Abstract
Synthesizing organocatalysts is often a long and cost-intensive process, therefore, the recovery and reuse of the catalysts are particularly important to establish sustainable organocatalytic transformations. In this work, we demonstrate the synthesis, application, and recycling of a new lipophilic cinchona squaramide organocatalyst. The synthesized lipophilic organocatalyst was applied in Michael additions. The catalyst was utilized to promote the Michael addition of cyclohexyl Meldrum’s acid to 4-chloro-trans-β-nitrostyrene (quantitative yield, up to 96% ee). Moreover, 1 mol % of the catalyst was feasible to conduct the gram-scale preparation of baclofen precursor (89% yield, 96% ee). Finally, thanks to the lipophilic character of the catalyst, it was easily recycled after the reaction by replacing the non-polar reaction solvent with a polar solvent, acetonitrile, with 91–100% efficiency, and the catalyst was reused in five reaction cycles without the loss of activity and selectivity.
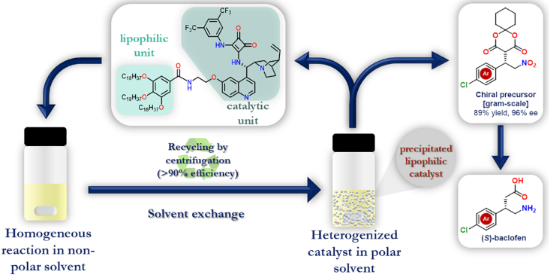
Graphical Abstract
Introduction
In today’s chemical industry, catalytic processes are of paramount importance. In particular, the application of asymmetric organocatalysts is receiving increased attention [1-4]. This is illustrated by the fact that in 2021 the Nobel Prize in Chemistry was awarded for the discovery of asymmetric organocatalysis [5]. The use of organocatalysts has been a major breakthrough in the realization of enantioselective transformations. Stereoselective synthesis is essential in the pharmaceutical industry, as the development of drugs often requires the production of enantiomerically pure chiral compounds [6-8].
The application of organocatalysts is well-established in several organic reactions, including but not limited to aldol reactions [6], Michael additions [9], Mannich reactions [10], aza-Henry reactions [11], and Diels–Alder cycloadditions [12,13]. Although the benefits of organocatalysts are undoubted, their synthesis is often a long and expensive process. Therefore, for sustainable application, the cost-efficient recovery and reuse of organocatalysts are critical issues. Fortunately, a wide range of recycling options are known in the literature, often based on liquid–solid phase separation [14].
Catalyst recycling can be achieved, for example, by immobilizing the catalysts to a solid support [15], e.g., silica gel [16-18], organic polymers [19-21], magnetic nanoparticles [22,23], or by membrane separation, e.g., using organic solvent nanofiltration (OSN) [24-26], which methods can be easily implemented in flow systems. Accordingly, the main recycling methods rely on the immobilization of catalysts on heterogeneous supports, however, this could often lead to the deterioration of activity and/or selectivity [27]. A possible solution to avoid these drawbacks is the heterogenization of the catalyst after a homogeneous reaction.
For example, by incorporating a lipophilic side chain [28] on the organocatalyst that does not interfere with its catalytic activity thanks to a linker between the catalyst and lipophilic units. In this way, a significant difference in polarity can be achieved between the catalyst and the other components of the reaction mixture. The lipophilic O-alkylated gallic acid unit increases the solubility of the organocatalyst in less polar solvents, such as DCM or toluene but leads to the precipitation of the organocatalyst in polar solvents, including MeOH or MeCN. As a result, the recycling of the organocatalysts can be achieved in a simple step by centrifugation or filtration.
Previously, we have demonstrated the homogeneous and heterogeneous recycling of cinchona-based organocatalysts [20,25,26,29]. Continuing our work, we aimed to synthesize a novel, recyclable lipophilic cinchona squaramide organocatalyst. Its catalytic activity and recyclability were examined in a new stereoselective synthesis of baclofen, which is used to treat muscle spasms [30]. Finally, the catalyst was easily recycled by centrifugation over five reaction cycles without significant loss of activity (Figure 1).
