containing a small amount of azo radical initiator (AIBN), was suspended in dilute aqueous polyvinyl alcohol (PVA) and heated under vigorous agitation. The suspended and stabilized droplets then converted to spherical and gel-type polymer beads. An overview of the immobilized Cinchona organocatalysts, and their constituents, is provided in Scheme 1.
![[1860-5397-8-125-i1]](/bjoc/content/inline/1860-5397-8-125-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Combined bead polymerization and Cinchona organocatalyst immobilization by thiol–ene addition.
Scheme 1: Combined bead polymerization and Cinchona organocatalyst immobilization by thiol–ene addition.
After polymerization, the polymer beads were filtered and purified by Soxhlet extraction, and their organocatalyst loadings were determined on the basis of CHN analysis, as only the organocatalytic moiety contains nitrogen. Generally, the yield of polymer beads, calculated on the basis of recovered material versus the combined mass of starting materials, varied between ca. 60–80%. Only an azo initiator, such as AIBN, could be utilized, because peroxide initiators, such as dibenzoyl peroxide, oxidise thiols.
The thiol-ene polymer beads 10–12 had a distinctively soft and gel-like appearance, but were easily handled like conventional microporous beads. They had favourable swelling characteristics in several organic solvents, particularly in THF and CH2Cl2, despite their significant degree of cross-linking. As such, they have much in common with the CLEAR (cross-linked ethoxylate acrylate resin) resins, a type of polymer support with all the characteristics of a microporous polymer that also has an unusually high degree of cross-linking [16]. Unlike vinyl ether 6 and allyl ether 9, acrylic building blocks such as 7 and 8 may undergo some degree of acrylic homopolymerization during network formation, although the thiol–ene addition is usually more rapid than the polymerization.
The ratio of thiol and alkene functionalities was adjusted to be close to unity, and the thiol–ene reaction usually has a high degree of conversion; however, the presence of free thiol groups is probably unavoidable. We were, nevertheless, curious to investigate how these supported organocatalysts would function in asymmetric transformations when compared to the unsupported catalysts.
Asymmetric organocatalytic transformations using immobilized Cinchona organocatalysts
With supported Cinchona organocatalysts 10–12 available, numerous organocatalytic transformations were potentially available for benchmarking. As a rough indication of activity, we started out by investigating quinine (1) and supported catalyst 10a–d in the Michael addition of 3-methoxythiophenol and cyclohex-2-enone (Table 1) [17]. Although the performance of quinine in this reaction is poor with regards to selectivity (providing only 23% ee), and probably not very useful for benchmarking, the supported catalysts 10a–d were obviously catalytically active, albeit modestly selective compared to the free catalyst, giving quantitative yields and a selectivity of 11–14% ee.
Table 1: Polymer-supported quinines in asymmetric Michael addition.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-125-i2.svg?max-width=637&scale=1.0)
|
||
| Catalyst | Yield [%]a | ee [%]b |
|---|---|---|
| 1 | >95 | 23 |
| 10a | >95 | 14 |
| 10b | >95 | 11 |
| 10c | 92 | 14 |
| 10d | >95 | 12 |
aIsolated yield. bDetermined by HPLC analysis.
Of greater interest was the performance of the primary amine organocatalysts 11. Polymer-supported catalysts 11a,b were tried out in the asymmetric preparation of the anticoagulant warfarin from benzylideneacetone and 4-hydroxycoumarin, a transformation that we have investigated in our group as part of developmental work in primary amine organocatalysis on a previous occasion (Table 2) [18]. Compared to the free catalyst 2, the yields obtained by using supported catalysts 11a,b are inferior, but this is most probably due to the lack of solubility of the hydroxycoumarin, a very insoluble compound, in the reaction medium (CH2Cl2) and probably not so much a lack of inherent activity. Interestingly, catalyst 11b, made by using dithiol 5 and trivinyl ether 6 (in addition to a few mol % of trithiol 4 to increase cross-linking slightly, giving beads of better quality), exhibited markedly improved selectivity compared to catalyst 11a, even matching that of the free catalyst 2. This may be connected to the fact that the Cinchona moiety becomes bound to the polymer network through the thiol, and use of a difunctional thiol then improves the mobility of this catalytic unit as it is now positioned at the end of a linker of greater length. This effect does not seem to have played out for catalyst 10d though. Catalysts 10a–d all seemed to behave much the same. Catalyst 11b could be recycled, but both yield and selectivity quickly eroded (Table 2).
