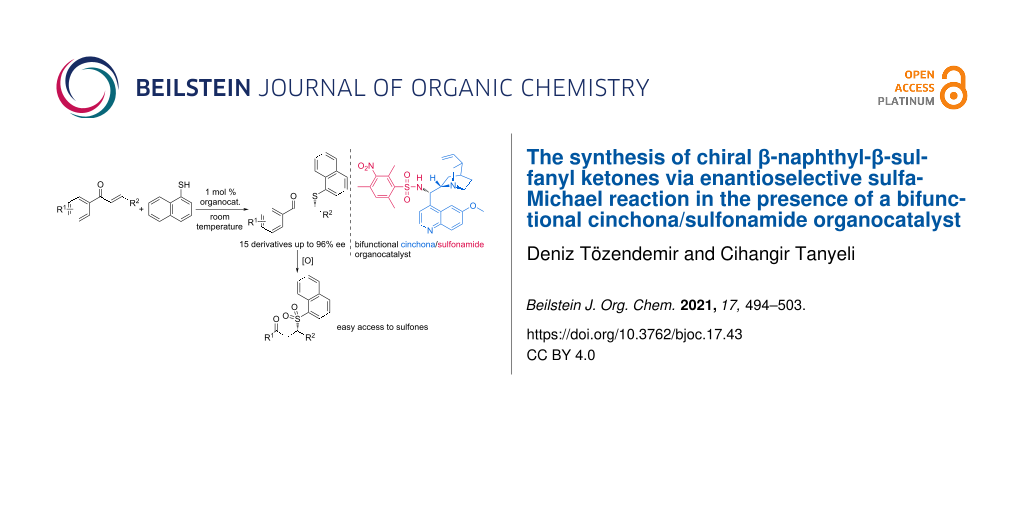
Abstract
Cinchona alkaloid-derived organocatalysts are widely employed in various asymmetric transformations, yielding products with high enantiopurity. In this respect, a bifunctional quinine-derived sulfonamide organocatalyst was developed to catalyze the asymmetric sulfa-Michael reaction of naphthalene-1-thiol with trans-chalcone derivatives. The target sulfa-Michael adducts were obtained with up to 96% ee under mild conditions and with a low (1 mol %) catalyst loading. Selected enantiomerically enriched sulfa-Michael addition products were subjected to oxidation to obtain the corresponding sulfones.

Graphical Abstract
Introduction
Derivatives of the naturally occurring cinchona alkaloids have shown remarkable performance as organocatalysts for stereoselective synthesis in the past decade [1-6]. Among them, quinine-derived organocatalysts make a noteworthy appearance in the formation of new stereogenic centres, which can serve as valuable building blocks for the construction of more elaborate structures [7-11]. An outstanding class of quinine derived organocatalysts exhibits a bifunctional mode of activation by the incorporation of an acidic unit, such as urea, thiourea, squaramide or sulfonamide moieties, giving rise to the simultaneous activation of both the nucleophile and the electrophile [12-15]. Quinine derived sulfonamides were first introduced to the literature by Song et al. [16]. Since then, many contributions were made regarding their applications in a variety of reaction types [17-20]. However, sulfa-Michael addition (SMA) reactions remain a rather less explored reaction among asymmetric organocatalytic transformations, mainly because of the high nucleophilicity of thiols causing difficulties in controlling the stereoselectivity [21], despite C–S bond-forming reactions are of great interest in synthetic organic chemistry [22]. Thus, the SMA with thiols and α,β-unsaturated ketones are generally carried out at low temperatures and with high catalyst loading [23-26]. The studies that employ mild conditions and low catalyst loading use thiophenol derivatives or simple alkylthiols as nucleophiles [27-30]. Thionaphthols, however, are overlooked in sulfa-Michael addition reactions. And to our best knowledge, no study is present concerned with SMAs with naphthalene-1-thiol as the nucleophile for the addition to enones.
Encouraged by the good results obtained with enantioselective sulfa-Michael additions of thiols to chalcones with sulfonamide-type organocatalysts in the literature [30,31], in this study, a new quinine sulfonamide organocatalyst derivative was developed to catalyze the enantioselective SMA of naphthalene-1-thiol to trans-chalcones under mild conditions and with a low (1 mol %) catalyst loading, to obtain enantiomerically enriched β-naphthyl-β-sulfanyl ketones with up to 96% ee. The target adducts are the core structure of seco-raloxifene derivatives, which are potent anti-breast cancer agents [32]. In addition, the same scaffold has also shown urease inhibitor activity [33]. Due to the shown biological attractiveness of those 1,3-biarylsulfanyl derivatives, the enantioenriched products can serve as important building blocks for new drugs. The sulfide moiety of β-naphthyl-β-sulfanyl ketones can be oxidized to form sulfones. Despite that sulfones were outshined by sulfonamides in medicinal chemistry, they have a large array of biologic activities that show promising effects as potent anti-HIV-1 [34], anti-hepatitis C [35], antifungal [36], insecticidal/acaricial [37] and antimalarial [38] agents.
Results and Discussion
We have previously reported the synthesis of new amino-substituted-DMAP-based sulfonamides [39] and quinine-based squaramide-type organocatalysts [40]. Motivated by the excellent results obtained with our aforementioned catalysts, we developed a new chiral bifunctional sulfonamide–quinine organocatalyst that unites both classes. The synthesis of the basic part was initiated by converting quinine to quinineamine via a Mitsunobu reaction, followed by a Staudinger reduction [40]. Then it was coupled with the acidic part, which was obtained by the nitration of 2,4,6-trimethylbenzenesulfonyl chloride [39] to obtain organocatalyst 5 (Scheme 1).
![[1860-5397-17-43-i1]](/bjoc/content/inline/1860-5397-17-43-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
This new organocatalyst was employed in the model asymmetric sulfa-Michael reaction of naphthalene-1-thiol and trans-chalcone, in addition to the amino-substituted DMAP and quinine-based organocatalysts (6, 7a–c and 8a–c) in our library (Figure 1), as well as previously reported quinine derived organocatalysts 8d and 9a,b in the literature [30,41].