![[1860-5397-19-133-1]](/bjoc/content/figures/1860-5397-19-133-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Application of cinchona squaramide 1 and recyclable, lipophilic cinchona squaramide organocatalysts 2 in a new, gram-scale stereoselective synthesis of baclofen precursor.
Figure 1: Application of cinchona squaramide 1 and recyclable, lipophilic cinchona squaramide organocatalysts ...
Results and Discussion
Synthesis of the lipophilic cinchona squaramide organocatalyst
Previously, we successfully applied quinine-derived squaramide (SQ) organocatalyst 1 in stereoselective Michael and aza-Michael additions with excellent enantiomeric excess values [26]. Our aim was to recycle this catalyst easily by incorporating a lipophilic unit, which leads to a drastic increase (5.78 to 28.8) in the logP value of the organocatalyst 2. The recyclable organocatalyst can be divided into three units: the catalytic unit, the linker, and the lipophilic tag with octadecyl chains (Figure 1).
The cinchona amine 3 was prepared starting from the naturally occurring quinine [31]. The gained catalyst was demethylated using BBr3 to give alcohol 4. The demethylated cinchona amine was reacted with half-squaramide [9] 5, resulting in demethylated squaramide 6. A short and flexible linker was applied between the catalytic and lipophilic units to avoid a decrease in the catalytic activity. The demethylated cinchona squaramide 6 was reacted with O-p-toluenesulfonyl-N-Boc-ethanolamine. The protecting group was removed using trifluoroacetic acid, followed by a neutralization step, gaining the cinchona squaramide organocatalyst 7 with a linker (Scheme 1).
![[1860-5397-19-133-i1]](/bjoc/content/inline/1860-5397-19-133-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of demethylated cinchona squaramide organocatalyst and the incorporation of the flexible 2-aminoethylene linker.
Scheme 1: Synthesis of demethylated cinchona squaramide organocatalyst and the incorporation of the flexible ...
The lipophilic unit from methyl gallate (8) was gained by following a literature procedure [32] with minor modifications. The octadecyl groups were attached to the hydroxy groups using Williamson-type ether synthesis. The octadecylated gallic acid ester 9 was hydrolyzed under basic conditions in an ethanol/water mixture. After the reaction, the pH of the mixture was adjusted to 4 with hydrochloric acid, which resulted in the precipitation of the product 10 in excellent yield (95%). Next, carboxylic acid 10 was converted into the corresponding acyl chloride 11 with thionyl chloride. Finally, the cinchona squaramide with linker 7 and the octadecylated gallic acid chloride 11 were coupled to form an amide using triethylamine as a base. The crude product was purified by chromatography and precipitated from acetonitrile to gain the lipophilic organocatalyst 2 (Scheme 2).
![[1860-5397-19-133-i2]](/bjoc/content/inline/1860-5397-19-133-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the lipophilic tag from methyl gallate (8) and attachment to the cinchona squaramide.
Scheme 2: Synthesis of the lipophilic tag from methyl gallate (8) and attachment to the cinchona squaramide.
Application and recycling of the lipophilic cinchona-squaramide organocatalyst in the stereoselective Michael addition
To prove that the previously applied catalytic unit kept its activity, the lipophilic organocatalyst 2 was applied in the stereoselective Michael addition of trans-β-nitrostyrene (12) and acetylacetone (13). Choosing the best solvent for the reaction is crucial, thus, solubility tests were carried out (Table 1). Since homogeneous catalysts usually exhibit higher activity and selectivity than their heterogeneous counterparts [27], our aim was to carry out the catalytic reaction homogeneously. The solubility of the lipophilic catalyst 2 was investigated in ten solvents with low polarity, including a new, bio-based polar aprotic solvent, MeSesamol [33]. The catalyst’s precipitation – which is necessary for its recycling – was tested by adding a polar solvent, i.e., methanol, to its solution.