Table 2: Polymer-supported primary amine Cinchona organocatalysts in asymmetric preparation of warfarin.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-125-i3.svg?max-width=637&scale=1.0)
|
||
| Catalyst | Yield [%]a | ee [%]b |
|---|---|---|
| 2 | 75 | 92 |
| 11a | 15 | 77 |
| 11bc | 25 | 94 |
| 11bd | 15 | 84 |
| 11be | — | — |
aIsolated yield. bDetermined by HPLC analysis. cFirst cycle. dSecond cycle. eThird cycle.
Having confidence in the overall validity of our approach to polymer-supported Cinchona organocatalysts, we tested the supported thiourea catalyst 12 in the Michael addition of thiophenol and cyclohex-2-enone [17], a transformation known to be very efficiently catalysed by the free thiourea catalyst 3 (Table 3) [19,20]. The immobilized catalyst 12 proved highly active (the uncatalysed reaction is very slow), rapidly giving quantitative yield, but somewhat reduced selectivity of the addition product compared to the free thiourea catalyst 3. To investigate the extent to which free thiol groups could influence the reaction by catalyzing a racemic pathway, we also tested polymer beads without any Cinchona moiety present in this transformation. Undeniably, polymer beads prepared without any Cinchona organocatalyst present did also catalyze the reaction. However, we found that several other polymer resins, such as unmodified Merrifield resin (chloromethylated and cross-linked polystyrene), influence the reaction in the same manner. Consequently, we do not believe this to be an intrinsic property of our thiol–ene polymer beads, connected to the effect of free thiol groups. In addition, capping free thiol groups on beforehand by treatment with excess methyl acrylate did not affect the performance in this transformation.
Table 3: Polymer-supported thiourea Cinchona organocatalyst in the asymmetric Michael addition.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-125-i4.svg?max-width=637&scale=1.0)
|
||
| Catalyst | Yield [%]a | ee [%]b |
|---|---|---|
| 3 | >95 | 83 |
| 12 | >95 | 69 |
aIsolated yield. bDetermined by HPLC analysis.
Catalyst 12 was also tested in the Michael addition of methyl malonate to trans-β-nitrostyrene (Table 4) [21]. The catalyst gave quantitative yield and excellent enantioselectivity after 3–4 days reaction time. However, the catalyst exhibited poor recycling properties as yields fell sharply after the second reaction cycle; but selectivity remained largely untouched. At this point, we suspected that loss of the catalytic entities from the polymer resin may explain this, and indeed, CHN analysis of polymer resins after recycling verified that a loss of nitrogen content, meaning leaching of the active species, had occurred.
Table 4: Polymer-supported thiourea Cinchona organocatalyst in the asymmetric Michael addition.
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-125-i5.svg?max-width=637&scale=1.0)
|
||
| Catalyst | Conversion [%]a | ee [%]b |
|---|---|---|
| 12c | >95 | 92 |
| 12d | >95 | 92 |
| 12e | 34 | 92 |
| 12f | trace | — |
aDetermined by 1H NMR analysis of crude reaction mixture. bDetermined by HPLC analysis. cFirst cycle, 3 d reaction time. dSecond cycle, 4 d reaction time. eThird cycle, 4 d reaction time. fFourth cycle, 4 d reaction time.
Conclusion
We have developed an unusually concise immobilization of Cinchona organocatalysts by thiol–ene suspension copolymerization of polyfunctional thiols and alkenes together with unmodified Cinchona organocatalyst precursors. As such, bead polymerization and catalyst immobilization is combined in a single step. Vinyl ethers, allyl ethers, acrylates and methacrylates can all be effectively incorporated as part of such thiol–ene networks. The supported organocatalysts have been tried out successfully in several asymmetric transformations, but catalyst recycling so far is relatively poor. Hopefully, this expedient method for immobilization of Cinchona derivatives can be further developed in the future to improve activity and selectivity, and also be widened to include other useful Cinchona-derived species.