![[1860-5397-17-43-1]](/bjoc/content/figures/1860-5397-17-43-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of the screened organocatalysts.
Figure 1: Structures of the screened organocatalysts.
Among the 11 screened organocatalysts, the ones with amino-substituted DMAP cores gave unimpressive ee values (Table 1, entries 2–5). The quinine-derived organocatalysts 8a–d failed to attain striking stereoselectivity (25–43% ee). The popular urea–quinine and thiourea–quinine organocatalysts both gave the target compound with only 41% ee, which was well below satisfactory (Table 1, entries 10 and 11). The best catalyst in terms of enantioselectivity proved to be the newly designed catalyst 5, which gave the desired product with 63% ee in 1 hour (Table 1, entry 1). After selecting the best-working catalyst, optimization studies were initiated on the model reaction to determine the conditions to achieve the best enantioselectivity.
Table 1: Catalyst and solvent screening.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-43-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | Catalyst | Solvent | Time (h) | Yieldb (%) | eec (%) |
| 1 | 5 | toluene | 1 | 98 | 63 |
| 2 | 6 | toluene | 2.5 | >99 | 30 |
| 3 | 7a | toluene | 0.5 | 92 | 46 |
| 4 | 7b | toluene | 0.5 | 63 | 40 |
| 5 | 7c | toluene | 0.5 | 79 | 52 |
| 6 | 8a | toluene | 0.5 | 72 | 41 |
| 7 | 8b | toluene | 0.5 | 96 | 43 |
| 8 | 8c | toluene | 0.5 | 90 | 31 |
| 9 | 8d | toluene | 0.5 | 95 | 25 |
| 10 | 9a | toluene | 0.5 | 71 | 41 |
| 11 | 9b | toluene | 0.5 | 94 | 41 |
| 12 | 5 | DCM | 2.5 | 42 | 65 |
| 13 | 5 | hexane | 1 | >99 | 6 |
| 14 | 5 | THF | 4 | 85 | 79 |
| 15 | 5 | CHCl3 | 2 | 50 | 65 |
| 16 | 5 | dioxane | 4 | 90 | 75 |
| 17 | 5 | TBME | 1 | 92 | 57 |
| 18 | 5 | EtOAc | 2 | 49 | 65 |
| 19 | 5 | MeCN | 1 | 81 | 55 |
| 20 | 5 | Et2O | 3 | 28 | 57 |
aUnless stated otherwise, all reactions were performed with 0.10 mmol trans-chalcone and 0.20 mmol naphthalene-1-thiol in 0.5 mL of solvent, in the presence of 2 mol % organocatalyst at rt. bIsolated yields. cDetermined by chiral HPLC analysis, AD-H column, hexane/isopropanol (99:1), 0.8 mL/min, 220 nm.
The first parameter screened was the effect of the solvent. Using THF and dioxane (Table 1, entries 14 and 16, respectively) gave the two highest results, but the use of dioxane is best avoided due to its toxicity. Except for hexane (only 6% ee, Table 1, entry 13), which resulted in an almost racemic product presumably due to solubility issues, all other solvents afforded the target SMA adduct with similar moderate ee values. Thus, THF was selected as the best solvent despite the longer reaction duration.
Then, the catalyst loading was varied between 0.1 and 10 mol % to investigate its effect on the enantioselectivity (Table 2, entries 1–6). At an extremely low catalyst loading of 0.1 mol %, the reaction was too sluggish; the amount of the product was too small and it was not isolated. Using 0.5 mol % of 5 gave rise to 82% ee (Table 2, entry 2), however, the outcome of the reaction with 1 mol % of 5 (83%, Table 2, entry 3) was slightly better than the former and was completed in a shorter time (40 hours, compared to 23 hours, respectively). Thus, that part of the optimization was continued with 1 mol % catalyst loading.
Table 2: Further screening results of the model reaction.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-43-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | Catalyst loading (mol %) | Conc. (M) | T (°C) | Time (h) | Yieldb (%) | eec (%) |
| 1 | 0.1 | 0.2 | rt | 72 | – | – |
| 2 | 0.5 | 0.2 | rt | 40 | 81 | 82 |
| 3 | 1 | 0.2 | rt | 23 | 93 | 83 |
| 4 | 2 | 0.2 | rt | 4 | 85 | 79 |
| 5 | 5 | 0.2 | rt | 6 | 95 | 76 |
| 6 | 10 | 0.2 | rt | 3 | 83 | 69 |
| 7 | 1 | 0.05 | rt | 72 | – | – |
| 8 | 1 | 0.1 | rt | 72 | – | – |
| 9 | 1 | 0.15 | rt | 26 | >99 | 79 |
| 10 | 1 | 0.25 | rt | 25 | >99 | 78 |
| 11 | 1 | 0.3 | rt | 20 | 91 | 79 |
| 12 | 1 | 0.4 | rt | 19 | >99 | 75 |
| 13d | 1 | 0.2 | rt | 72 | – | – |
| 14e | 1 | 0.2 | rt | 41 | 91 | 79 |
| 15f | 1 | 0.2 | rt | 19 | 90 | 78 |
| 16 | 1 | 0.2 | 0 | 40 | 96 | 73 |
| 17 | 1 | 0.2 | −20 | 41 | 97 | 68 |
| 18 | 1 | 0.2 | −40 | 49 | 90 | 62 |
aUnless stated otherwise, all reactions were performed with a 1:2 ratio of trans-chalcone/naphthalene-1-thiol in THF, in the presence of organocatalyst 5 at the indicated temperature. bIsolated yields. cDetermined by chiral HPLC analysis, AD-H column, 99:1 hexane/isopropanol, 0.8 mL/min, 220 nm. dThe reaction was carried out using a 1:1 ratio of trans-chalcone/naphthalene-1-thiol. eThe reaction was carried out using a 1:1.5 ratio of trans-chalcone/naphthalene-1-thiol. fThe reaction was carried out using a 1:3 ratio of trans-chalcone/naphthalene-1-thiol.