Table 1: Catalyst 2 solubility in solvents with low polarity.a
| Solvent | Dissolved? | Precipitated by MeOH? |
| anisole |
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-133-i5.png?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-133-i6.png?max-width=637&scale=1.0)
|
| butyl acetate |
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-133-i7.png?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-19-133-i8.png?max-width=637&scale=1.0)
|
| cyclohexaneb |
![[Graphic 5]](/bjoc/content/inline/1860-5397-19-133-i9.png?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-19-133-i10.png?max-width=637&scale=1.0)
|
| cyclopentyl methyl ether (CPME) |
![[Graphic 7]](/bjoc/content/inline/1860-5397-19-133-i11.png?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-19-133-i12.png?max-width=637&scale=1.0)
|
| dichloromethane (DCM) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-19-133-i13.png?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-19-133-i14.png?max-width=637&scale=1.0)
|
| dimethyl carbonate (DMC) |
![[Graphic 11]](/bjoc/content/inline/1860-5397-19-133-i15.png?max-width=637&scale=1.0)
|
– |
| heptane |
![[Graphic 12]](/bjoc/content/inline/1860-5397-19-133-i16.png?max-width=637&scale=1.0)
|
– |
| MeSesamol [33] |
![[Graphic 13]](/bjoc/content/inline/1860-5397-19-133-i17.png?max-width=637&scale=1.0)
|
– |
| 2-methyltetrahydrofuran (2-MeTHF) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-19-133-i18.png?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-19-133-i19.png?max-width=637&scale=1.0)
|
| toluene |
![[Graphic 16]](/bjoc/content/inline/1860-5397-19-133-i20.png?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-19-133-i21.png?max-width=637&scale=1.0)
|
aTo the lipophilic catalyst (2 mg) in a vial the appropriate solvent (200 µL) was added. Then, to check the precipitability of the catalyst, MeOH (800 µL) was added. bLarger amounts of cyclohexane (1 mL) was needed to dissolve the catalyst.
The main requirement for the polar solvent is that it should not dissolve the catalyst while it should completely dissolve the product. Therefore, we investigated the solubility of the Michael adduct 14 in methanol, ethanol, propan-2-ol, Patosolv® (a mixture of 85% of ethanol and 15% of propan-2-ol), and acetonitrile. The highest solubility of 14 was found in acetonitrile (63 mg mL−1) and methanol (17 mg mL−1). In both of these solvents, a low solubility of the lipophilic catalyst 2 was measured (<0.5 mg mL−1). Based on these results, acetonitrile was chosen as a precipitating solvent for the catalyst recycling.
To investigate the solvent effect, the stereoselective Michael addition reaction was carried out in those solvents that dissolved the catalyst and from which the catalyst was successfully precipitated by adding methanol. Furthermore, a reaction in which acetylacetone (13) did not only act as a reactant but also as a solvent was examined (Table 2).
Table 2: Solvent screening in the Michael addition of acetylacetone (13) to trans-β-nitrostyrene (12).a
![[Graphic 18]](/bjoc/content/inline/1860-5397-19-133-i22.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Yieldb [%] | eec [%] |
| 1 | CPME | 85 | 91 |
| 2 | toluene | 88 | 93 |
| 3 | DCM | 91 | 93 |
| 4 | acetylacetone | 89 | 88 |
| 5 | butyl acetate | 22 | 94 |
| 6 | 2-MeTHF | 94 | 87 |
| 7 | anisole | 84 | 89 |
aReaction conditions: acetylacetone (13, 0.21 mmol) was added to the solution of trans-β-nitrostyrene (12, 0.08 mmol) and 5 mol % of catalyst 2 in 0.5 mL of solvent, then, the resulting mixture was stirred at room temperature for 24 hours. bIsolated yields. cDetermined by chiral HPLC ((S)-enantiomer).