Experimental
General: 1H NMR and 13C NMR spectra were recorded on a Bruker AV 600 (600/150 MHz), Bruker Advance DRX 500 (500/125 MHz), Bruker DPX 300 (300/75 MHz) or Bruker DPX 200 (200/50 MHz) spectrometer. Dry THF was obtained from a solvent purification system (MB SPS-800 from MBraun). All other reagents and solvents were used as received. CHN analyses were carried out in the School of Chemistry, at the University of Birmingham, UK. For flash chromatography, silica gel from SdS (60 A, 40–63 μm, 550 m2/g, pH 7) and Merck (silica gel 60, 0.40–0.63 mm, 480–540 m2/g, pH 6.5–7.5) were used, either manually or with an automated system (Isco Inc. CombiFlash Companion with PeakTrak software), with EtOAc/hexanes of technical quality. Enantiomeric excess was determined by HPLC analysis using analytical columns (Chiralpak AS-H or AD-H from Daicel Chemical Industries).
The Cinchona organocatalysts 2 and 3 were prepared from quinine (1) as described in the literature [15]. Dithiol 5 was prepared as described in the literature [14], and this compound was kept refrigerated and protected from light in order to avoid deterioration on storage.
Preparation of polymer-supported quinines 10a–d: Quinine (1, 2.10 mmol for 10a or 2.00 mmol for 10b/10c or 1.50 mmol for 10d), trithiol 4 (6.20 mmol for 10a/10c or 6.00 mmol for 10b or 0.18 mmol for 10d), dithiol 5 (4.00 mmol for 10d), dimethacrylate 7 (8.90 mmol for 10a), diacrylate 8 (8.10 mmol for 10b), diallyl ether 9 (8.10 mmol for 10c), trivinyl ether 6 (2.43 mmol for 10d) and AIBN (5 wt % relative to monomers) were dissolved in a monomer diluent (13 mL PhCl for 10a or 15 mL PhCl for 10b/10c or 5 mL PhCl for 10d). Aqueous PVA (100 mL for 10a or 70 mL for 10b/10c or 39 mL for 10d, 0.5% Mowiol 40-88) was added under stirring to give an oil-in-water type emulsion. The system was flushed with argon for 5 min. The suspension was heated to 70 °C and kept at this temperature for 3 h under stirring, allowed to cool to room temperature, and poured into a beaker containing MeOH (250 mL). The suspension was stirred for 15 min, and the polymer beads were allowed to settle by gravity for 10 min. The supernatant was removed by decantation and the process was repeated until the supernatant was transparent (1–2 repetitions). CH2Cl2 (50 mL) was added, the suspension was filtered by vacuum, and the polymer beads were washed with water (1000 mL), THF–H2O (200 mL, 1:1), MeOH (200 mL) and CH2Cl2 (50 mL). The polymer beads were then transferred to a cellulose paper thimble and purified by Soxhlet extraction with CH2Cl2 (70 mL) for 12 h, and then the purified beads were left to dry at room temperature for 24 h (67% yield for 10a, 67% yield for 10b, 63% yield for 10c, 64% yield for 10d). Catalyst loadings were determined by CHN analysis.
Preparation of polymer-supported primary amine Cinchona organocatalysts 11a,b: Cinchona derivative 2 (2.00 mmol for 11a or 1.20 mmol for 11b), trithiol 4 (5.00 mmol for 11a or 0.16 mmol for 11b), dithiol 5 (4.20 mmol for 11b), dimethacrylate 7 (6.24 mmol for 11a), trivinyl ether 6 (2.51 mmol for 11b) and AIBN (5 wt % relative to monomers) were dissolved in a monomer diluent (12 mL PhCl for 11a or 6 mL PhMe for 11b). Aqueous PVA (70 mL for 11a or 40 mL for 11b, 0.5% Mowiol 40-88) and potassium iodide (to inhibit polymerization in the aqueous phase, 23 mg for 11b) was added under stirring to give an oil-in-water type emulsion. The system was flushed with argon for 5 min. The suspension was heated to 70 °C and kept at this temperature for 3 h under stirring, allowed to cool to room temperature, and then poured into a beaker containing MeOH (250 mL). The suspension was stirred for 15 min, and the polymer beads were allowed to settle by gravity for 10 min. The supernatant was removed by decantation, and the process was repeated until the supernatant was transparent (1–2 repetitions). CH2Cl2 (50 mL) was added, the suspension was filtered by vacuum, and the polymer beads washed with water (1000 mL), THF–H2O (200 mL, 1:1), MeOH (200 mL) and CH2Cl2 (50 mL). The polymer beads were then transferred to a cellulose paper thimble and purified by Soxhlet extraction with CH2Cl2 (70 mL) for 12 h, and then the purified beads were left to dry at room temperature for 24 h (75% yield for 11a, 85% yield for 11b). Catalyst loadings were determined by CHN analysis.