The effect of the concentration of the reaction mixture was investigated by changing the chalcone concentration gradually from 0.05 to 0.4 M (Table 2, entries 3 and 7–12). The best selectivity (83% ee) was obtained at a 0.2 M chalcone concentration (Table 2, entry 3). Diluting the reaction mixture further than 0.15 M had decreased the rate of reaction considerably and the amounts of the products were not sufficient to be isolated. Increasing the concentration led to a small decrease in ee. Using an equimolar mixture of chalcone and naphthalene-1-thiol had a similar outcome on the progress of the reaction as dilution (Table 2, entry 14). Changing the chalcone/naphthalene-1-thiol ratios to 1:1.5 or 1:3 resulted in small losses in enantioselectivity. Hence, the studies were continued with 2 molar equivalents of naphthalene-1-thiol to trans-chalcone.
The optimization studies were concluded by investigating the effect of the temperature on the asymmetric induction (Table 2, entries 16–18). Lowering the temperature gradually to −40 °C caused a significant loss in ee, allowing the synthesis of the product 12a with a final ee of 62% (Table 2, entry 18). This unexpected phenomenon could be linked to an enthalpic factor that favors the formation of the major enantiomer at higher temperature, or due to a change in the reaction mechanism when the temperature was altered. The most enantioenriched product was ultimately obtained at room temperature (83% ee, Table 2, entry 3).
The absolute configuration of the product was assigned as “S” by comparing the obtained optical rotation value with the values in the literature for the organocatalytic SMA of thiols to trans-chalcone derivatives [29,42]. A transition state model to explain the origin of the stereoinduction was proposed (Scheme 2), according to the Houk’s Brønsted acid hydrogen bonding model. Guo’s computational work in 2017 on the sulfa-Michael addition of thiols to enones in the presence of cinchona alkaloid-type organocatalysts showed that Houk’s mode of activation was of lower energy than Wynberg’s activation mode, in which the activation and orientation of the nucleophile is done by the quinuclidine core [43]. According to our proposed model, the protonated quinuclidinium ion stabilizes the newly forming alkoxide on the electrophile while the deprotonated nucleophile is oriented by the Brønsted acid moiety.
![[1860-5397-17-43-i2]](/bjoc/content/inline/1860-5397-17-43-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed transition state for the SMA of 1-thionaphthol to trans-chalcones.
Scheme 2: Proposed transition state for the SMA of 1-thionaphthol to trans-chalcones.
The substrate scope was extended to substituted chalcones, under the optimized conditions (Table 3). The chalcone derivatives used in this work were obtained by Claisen–Schmidt condensation, using known procedures [44]. Among the chalcone derivatives employed in the model reaction, the best result in terms of enantioselectivity was attained with 4-methyl-substituted chalcone, which allowed the synthesis of the corresponding SMA adduct 12d with an excellent ee of 91% (Table 3, entry 4). Good to moderate results were obtained with chalcone derivatives possessing either electron-donating or -withdrawing substituents. Compared to the unsubstituted trans-chalcone, an unexpected and drastic decrease in enantioselectivity was observed with chalcone derivative 11c, however (23% ee, Table 3, entry 3).
Table 3: Results of the SMA of naphthalene-1-thiol to substituted trans-chalcones in THF.
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-43-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | 11 | R1, R2 | 12 | Time (h) | Yieldb (%) | eec (%) |
| 1 | 11a | Ph, Ph | 12a | 23 | 93 | 83 |
| 2 | 11b | Ph, 3-MeC6H4 | 12b | 46 | 87 | 78 |
| 3 | 11c | Ph, 4-MeC6H4 | 12c | 20 | >99 | 23 |
| 4 | 11d | 4-MeC6H4, Ph | 12d | 42 | >99 | 91 |
| 5 | 11e | Ph, 3-OMeC6H4 | 12e | 42 | 83 | 58 |
| 6 | 11f | Ph, 4-OMeC6H4 | 12f | 20 | 88 | 63 |
| 7 | 11g | Ph, 3,4,5-(OMe)3C6H2 | 12g | 21 | 94 | 50 |
| 8 | 11h | Ph, 2-ClC6H4 | 12h | 23 | 84 | 66 |
| 9 | 11i | Ph, 3-ClC6H4 | 12i | 22 | 79 | 85 |
| 10 | 11j | Ph, 4-ClC6H4 | 12j | 21 | >99 | 71 |
| 11 | 11k | Ph, 3-BrC6H4 | 12k | 40 | 66 | 51 |
| 12 | 11l | 4-BrC6H4, Ph | 12l | 23 | >99 | 82 |
| 13 | 11m | Ph, 4-CF3C6H4 | 12m | 19 | 79 | 67 |
| 14 | 11n | 2-NO2C6H4, Ph | 12n | 21 | 84 | 82 |
| 15 | 11o | Ph, 4-NO2C6H4 | 12o | 21 | 81 | 68 |
aUnless stated otherwise, all reactions were performed with 0.20 mmol trans-chalcone and 0.40 mmol naphthalene-1-thiol in 1.0 mL of THF, in the presence of 1 mol % 5 at rt. bIsolated yields. cDetermined by chiral HPLC analysis.
Intrigued by this unexpected result, we decided to revisit the solvent screening. For this purpose, the sulfa-Michael addition of naphthalene-1-thiol to 11c was carried out again in toluene, dioxane, DCM and THF (Table 4).
Table 4: Solvent screening results for the SMA of naphthalene-1-thiol to chalcone derivative 11c.
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-43-i6.svg?max-width=637&scale=1.0)
|
||||
| Entrya | Solvent | Time (h) | Yieldb (%) | eec (%) |
| 1 | THF | 20 | >99 | 23 |
| 2 | toluene | 6 | 95 | 78 |
| 3 | dioxane | 20 | 78 | 76 |
| 4 | DCM | 6 | 94 | 93 |
aUnless stated otherwise, all reactions were performed with 0.20 mmol 11c and 0.40 mmol naphthalene-1-thiol in 1.0 mL of solvent, in the presence of 1 mol % 5 at rt. bIsolated yields. cDetermined by chiral HPLC analysis.