The highest yields and enantiomeric excess values were reached in CPME, toluene, and dichloromethane (Table 2, entries 1–3). When acetylacetone was used as solvent, the enantiomeric excess was slightly lower because the catalyst was not completely dissolved in acetylacetone, which resulted in a heterogeneous reaction mixture (Table 2, entry 4). The importance of correct solvent selection is illustrated by the case of butyl acetate, in which only a low yield was observed (Table 2, entry 5). During solvent selection, both the catalytic performance and green chemistry aspects were addressed. For this purpose, we followed GSK’s solvent sustainability guide [34], which ranks solvents according to their properties, such as waste generation, environmental and health impacts, and boiling point (Figure 2).
![[1860-5397-19-133-2]](/bjoc/content/figures/1860-5397-19-133-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Classification of the tested non-polar solvents according to the GSK’s solvent sustainability guide [34].
Figure 2: Classification of the tested non-polar solvents according to the GSK’s solvent sustainability guide ...
Considering the three factors mentioned above (yield, enantiomeric excess, and green chemistry), toluene was chosen as a solvent for the recycling reactions. The schematic for the recycling by solvent replacement is shown in Figure 3.
![[1860-5397-19-133-3]](/bjoc/content/figures/1860-5397-19-133-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Recycling of the lipophilic organocatalyst in the stereoselective Michael addition by replacing the solvent.
Figure 3: Recycling of the lipophilic organocatalyst in the stereoselective Michael addition by replacing the...
After the stereoselective Michael addition was completed in non-polar toluene, toluene was evaporated in vacuo and then a polar solvent, acetonitrile was added, leading to the precipitation of the lipophilic catalyst but dissolution of the other reaction components. The reaction mixture was then transferred to Eppendorf vials and centrifuged (8 min, 13500 rpm). After phase separation, the product was isolated from the supernatant by preparative thin-layer chromatography, while the catalyst was reused in subsequent reaction cycles. The precipitated catalyst was further washed twice with acetonitrile, and dried in vacuo. The results are collected in Table 3.
Table 3: Recycling of lipophilic organocatalyst 2 in the Michael addition of acetylacetone (13) to trans-β-nitrostyrene (12).a
![[Graphic 19]](/bjoc/content/inline/1860-5397-19-133-i23.svg?max-width=637&scale=1.0)
|
|||
| Round | Yieldb [%] | eec [%] | Catalyst recycling efficiency [%] |
| 1 | 84 | 91 | 91 |
| 2 | 89 | 90 | 99 |
| 3 | 94 | 91 | 92 |
| 4 | 93 | 91 | 99 |
| 5 | 96 | 92 | 97 |
aReaction conditions: acetylacetone (13, 0.96 mmol) was added to the solution of trans-β-nitrostyrene (12, 0.38 mmol) and 5 mol % of catalyst 2 in 2 mL of toluene, then, the resulting mixture was stirred at room temperature for 24 hours. After the reaction was completed, the volatile components were evaporated, and acetonitrile was added for the recycling of the catalyst 2. bIsolated yields. cDetermined by chiral HPLC ((S)-enantiomer).
Application and recycling of the lipophilic cinchona-squaramide organocatalyst in the synthesis of a baclofen precursor
After the successful application of the lipophilic catalyst, its catalytic activity was also investigated in another industrially relevant stereoselective Michael addition. This type of reaction could be used in the synthesis of several drugs to form a carbon–carbon bond in a stereoselective manner [6,35]. Our goal was to synthesize the chiral precursor 17 of baclofen (Scheme 3).
![[1860-5397-19-133-i3]](/bjoc/content/inline/1860-5397-19-133-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: A new, stereoselective synthetic route for baclofen.
Scheme 3: A new, stereoselective synthetic route for baclofen.
To achieve this objective, we first planned to use Meldrum’s acid and 4-chloro-trans-β-nitrostyrene (16). Based on the literature [36], Meldrum’s acid has a low solubility in non-polar solvents, resulting in diminished enantioselectivity. Furthermore, in our case, the application of polar solvents would not be favorable due to the low solubility of the lipophilic organocatalyst in these solvents. Consequently, we applied the cyclohexyl derivative of Meldrum’s acid 15, which exhibits enhanced solubility in non-polar solvents [36]. The Meldrum’s acid derivative 15 was synthesized from malonic acid and cyclohexanone using acetic anhydride and sulfuric acid as a catalyst (see Supporting Information File 1) [37].