Preparation of polymer-supported thiourea Cinchona organocatalyst 12: Cinchona derivative 3 (0.538 mmol), trithiol 4 (0.17 mmol), dithiol 5 (3.48 mmol), trivinyl ether 6 (2.32 mmol) and AIBN (15 wt % relative to monomers) were dissolved in PhMe (7 mL). Aqueous PVA (50 mL, 0.5% Mowiol 40-88) and potassium iodide (to inhibit polymerization in the aqueous phase, 20 mg) was added under stirring to give an oil-in-water type emulsion. The system was flushed with argon for 5 min. The suspension was heated to 70 °C and kept at this temperature for 1 h under stirring, allowed to cool to room temperature, and poured into a beaker containing MeOH (250 mL). The suspension was stirred for 15 min, and the polymer beads were allowed to settle by gravity for 10 min. The supernatant was removed by decantation, and the process was repeated until the supernatant was transparent (1–2 repetitions). CH2Cl2 (50 mL) was added, the suspension was filtered by vacuum, and the polymer beads washed with water (1000 mL), THF–H2O (200 mL, 1:1), MeOH (200 mL) and CH2Cl2 (50 mL). The polymer beads were then transferred to a cellulose paper thimble and purified by Soxhlet extraction with CH2Cl2 (70 mL) for 12 h, and the purified beads were left to dry at room temperature for 24 h (62% yield). Catalyst loadings were determined by CHN analysis.
General procedure for asymmetric Michael addition of 3-methoxythiophenol to cyclohex-2-enone: 3-Methoxythiophenol (0.30 mL, 3.10 mmol), and catalyst (1 mol %) were dissolved in CH2Cl2 (6 mL). Cyclohex-2-enone (0.50 mL, 4.03 mmol) was then added in one portion, and the resulting mixture was stirred at room temperature for 2 h. The crude reaction mixture was filtered, and the polymer beads were washed with CH2Cl2 (50 mL). The combined organic phase was evaporated in vacuo, and the crude product was purified by flash chromatography on silica gel (10% EtOAc in hexanes) to give the product as a colorless oil. This is a known compound [17]. Enantiomeric excess was determined by HPLC analysis (Chiralpak AS-H, 50% iPrOH in isohexane, 0.3 mL/min): tR = 25.2 min and 38.2 min.
General procedure for asymmetric Michael addition of 4-hydroxycoumarin to benzylideneacetone: To a vial containing 4-hydroxycumarin (0.35 g, 2.17 mmol), benzylideneacetone (0.56 g, 3.82 mmol) and catalyst (20 mol %), was added CH2Cl2 (18 mL) and CF3CO2H (60 μL, 40 mol %). The reaction mixture was stirred at room temperature for 72 h, diluted with CH2Cl2 and filtered. The polymer beads were washed with CH2Cl2 (70 mL). The combined organic phase was evaporated in vacuo, and the crude product was purified by flash chromatography on silica gel (10% EtOAc in hexanes and then 25% EtOAc in hexanes) to give the product as a colorless solid. This is a known compound [18]. Enantiomeric excess was determined by HPLC analysis (Chiralpak AD-H, 20% iPrOH in isohexane, 1.0 mL/min): tR = 7.3 min and 14.7 min.
General procedure for asymmetric Michael addition of thiophenol to cyclohex-2-enone: Thiophenol (0.20 mL, 1.95 mmol) and catalyst (1 mol %) were dissolved in CH2Cl2 (1.7 mL). Cyclohex-2-enone (0.15 g, 1.52 mmol) was added in one portion, and the resulting reaction mixture was stirred at room temperature for 2.5 h. The crude reaction mixture was diluted with CH2Cl2 and filtered, and the polymer beads were washed with CH2Cl2 (70 mL). The combined organic phase was evaporated in vacuo, and the crude product was purified by flash chromatography on silica gel (5% EtOAc in hexanes) to give the product as a colorless oil. This is a known compound [17]. Enantiomeric excess was determined by HPLC analysis (Chiralpak AD-H, 2% iPrOH in isohexane, 1.0 mL/min): tR = 19.2 min and 26.2 min.