In DCM, 93% ee was attained for adduct 12c. In the light of this striking result, we decided to repeat the derivatization studies with DCM (Table 5).
Table 5: Results for the SMA of naphthalene-1-thiol to substituted trans-chalcones in DCM.
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-43-i7.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | 11 | R1, R2 | Product | Time (h) | Yieldb (%) | eec (%) |
| 1 | 11a | Ph, Ph | 12a | 21 | 90 | 66 |
| 2 | 11b | Ph, 3-MeC6H4 | 12b | 22 | 77 | 70 |
| 3 | 11c | Ph, 4-MeC6H4 | 12c | 6 | 94 | 94 |
| 4 | 11d | 4-MeC6H4, Ph | 12d | 21 | >99 | 86 |
| 5 | 11e | Ph, 3-OMeC6H4 | 12e | 24 | 73 | 63 |
| 6 | 11f | Ph, 4-OMeC6H4 | 12f | 23 | 93 | 84 |
| 7 | 11g | Ph, 3,4,5-(OMe)3C6H2 | 12g | 24 | >99 | 96 |
| 8 | 11h | Ph, 2-ClC6H4 | 12h | 23 | 85 | 44 |
| 9 | 11i | Ph, 3-ClC6H4 | 12i | 21 | 71 | 15 |
| 10 | 11j | Ph, 4-ClC6H4 | 12j | 21 | 86 | 51 |
| 11 | 11k | Ph, 3-BrC6H4 | 12k | 23 | 84 | 26 |
| 12 | 11l | 4-BrC6H4, Ph | 12l | 23 | 57 | 74 |
| 13 | 11m | Ph, 4-CF3C6H4 | 12m | 2 | 88 | 2 |
| 14 | 11n | 2-NO2C6H4, Ph | 12n | 21 | 91 | 74 |
| 15 | 11o | Ph, 4-NO2C6H4 | 12o | 23 | 92 | 6 |
aUnless stated otherwise, all reactions were performed with 0.20 mmol trans-chalcone and 0.40 mmol naphthalene-1-thiol in 1.0 mL of DCM, in the presence of 1 mol % 5 at rt. bIsolated yields. cDetermined by chiral HPLC analysis.
Employing DCM as the solvent showed significant improvements in the asymmetric induction for the chalcone derivatives having electron-donating methyl and methoxy substituents (Table 5, entries 3–7), especially with 4-tolyl- and 3,4,5-trimethoxyphenyl substituents. For the 4-tolyl derivative, the 23% ee value obtained with THF (Table 3, entry 3) was increased to 94% when switched to DCM (Table 5, entry 3). For the latter case, a significant improvement in the ee was observed from 50% (Table 3, entry 7) to 96% ee (Table 5, entry 7). The only exception to this pattern was with the 3-methyl derivative of chalcone, which resulted in a small decrease in enantioselectivity (78% to 70% ee, entry 2 in Table 3 and Table 5) when THF was changed to DCM. For halogens and electron-withdrawing substituents, an opposite behavior was observed. The use of DCM instead of THF led to lower ee values for the chalcone derivatives having the aforementioned substituents (Table 5, entries 8–15).
The most dramatic decreases in the selectivity were observed for derivatives 12m and 12o, for which the outcomes of the reactions were almost racemic (Table 5, entries 13 and 15). The solvent effects on the SMA of the chalcone derivatives and naphthalene-1-thiol are summarized in Figure 2. This behavior might be related to the better stabilization of the transition state of the substrates containing electron-withdrawing substituents or halogen atoms with THF, or vice versa.
![[1860-5397-17-43-2]](/bjoc/content/figures/1860-5397-17-43-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Comparison of the ee values of SMA in the presence of THF and DCM as solvent.
Figure 2: Comparison of the ee values of SMA in the presence of THF and DCM as solvent.
In order to enhance the potential bioactivity of the obtained enantioenriched products, selected SMA adducts (β-naphthyl-β-sulfanyl ketones) were subjected to oxidation with m-CPBA (Table 6) [45].
Table 6: Synthesis of enantioenriched sulfones from β-naphthyl-β-sulfanyl ketonesa.
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-43-i8.svg?max-width=637&scale=1.0)
|
||||
| Entrya | R | Product | Yieldb (%) | eec (%) |
| 1 | Ph | 13a | 43 | 86 |
| 2 | 3-OMeC6H4 | 13b | 45 | 68 |
| 3 | 4-OMeC6H4 | 13c | 62 | 86 |
| 4 | 4-ClC6H4 | 13d | 28 | 66 |
aUnless stated otherwise, all reactions were performed with 1 equiv β-aryl-β-sulfanyl ketone derivative and 2.2 equiv m-CPBA in DCM.
During the oxidation reaction, it was seen that the enantioenriched sulfa-Michael adducts undergo retro-sulfa-Michael reaction. The low yields of the oxidation and the fluctuations in enantioselectivity compared to the starting sulfa-Michael adducts can be attributed to this unpreventable retro-reaction. Despite this setback, the target sulfones were obtained with moderate to good ee values.
Conclusion
In conclusion, we report the enantioselective organocatalytic sulfa-Michael addition reaction of naphthalene-1-thiol to trans-chalcones, in the presence of a new bifunctional quinine derived sulfonamide organocatalyst. The adducts obtained with moderate to excellent ee values are β-naphthyl-β-sulfanyl ketones, which have potent activity against breast cancer. The easy access to the corresponding sulfones presents a versatile route for the implementation of a new biologically active moiety, the sulfone, to the β-naphthyl-β-sulfanyl ketones. The enantioenriched products of both classes can be evaluated as building blocks of new potential drug molecules.