In a similar manner as the former Michael addition, a solvent screening was carried out in the previously well-established four solvents: CPME, 2-MeTHF, anisole, and toluene (Table 4, entries 1–4).
Table 4: Solvent and catalyst amount screening in the Michael addition of the cyclohexyl derivative of Meldrum’s acid 15 to 4-chloro-trans-β-nitrostyrene (16).a
![[Graphic 20]](/bjoc/content/inline/1860-5397-19-133-i24.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Catalyst amount [mol%] | Yieldb [%] | eec [%] |
| 1 | CPME | 10 | 93 | 82 |
| 2 | 2-MeTHF | 10 | 93 | 29 |
| 3 | anisole | 10 | >99 | 93 |
| 4 | toluene | 10 | >99 | 92 |
| 5 | toluene | 5 | 97 | 93 |
| 6d | toluene | 1 | 94 | 96 |
aReaction conditions: Meldrum’s acid derivative 15 (0.064 mmol) was added to the solution of 4-chloro-trans-β-nitrostyrene (16, 0.096 mmol) and 1, 5 or 10 mol % of catalyst 2 in 470 µL of solvent, then, the resulting mixture was stirred at room temperature for 3 hours. bIsolated yields. cDetermined by chiral HPLC ((S)-enantiomer). dReaction time was 5 hours to achieve full conversion.
Based on the solvent screening, high yields and good enantioselectivity can be achieved in anisole and toluene, while using 2-MeTHF drastically decreased the enantioselectivity. From the perspective of green chemistry, anisole would be more favorable, but toluene is also a distinctly better alternative to the other solvents (e.g., DCM) commonly used in Michael additions. Another critical aspect that needs to be considered is how difficult the removal of the solvent is. In this respect, the boiling point of toluene (111 °C) is preferable to that of anisole, which has a boiling point of 154 °C. The results of catalyst recycling in anisole and toluene are shown in Table 5.
Table 5: Recycling of lipophilic organocatalyst 2 in the Michael addition of the cyclohexyl derivative of Meldrum’s acid 15 to 4-chloro-trans-β-nitrostyrene (16).a
![[Graphic 21]](/bjoc/content/inline/1860-5397-19-133-i25.svg?max-width=637&scale=1.0)
|
|||
| Round | Solvent | Yieldb [%] | eec [%] |
| 1 | anisole | 95 | 92 |
| 2 | >99 | 95 | |
| 3 | >99 | 94 | |
| 4 | >99 | 93 | |
| 5 | >99 | 93 | |
| 1 | toluene | 94 | 93 |
| 2 | >99 | 94 | |
| 3 | >99 | 94 | |
| 4 | >99 | 94 | |
| 5 | 97 | 92 | |
aReaction conditions: Meldrum’s acid derivative 15 (0.19 mmol) was added to the solution of 4-chloro-trans-β-nitrostyrene (16, 0.29 mmol) and 10 mol % of catalyst 2 in 1.4 mL of solvent, then, the resulting mixture was stirred at room temperature for 3 hours. After the reaction was completed, the volatile components were evaporated, and acetonitrile was added for the recycling of the catalyst 2. bIsolated yields. cDetermined by chiral HPLC ((S)-enantiomer).
The lipophilic organocatalyst maintained its activity in both solvents over the five reaction cycles while the catalyst loss was marginal (<10%). It is important to note that in an industrial-scale process, the catalyst loss could be further diminished, and the centrifugation could be replaced by a simple filtration. Furthermore, for large-scale applications, the effect of catalyst amount was also investigated (Table 4, entries 4–6). Yields and enantiomeric excess values did not change significantly when the catalyst amount was reduced from 10 mol % to 1 mol %, however, a longer reaction time (5 hours) was required for the 1 mol % catalyst loading.