General procedure for asymmetric Michael addition of methyl malonate to trans-β-nitrostyrene: trans-β-Nitrostyrene (83.4 mg, 0.56 mmol) and methyl malonate (0.23 g, 1.78 mmol) were dissolved in toluene (1 mL). Catalyst 12 (10 mol %) was added, and the reaction mixture was left at −30 °C for 72 h. The crude reaction mixture was diluted with CH2Cl2 and filtered, and the polymer beads were washed with CH2Cl2 (50 mL). The combined organic phase was evaporated in vacuo, and the crude product was purified by flash chromatography on silica gel (Gradient: 5–40% EtOAc in hexanes). This is a known compound [21]. The conversion of starting material was determined by 1H NMR analysis of the crude product. Enantiomeric excess was determined by HPLC analysis (Chiralpak AD-H, 30% iPrOH in isohexane, 1.0 mL/min): tR = 7.3 min and 9.3 min.
References
-
Kristensen, T. E.; Hansen, T. Synthesis of Chiral Catalysts Supported on Organic Polymers. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Gruttadauria, M.; Giacalone, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp 209–256. doi:10.1002/9781118087992.ch4
Return to citation in text: [1] [2] [3] [4] [5] -
Gruttadauria, M.; Giacalone, F.; Noto, R. Adv. Synth. Catal. 2009, 351, 33–57. doi:10.1002/adsc.200800731
Return to citation in text: [1] -
Alza, E.; Rodríguez-Escrich, C.; Sayalero, S.; Bastero, A.; Pericàs, M. A. Chem.–Eur. J. 2009, 15, 10167–10172. doi:10.1002/chem.200901310
Return to citation in text: [1] -
Cambeiro, X. C.; Martín-Rapún, R.; Miranda, P. O.; Sayalero, S.; Alza, E.; Llanes, P.; Pericàs, M. A. Beilstein J. Org. Chem. 2011, 7, 1486–1493. doi:10.3762/bjoc.7.172
Return to citation in text: [1] -
Alza, E.; Sayalero, S.; Cambeiro, X. C.; Martín-Rapún, R.; Miranda, P. O.; Pericàs, M. A. Synlett 2011, 464–468. doi:10.1055/s-0030-1259528
Return to citation in text: [1] -
Kristensen, T. E.; Vestli, K.; Fredriksen, K. A.; Hansen, F. K.; Hansen, T. Org. Lett. 2009, 11, 2968–2971. doi:10.1021/ol901134v
Return to citation in text: [1] [2] [3] -
Kristensen, T. E.; Vestli, K.; Jakobsen, M. G.; Hansen, F. K.; Hansen, T. J. Org. Chem. 2010, 75, 1620–1629. doi:10.1021/jo902585j
Return to citation in text: [1] [2] [3] -
Kobayashi, N.; Iwai, K. J. Am. Chem. Soc. 1978, 100, 7071–7072. doi:10.1021/ja00490a053
Return to citation in text: [1] -
Hedge, P.; Khoshdel, E.; Waterhouse, J.; Fréchet, J. M. J. J. Chem. Soc., Perkin Trans. 1 1985, 2327–2331. doi:10.1039/P19850002327
Return to citation in text: [1] -
Posner, T. Ber. Dtsch. Chem. Ges. 1905, 38, 646–657. doi:10.1002/cber.190503801106
Return to citation in text: [1] -
Hoyle, C. E.; Lee, T. Y.; Roper, T. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5301–5338. doi:10.1002/pola.20366
Return to citation in text: [1] [2] -
Kade, M. J.; Burke, D. J.; Hawker, C. J. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 743–750. doi:10.1002/pola.23824
Return to citation in text: [1] -
Hoyle, C. E.; Bowman, C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. doi:10.1002/anie.200903924
Return to citation in text: [1] [2] -
Song, J.; Chen, L.; Wang, Y.; Chen, W.; Wang, R. Front. Chem. Eng. China 2008, 2, 390–395. doi:10.1007/s11705-008-0080-6