Experimental
Materials and methods
All chemicals were purchased from Sigma-Aldrich or Acros Organics. Column chromatography was performed using silica gel 60 (200–300 mesh) as supporting material. All eluents were distilled prior to use. 1H NMR and 13C NMR spectra were recorded on a 400 MHz spectrometer, using CDCl3 as solvent. Chemical shift values are reported in ppm with TMS as standard, J values are given in Hertz. Optical rotations were determined by a polarimeter and reported as [α]DT (c in g/100 mL solvent). Enantiomeric excess values were determined by chiral HPLC chromatography using Agilent instrument. All new products were further analyzed by LC/MS–HRMS–TOF or MALDI–ESI–TOFMS.
Synthesis of organocatalyst 5
A solution of quinineamine 2 (226.40 mg, 0.70 mmol) and triethylamine (107 μL, 0.77 mmol) in CH2Cl2 was added to a screw-capped reaction vial. To this mixture, 2,4,6-trimethyl-3-nitrobenzenesulfonyl chloride 4 (184.59 mg, 0.70 mmol) was added at 0 °C. The mixture was then allowed to warm up to room temperature and stirred overnight. The crude product was directly loaded on a silica gel column, with ethyl acetate/triethylamine (98:2) as the eluent to afford the corresponding sulfonamide 5.
General procedure for the synthesis of 12a–o
A screw-capped reaction vial was charged with trans-chalcone derivative 11a–o (0.20 mmol) and organocatalyst 5 (1.10 mg, 0.0020 mmol) and 0.5 mL of THF or DCM. Then a solution of naphthalene-1-thiol (10, 55.4 µL, 0.40 mmol) in 1.0 mL of the selected solvent was introduced slowly into the vial with stirring. The mixture was stirred at room temperature and the progress of the reaction was monitored by TLC. Upon the consumption of the chalcone derivative, the reaction mixture was directly subjected to flash column chromatography, using a n-hexane/ethyl acetate mixture as eluent to yield the products 12a–o.
General procedure for the synthesis of sulfones
A solution of β-naphthyl-β-sulfanyl ketone (0.1 mmol) in 0.8 mL DCM was cooled to 0 °C. m-CPBA (0.22 mmol, 37.97 mg) was added to this stirred solution portionwise in 15 minutes. Then, the mixture was allowed to warm up to room temperature and stirred for a total of 30 minutes. After the completion of the reaction, the reaction mixture was diluted with 0.8 mL of DCM. Then washed with 3 × 0.8 mL of 5% K2CO3 (aq) and 3 × 1 mL of 5% NaHCO3 (aq) to remove the excess m-CPBA. The aqueous layer was extracted with DCM (3 × 1 mL). The organic layers were then combined, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica with using n-hexane/ethyl acetate as eluent to afford the target sulfones.
Supporting Information
| Supporting Information File 1: Copies of 1H and 13C NMR spectra, HPLC chromatograms and characterization data of the products. | ||
| Format: PDF | Size: 5.5 MB | Download |
References
-
Marcelli, T.; Hiemstra, H. Synthesis 2010, 1229–1279. doi:10.1055/s-0029-1218699
Return to citation in text: [1] -
Bae, H. Y.; Song, C. E. Bull. Korean Chem. Soc. 2014, 35, 1590–1600. doi:10.5012/bkcs.2014.35.6.1590
Return to citation in text: [1] -
Moran, A.; Hamilton, A.; Bo, C.; Melchiorre, P. J. Am. Chem. Soc. 2013, 135, 9091–9098. doi:10.1021/ja404784t
Return to citation in text: [1] -
Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036
Return to citation in text: [1] -
Chauhan, P.; Chimni, S. S. Tetrahedron: Asymmetry 2013, 24, 343–356. doi:10.1016/j.tetasy.2013.03.002
Return to citation in text: [1] -
Pellissier, H. Synthesis 2019, 51, 1311–1318. doi:10.1055/s-0037-1610350
Return to citation in text: [1] -
Montesinos-Magraner, M.; Vila, C.; Cantón, R.; Blay, G.; Fernández, I.; Muñoz, M. C.; Pedro, J. R. Angew. Chem., Int. Ed. 2015, 54, 6320–6324. doi:10.1002/anie.201501273
Return to citation in text: [1] -
Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Di Sabato, A.; Salvio, R.; Bella, M. Angew. Chem., Int. Ed. 2016, 55, 6525–6529. doi:10.1002/anie.201601660
Return to citation in text: [1] -
Yang, W.; He, H.-X.; Gao, Y.; Du, D.-M. Adv. Synth. Catal. 2013, 355, 3670–3678. doi:10.1002/adsc.201300670
Return to citation in text: [1] -
Zhao, Y.-L.; Fei, X.-H.; Tang, Y.-Q.; Xu, P.-F.; Yang, F.-F.; He, B.; Fu, X.-Z.; Yang, Y.-Y.; Zhou, M.; Mao, Y.-H.; Dong, Y.-X.; Li, C. J. Org. Chem. 2019, 84, 8168–8176. doi:10.1021/acs.joc.9b01142
Return to citation in text: [1] -
Karahan, S.; Tanyeli, C. New J. Chem. 2017, 41, 9192–9202. doi:10.1039/c7nj01395g
Return to citation in text: [1] -
Rana, N. K.; Singh, V. K. Org. Lett. 2011, 13, 6520–6523. doi:10.1021/ol202808n
Return to citation in text: [1] -
Connon, S. J. Chem. Commun. 2008, 2499. doi:10.1039/b719249e
Return to citation in text: [1] -
Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem. – Eur. J. 2011, 17, 6890–6899. doi:10.1002/chem.201003694
Return to citation in text: [1] -
Malerich, J. P.; Hagihara, K.; Rawal, V. H. J. Am. Chem. Soc. 2008, 130, 14416–14417. doi:10.1021/ja805693p
Return to citation in text: [1] -
Oh, S. H.; Rho, H. S.; Lee, J. W.; Lee, J. E.; Youk, S. H.; Chin, J.; Song, C. E. Angew. Chem., Int. Ed. 2008, 47, 7872–7875. doi:10.1002/anie.200801636
Return to citation in text: [1] -
Bae, H. Y.; Sim, J. H.; Lee, J.-W.; List, B.; Song, C. E. Angew. Chem., Int. Ed. 2013, 52, 12143–12147. doi:10.1002/anie.201306297
Return to citation in text: [1] -
Luo, J.; Xu, L.-W.; Hay, R. A. S.; Lu, Y. Org. Lett. 2009, 11, 437–440. doi:10.1021/ol802486m
Return to citation in text: [1] -
Park, S. E.; Nam, E. H.; Jang, H. B.; Oh, J. S.; Some, S.; Lee, Y. S.; Song, C. E. Adv. Synth. Catal. 2010, 352, 2211–2217. doi:10.1002/adsc.201000289
Return to citation in text: [1] -
Blise, K.; Cvitkovic, M. W.; Gibbs, N. J.; Roberts, S. F.; Whitaker, R. M.; Hofmeister, G. E.; Kohen, D. J. Org. Chem. 2017, 82, 1347–1355. doi:10.1021/acs.joc.6b02320
Return to citation in text: [1] -
Enders, D.; Lüttgen, K.; Narine, A. A. Synthesis 2007, 959–980. doi:10.1055/s-2007-965968
See for a comprehensive review on asymmetric SMA.
Return to citation in text: [1] -
Chauhan, P.; Mahajan, S.; Enders, D. Chem. Rev. 2014, 114, 8807–8864. doi:10.1021/cr500235v
See for a comprehensive review on organocatalytic C–S bond forming reactions.
Return to citation in text: [1] -
Marigo, M.; Schulte, T.; Franzén, J.; Jørgensen, K. A. J. Am. Chem. Soc. 2005, 127, 15710–15711. doi:10.1021/ja055291w
Return to citation in text: [1] -
Liu, Y.; Sun, B.; Wang, B.; Wakem, M.; Deng, L. J. Am. Chem. Soc. 2009, 131, 418–419. doi:10.1021/ja8085092
Return to citation in text: [1] -
Fang, X.; Li, J.; Wang, C.-J. Org. Lett. 2013, 15, 3448–3451. doi:10.1021/ol4015305
Return to citation in text: [1] -
Skarżewski, J.; Zielińska-Błajet, M.; Turowska-Tyrk, I. Tetrahedron: Asymmetry 2001, 12, 1923–1928. doi:10.1016/s0957-4166(01)00330-5
Return to citation in text: [1] -
McDaid, P.; Chen, Y.; Deng, L. Angew. Chem., Int. Ed. 2002, 41, 338–340. doi:10.1002/1521-3773(20020118)41:2<338::aid-anie338>3.0.co;2-m
Return to citation in text: [1] -
Hiemstra, H.; Wynberg, H. J. Am. Chem. Soc. 1981, 103, 417–430. doi:10.1021/ja00392a029
Return to citation in text: [1] -
Rana, N. K.; Selvakumar, S.; Singh, V. K. J. Org. Chem. 2010, 75, 2089–2091. doi:10.1021/jo902634a
Return to citation in text: [1] [2] -
Dai, L.; Wang, S.-X.; Chen, F.-E. Adv. Synth. Catal. 2010, 352, 2137–2141. doi:10.1002/adsc.201000334
Return to citation in text: [1] [2] [3] -
Corti, V.; Camarero Gonzalez, P.; Febvay, J.; Caruana, L.; Mazzanti, A.; Fochi, M.; Bernardi, L. Eur. J. Org. Chem. 2017, 49–52. doi:10.1002/ejoc.201601364
Return to citation in text: [1] -
Kumar, A.; Tripathi, V. D.; Kumar, P.; Gupta, L. P.; Akanksha; Trivedi, R.; Bid, H.; Nayak, V. L.; Siddiqui, J. A.; Chakravarti, B.; Saxena, R.; Dwivedi, A.; Siddiquee, M. I.; Siddiqui, U.; Konwar, R.; Chattopadhyay, N. Bioorg. Med. Chem. 2011, 19, 5409–5419. doi:10.1016/j.bmc.2011.07.056
Return to citation in text: [1] -
Ahari-Mostafavi, M. M.; Sharifi, A.; Mirzaei, M.; Amanlou, M. J. Iran. Chem. Soc. 2014, 11, 1113–1119. doi:10.1007/s13738-013-0379-1
Return to citation in text: [1] -
Meadows, D. C.; Sanchez, T.; Neamati, N.; North, T. W.; Gervay-Hague, J. Bioorg. Med. Chem. 2007, 15, 1127–1137. doi:10.1016/j.bmc.2006.10.017
Return to citation in text: [1] -
Velázquez, F.; Sannigrahi, M.; Bennett, F.; Lovey, R. G.; Arasappan, A.; Bogen, S.; Nair, L.; Venkatraman, S.; Blackman, M.; Hendrata, S.; Huang, Y.; Huelgas, R.; Pinto, P.; Cheng, K.-C.; Tong, X.; McPhail, A. T.; Njoroge, F. G. J. Med. Chem. 2010, 53, 3075–3085. doi:10.1021/jm9016027
Return to citation in text: [1] -
Otzen, T.; Wempe, E. G.; Kunz, B.; Bartels, R.; Lehwark-Yvetot, G.; Hänsel, W.; Schaper, K.-J.; Seydel, J. K. J. Med. Chem. 2004, 47, 240–253. doi:10.1021/jm030931w
Return to citation in text: [1] -
Yu, X.; Liu, Y.; Li, Y.; Wang, Q. J. Agric. Food Chem. 2016, 64, 3034–3040. doi:10.1021/acs.jafc.6b00645
Return to citation in text: [1] -
Galli, U.; Lazzarato, L.; Bertinaria, M.; Sorba, G.; Gasco, A.; Parapini, S.; Taramelli, D. Eur. J. Med. Chem. 2005, 40, 1335–1340. doi:10.1016/j.ejmech.2005.05.001
Return to citation in text: [1] -
Işık, M.; Tanyeli, C. J. Org. Chem. 2013, 78, 1604–1611. doi:10.1021/jo302713b
Return to citation in text: [1] [2] -
Kanberoğlu, E.; Tanyeli, C. Asian J. Org. Chem. 2016, 5, 114–119. doi:10.1002/ajoc.201500339
Return to citation in text: [1] [2] -
Bernardi, L.; Fini, F.; Herrera, R. P.; Ricci, A.; Sgarzani, V. Tetrahedron 2006, 62, 375–380. doi:10.1016/j.tet.2005.09.076
Return to citation in text: [1] -
Ricci, P.; Carlone, A.; Bartoli, G.; Bosco, M.; Sambri, L.; Melchiorre, P. Adv. Synth. Catal. 2008, 350, 49–53. doi:10.1002/adsc.200700382