Return to citation in text: [1] [2] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s
Return to citation in text: [1] [2] [3] -
Kempe, M.; Barany, G. J. Am. Chem. Soc. 1996, 118, 7083–7093. doi:10.1021/ja954196s
Return to citation in text: [1] -
Hiemstra, H.; Wynberg, H. J. Am. Chem. Soc. 1981, 103, 417–430. doi:10.1021/ja00392a029
Return to citation in text: [1] [2] [3] [4] -
Kristensen, T. E.; Vestli, K.; Hansen, F. K.; Hansen, T. Eur. J. Org. Chem. 2009, 5185–5191. doi:10.1002/ejoc.200900664
Return to citation in text: [1] [2] -
Jang, H. B.; Rho, H. S.; Oh, J. S.; Nam, E. H.; Park, S. E.; Bae, H. Y.; Song, C. E. Org. Biomol. Chem. 2010, 8, 3918–3922. doi:10.1039/c0ob00047g
Return to citation in text: [1] -
Li, B.-J.; Jiang, L.; Liu, M.; Chen, Y.-C.; Ding, L.-S.; Wu, Y. Synlett 2005, 603–606. doi:10.1055/s-2005-863710
Return to citation in text: [1] -
McCooey, S. H.; Connon, S. J. Angew. Chem. 2005, 117, 6525–6528. doi:10.1002/ange.200501721
Return to citation in text: [1] [2]
| 17. | Hiemstra, H.; Wynberg, H. J. Am. Chem. Soc. 1981, 103, 417–430. doi:10.1021/ja00392a029 |
| 21. | McCooey, S. H.; Connon, S. J. Angew. Chem. 2005, 117, 6525–6528. doi:10.1002/ange.200501721 |
| 1. | Kristensen, T. E.; Hansen, T. Synthesis of Chiral Catalysts Supported on Organic Polymers. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Gruttadauria, M.; Giacalone, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp 209–256. doi:10.1002/9781118087992.ch4 |
| 6. | Kristensen, T. E.; Vestli, K.; Fredriksen, K. A.; Hansen, F. K.; Hansen, T. Org. Lett. 2009, 11, 2968–2971. doi:10.1021/ol901134v |
| 7. | Kristensen, T. E.; Vestli, K.; Jakobsen, M. G.; Hansen, F. K.; Hansen, T. J. Org. Chem. 2010, 75, 1620–1629. doi:10.1021/jo902585j |
| 11. | Hoyle, C. E.; Lee, T. Y.; Roper, T. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5301–5338. doi:10.1002/pola.20366 |
| 6. | Kristensen, T. E.; Vestli, K.; Fredriksen, K. A.; Hansen, F. K.; Hansen, T. Org. Lett. 2009, 11, 2968–2971. doi:10.1021/ol901134v |
| 7. | Kristensen, T. E.; Vestli, K.; Jakobsen, M. G.; Hansen, F. K.; Hansen, T. J. Org. Chem. 2010, 75, 1620–1629. doi:10.1021/jo902585j |
| 15. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 3. | Alza, E.; Rodríguez-Escrich, C.; Sayalero, S.; Bastero, A.; Pericàs, M. A. Chem.–Eur. J. 2009, 15, 10167–10172. doi:10.1002/chem.200901310 |
| 4. | Cambeiro, X. C.; Martín-Rapún, R.; Miranda, P. O.; Sayalero, S.; Alza, E.; Llanes, P.; Pericàs, M. A. Beilstein J. Org. Chem. 2011, 7, 1486–1493. doi:10.3762/bjoc.7.172 |
| 5. | Alza, E.; Sayalero, S.; Cambeiro, X. C.; Martín-Rapún, R.; Miranda, P. O.; Pericàs, M. A. Synlett 2011, 464–468. doi:10.1055/s-0030-1259528 |
| 13. | Hoyle, C. E.; Bowman, C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. doi:10.1002/anie.200903924 |
| 2. | Gruttadauria, M.; Giacalone, F.; Noto, R. Adv. Synth. Catal. 2009, 351, 33–57. doi:10.1002/adsc.200800731 |
| 14. | Song, J.; Chen, L.; Wang, Y.; Chen, W.; Wang, R. Front. Chem. Eng. China 2008, 2, 390–395. doi:10.1007/s11705-008-0080-6 |
| 1. | Kristensen, T. E.; Hansen, T. Synthesis of Chiral Catalysts Supported on Organic Polymers. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Gruttadauria, M.; Giacalone, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp 209–256. doi:10.1002/9781118087992.ch4 |