Return to citation in text: [1] -
Guo, J.; Wong, M. W. J. Org. Chem. 2017, 82, 4362–4368. doi:10.1021/acs.joc.7b00388
Return to citation in text: [1] -
Wattanasin, S.; Murphy, W. S. Synthesis 1980, 647–650. doi:10.1055/s-1980-29155
Return to citation in text: [1] -
Konduru, N. K.; Dey, S.; Sajid, M.; Owais, M.; Ahmed, N. Eur. J. Med. Chem. 2013, 59, 23–30. doi:10.1016/j.ejmech.2012.09.004
Return to citation in text: [1]
| 1. | Marcelli, T.; Hiemstra, H. Synthesis 2010, 1229–1279. doi:10.1055/s-0029-1218699 |
| 2. | Bae, H. Y.; Song, C. E. Bull. Korean Chem. Soc. 2014, 35, 1590–1600. doi:10.5012/bkcs.2014.35.6.1590 |
| 3. | Moran, A.; Hamilton, A.; Bo, C.; Melchiorre, P. J. Am. Chem. Soc. 2013, 135, 9091–9098. doi:10.1021/ja404784t |
| 4. | Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036 |
| 5. | Chauhan, P.; Chimni, S. S. Tetrahedron: Asymmetry 2013, 24, 343–356. doi:10.1016/j.tetasy.2013.03.002 |
| 6. | Pellissier, H. Synthesis 2019, 51, 1311–1318. doi:10.1055/s-0037-1610350 |
| 17. | Bae, H. Y.; Sim, J. H.; Lee, J.-W.; List, B.; Song, C. E. Angew. Chem., Int. Ed. 2013, 52, 12143–12147. doi:10.1002/anie.201306297 |
| 18. | Luo, J.; Xu, L.-W.; Hay, R. A. S.; Lu, Y. Org. Lett. 2009, 11, 437–440. doi:10.1021/ol802486m |
| 19. | Park, S. E.; Nam, E. H.; Jang, H. B.; Oh, J. S.; Some, S.; Lee, Y. S.; Song, C. E. Adv. Synth. Catal. 2010, 352, 2211–2217. doi:10.1002/adsc.201000289 |
| 20. | Blise, K.; Cvitkovic, M. W.; Gibbs, N. J.; Roberts, S. F.; Whitaker, R. M.; Hofmeister, G. E.; Kohen, D. J. Org. Chem. 2017, 82, 1347–1355. doi:10.1021/acs.joc.6b02320 |
| 36. | Otzen, T.; Wempe, E. G.; Kunz, B.; Bartels, R.; Lehwark-Yvetot, G.; Hänsel, W.; Schaper, K.-J.; Seydel, J. K. J. Med. Chem. 2004, 47, 240–253. doi:10.1021/jm030931w |
| 16. | Oh, S. H.; Rho, H. S.; Lee, J. W.; Lee, J. E.; Youk, S. H.; Chin, J.; Song, C. E. Angew. Chem., Int. Ed. 2008, 47, 7872–7875. doi:10.1002/anie.200801636 |
| 37. | Yu, X.; Liu, Y.; Li, Y.; Wang, Q. J. Agric. Food Chem. 2016, 64, 3034–3040. doi:10.1021/acs.jafc.6b00645 |
| 12. | Rana, N. K.; Singh, V. K. Org. Lett. 2011, 13, 6520–6523. doi:10.1021/ol202808n |
| 13. | Connon, S. J. Chem. Commun. 2008, 2499. doi:10.1039/b719249e |
| 14. | Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem. – Eur. J. 2011, 17, 6890–6899. doi:10.1002/chem.201003694 |
| 15. | Malerich, J. P.; Hagihara, K.; Rawal, V. H. J. Am. Chem. Soc. 2008, 130, 14416–14417. doi:10.1021/ja805693p |
| 34. | Meadows, D. C.; Sanchez, T.; Neamati, N.; North, T. W.; Gervay-Hague, J. Bioorg. Med. Chem. 2007, 15, 1127–1137. doi:10.1016/j.bmc.2006.10.017 |
| 7. | Montesinos-Magraner, M.; Vila, C.; Cantón, R.; Blay, G.; Fernández, I.; Muñoz, M. C.; Pedro, J. R. Angew. Chem., Int. Ed. 2015, 54, 6320–6324. doi:10.1002/anie.201501273 |
| 8. | Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Di Sabato, A.; Salvio, R.; Bella, M. Angew. Chem., Int. Ed. 2016, 55, 6525–6529. doi:10.1002/anie.201601660 |
| 9. | Yang, W.; He, H.-X.; Gao, Y.; Du, D.-M. Adv. Synth. Catal. 2013, 355, 3670–3678. doi:10.1002/adsc.201300670 |
| 10. | Zhao, Y.-L.; Fei, X.-H.; Tang, Y.-Q.; Xu, P.-F.; Yang, F.-F.; He, B.; Fu, X.-Z.; Yang, Y.-Y.; Zhou, M.; Mao, Y.-H.; Dong, Y.-X.; Li, C. J. Org. Chem. 2019, 84, 8168–8176. doi:10.1021/acs.joc.9b01142 |
| 11. | Karahan, S.; Tanyeli, C. New J. Chem. 2017, 41, 9192–9202. doi:10.1039/c7nj01395g |
| 35. | Velázquez, F.; Sannigrahi, M.; Bennett, F.; Lovey, R. G.; Arasappan, A.; Bogen, S.; Nair, L.; Venkatraman, S.; Blackman, M.; Hendrata, S.; Huang, Y.; Huelgas, R.; Pinto, P.; Cheng, K.-C.; Tong, X.; McPhail, A. T.; Njoroge, F. G. J. Med. Chem. 2010, 53, 3075–3085. doi:10.1021/jm9016027 |
| 27. | McDaid, P.; Chen, Y.; Deng, L. Angew. Chem., Int. Ed. 2002, 41, 338–340. doi:10.1002/1521-3773(20020118)41:2<338::aid-anie338>3.0.co;2-m |
| 28. | Hiemstra, H.; Wynberg, H. J. Am. Chem. Soc. 1981, 103, 417–430. doi:10.1021/ja00392a029 |
| 29. | Rana, N. K.; Selvakumar, S.; Singh, V. K. J. Org. Chem. 2010, 75, 2089–2091. doi:10.1021/jo902634a |
| 30. | Dai, L.; Wang, S.-X.; Chen, F.-E. Adv. Synth. Catal. 2010, 352, 2137–2141. doi:10.1002/adsc.201000334 |
| 32. | Kumar, A.; Tripathi, V. D.; Kumar, P.; Gupta, L. P.; Akanksha; Trivedi, R.; Bid, H.; Nayak, V. L.; Siddiqui, J. A.; Chakravarti, B.; Saxena, R.; Dwivedi, A.; Siddiquee, M. I.; Siddiqui, U.; Konwar, R.; Chattopadhyay, N. Bioorg. Med. Chem. 2011, 19, 5409–5419. doi:10.1016/j.bmc.2011.07.056 |
| 23. | Marigo, M.; Schulte, T.; Franzén, J.; Jørgensen, K. A. J. Am. Chem. Soc. 2005, 127, 15710–15711. doi:10.1021/ja055291w |
| 24. | Liu, Y.; Sun, B.; Wang, B.; Wakem, M.; Deng, L. J. Am. Chem. Soc. 2009, 131, 418–419. doi:10.1021/ja8085092 |
| 25. | Fang, X.; Li, J.; Wang, C.-J. Org. Lett. 2013, 15, 3448–3451. doi:10.1021/ol4015305 |
| 26. | Skarżewski, J.; Zielińska-Błajet, M.; Turowska-Tyrk, I. Tetrahedron: Asymmetry 2001, 12, 1923–1928. doi:10.1016/s0957-4166(01)00330-5 |
| 33. | Ahari-Mostafavi, M. M.; Sharifi, A.; Mirzaei, M.; Amanlou, M. J. Iran. Chem. Soc. 2014, 11, 1113–1119. doi:10.1007/s13738-013-0379-1 |
| 22. |
Chauhan, P.; Mahajan, S.; Enders, D. Chem. Rev. 2014, 114, 8807–8864. doi:10.1021/cr500235v
See for a comprehensive review on organocatalytic C–S bond forming reactions. |
| 21. |
Enders, D.; Lüttgen, K.; Narine, A. A. Synthesis 2007, 959–980. doi:10.1055/s-2007-965968
See for a comprehensive review on asymmetric SMA. |
| 30. | Dai, L.; Wang, S.-X.; Chen, F.-E. Adv. Synth. Catal. 2010, 352, 2137–2141. doi:10.1002/adsc.201000334 |
| 31. | Corti, V.; Camarero Gonzalez, P.; Febvay, J.; Caruana, L.; Mazzanti, A.; Fochi, M.; Bernardi, L. Eur. J. Org. Chem. 2017, 49–52. doi:10.1002/ejoc.201601364 |
| 40. | Kanberoğlu, E.; Tanyeli, C. Asian J. Org. Chem. 2016, 5, 114–119. doi:10.1002/ajoc.201500339 |
| 38. | Galli, U.; Lazzarato, L.; Bertinaria, M.; Sorba, G.; Gasco, A.; Parapini, S.; Taramelli, D. Eur. J. Med. Chem. 2005, 40, 1335–1340. doi:10.1016/j.ejmech.2005.05.001 |
| 39. | Işık, M.; Tanyeli, C. J. Org. Chem. 2013, 78, 1604–1611. doi:10.1021/jo302713b |
| 45. | Konduru, N. K.; Dey, S.; Sajid, M.; Owais, M.; Ahmed, N. Eur. J. Med. Chem. 2013, 59, 23–30. doi:10.1016/j.ejmech.2012.09.004 |
| 43. | Guo, J.; Wong, M. W. J. Org. Chem. 2017, 82, 4362–4368. doi:10.1021/acs.joc.7b00388 |
| 44. | Wattanasin, S.; Murphy, W. S. Synthesis 1980, 647–650. doi:10.1055/s-1980-29155 |
| 30. | Dai, L.; Wang, S.-X.; Chen, F.-E. Adv. Synth. Catal. 2010, 352, 2137–2141. doi:10.1002/adsc.201000334 |
| 41. | Bernardi, L.; Fini, F.; Herrera, R. P.; Ricci, A.; Sgarzani, V. Tetrahedron 2006, 62, 375–380. doi:10.1016/j.tet.2005.09.076 |
| 29. | Rana, N. K.; Selvakumar, S.; Singh, V. K. J. Org. Chem. 2010, 75, 2089–2091. doi:10.1021/jo902634a |
| 42. | Ricci, P.; Carlone, A.; Bartoli, G.; Bosco, M.; Sambri, L.; Melchiorre, P. Adv. Synth. Catal. 2008, 350, 49–53. doi:10.1002/adsc.200700382 |
| 40. | Kanberoğlu, E.; Tanyeli, C. Asian J. Org. Chem. 2016, 5, 114–119. doi:10.1002/ajoc.201500339 |
| 39. | Işık, M.; Tanyeli, C. J. Org. Chem. 2013, 78, 1604–1611. doi:10.1021/jo302713b |
© 2021 Tözendemir and Tanyeli; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)