| 8. | Kobayashi, N.; Iwai, K. J. Am. Chem. Soc. 1978, 100, 7071–7072. doi:10.1021/ja00490a053 |
| 10. | Posner, T. Ber. Dtsch. Chem. Ges. 1905, 38, 646–657. doi:10.1002/cber.190503801106 |
| 1. | Kristensen, T. E.; Hansen, T. Synthesis of Chiral Catalysts Supported on Organic Polymers. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Gruttadauria, M.; Giacalone, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp 209–256. doi:10.1002/9781118087992.ch4 |
| 11. | Hoyle, C. E.; Lee, T. Y.; Roper, T. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5301–5338. doi:10.1002/pola.20366 |
| 12. | Kade, M. J.; Burke, D. J.; Hawker, C. J. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 743–750. doi:10.1002/pola.23824 |
| 13. | Hoyle, C. E.; Bowman, C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. doi:10.1002/anie.200903924 |
| 1. | Kristensen, T. E.; Hansen, T. Synthesis of Chiral Catalysts Supported on Organic Polymers. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Gruttadauria, M.; Giacalone, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp 209–256. doi:10.1002/9781118087992.ch4 |
| 6. | Kristensen, T. E.; Vestli, K.; Fredriksen, K. A.; Hansen, F. K.; Hansen, T. Org. Lett. 2009, 11, 2968–2971. doi:10.1021/ol901134v |
| 7. | Kristensen, T. E.; Vestli, K.; Jakobsen, M. G.; Hansen, F. K.; Hansen, T. J. Org. Chem. 2010, 75, 1620–1629. doi:10.1021/jo902585j |
| 1. | Kristensen, T. E.; Hansen, T. Synthesis of Chiral Catalysts Supported on Organic Polymers. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Gruttadauria, M.; Giacalone, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp 209–256. doi:10.1002/9781118087992.ch4 |
| 9. | Hedge, P.; Khoshdel, E.; Waterhouse, J.; Fréchet, J. M. J. J. Chem. Soc., Perkin Trans. 1 1985, 2327–2331. doi:10.1039/P19850002327 |
| 17. | Hiemstra, H.; Wynberg, H. J. Am. Chem. Soc. 1981, 103, 417–430. doi:10.1021/ja00392a029 |
| 15. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 16. | Kempe, M.; Barany, G. J. Am. Chem. Soc. 1996, 118, 7083–7093. doi:10.1021/ja954196s |
| 17. | Hiemstra, H.; Wynberg, H. J. Am. Chem. Soc. 1981, 103, 417–430. doi:10.1021/ja00392a029 |
| 18. | Kristensen, T. E.; Vestli, K.; Hansen, F. K.; Hansen, T. Eur. J. Org. Chem. 2009, 5185–5191. doi:10.1002/ejoc.200900664 |
| 15. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 14. | Song, J.; Chen, L.; Wang, Y.; Chen, W.; Wang, R. Front. Chem. Eng. China 2008, 2, 390–395. doi:10.1007/s11705-008-0080-6 |
| 19. | Jang, H. B.; Rho, H. S.; Oh, J. S.; Nam, E. H.; Park, S. E.; Bae, H. Y.; Song, C. E. Org. Biomol. Chem. 2010, 8, 3918–3922. doi:10.1039/c0ob00047g |
| 20. | Li, B.-J.; Jiang, L.; Liu, M.; Chen, Y.-C.; Ding, L.-S.; Wu, Y. Synlett 2005, 603–606. doi:10.1055/s-2005-863710 |
| 21. | McCooey, S. H.; Connon, S. J. Angew. Chem. 2005, 117, 6525–6528. doi:10.1002/ange.200501721 |
| 18. | Kristensen, T. E.; Vestli, K.; Hansen, F. K.; Hansen, T. Eur. J. Org. Chem. 2009, 5185–5191. doi:10.1002/ejoc.200900664 |
| 17. | Hiemstra, H.; Wynberg, H. J. Am. Chem. Soc. 1981, 103, 417–430. doi:10.1021/ja00392a029 |
© 2012 Fredriksen et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)