Gram-scale synthesis of baclofen
Finally, we planned to demonstrate the recyclability of our lipophilic organocatalyst 2 in the gram-scale synthesis of baclofen precursor (S)-17. The catalyst loading was set to 1 mol % to reduce the needed catalyst amount and the reaction time was increased to 5 hours to achieve full conversion. After the organocatalytic reaction in toluene, the volatile components were evaporated, and acetonitrile was added to precipitate the catalyst. In contrast to the small-scale recycling, in this case, we used filtration instead of centrifugation to recover the catalyst without significant loss (<5%). This demonstrates that we developed an organocatalytic reaction that can be easily scaled-up and the novel lipophilic catalyst can be recovered not only by centrifugation but also by filtration.
From the baclofen precursor (S)-17, baclofen can be synthesized in two steps. The nitro acid (S)-18 was obtained using HCl in THF in good yield (70%), which could be reduced to (S)-baclofen hydrochloride using Raney nickel as catalyst (Scheme 4) [38].
![[1860-5397-19-133-i4]](/bjoc/content/inline/1860-5397-19-133-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Gram-scale synthesis of (S)-baclofen hydrochloride.
Scheme 4: Gram-scale synthesis of (S)-baclofen hydrochloride.
Conclusion
In conclusion, we have prepared a new lipophilic cinchona squaramide organocatalyst 2 modified with octadecyl side chains. Thanks to the lipophilic unit, the catalyst can be easily precipitated by exchanging the non-polar solvent with a more polar one, and then its separation can be achieved by centrifugation. The lipophilic catalyst 2 demonstrated its excellent catalytic activity in two stereoselective Michael addition reactions. Homogeneous catalysis was carried out in non-polar solvents (i.e., toluene), which allows the high performance of the lipophilic organocatalyst in terms of yield and stereoselectivity. To facilitate the pharmaceutical use of the lipophilic organocatalyst, we investigated a new, industry-relevant synthesis route of baclofen in the gram-scale. The chiral precursor 17 of baclofen was obtained with quantitative yield and excellent enantiomeric excess (up to 96%). The catalyst was applied in five consecutive runs without a decrease in catalytic activity, moreover, the catalyst loss was also negligible (<10%). Overall, it can be concluded that the incorporation of the lipophilic unit does not affect the catalytic activity and selectivity but enables the facile recycling of the catalyst.
Experimental
General information
The starting materials and reagents were purchased from commercially available sources (Merck, TCI Europe, and VWR). Infrared (IR) spectra were recorded on a Bruker Alpha-T Fourier-transform IR (FTIR) spectrometer. Optical rotations were measured on a Perkin Elmer 241 polarimeter calibrated by measuring the optical rotations of both enantiomers of menthol. The reactions under pressure were carried out in a 150 mL pressure flask (Synthware Glass). Thin-layer chromatography (TLC) was performed using silica gel 60 F254 (Merck) plates. The spots of the materials on TLC plates were visualized by UV light at 254 nm. The reactions were monitored by TLC and high-performance liquid chromatography–mass spectrometry (HPLC–MS). The solvent ratios of the eluents are given in volume units (mL mL−1). Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DRX-500 Avance spectrometer (at 500 and 126 MHz for the 1H and 13C spectra, respectively) or on a Bruker 300 Avance spectrometer (at 300 and 75.5 MHz for the 1H and 13C spectra, respectively) or on a Bruker Avance III HD (at 600 MHz for 1H and at 150 MHz for 13C spectra) at specified temperatures. High-resolution MS was measured on a Bruker MicroTOF II instrument using positive electrospray ionization. HPLC–MS was performed on an HPLC system using a Shimadzu LCMS-2020 (Shimadzu Corp., Japan) device equipped with a Reprospher (Altmann Analytik Corp., Germany) 100 Å C18 (5 µm; 100 × 3 mm) column and a positive/negative double ion source with a quadrupole MS analyzer in the range of 50–1000 m/z. Further details are available in Supporting Information File 1. The enantiomeric ratios of the samples were determined by chiral high-performance liquid chromatography (HPLC) measurements. The exact conditions of chiral HPLC are indicated in the experimental section of the corresponding compound. MarvinSketch was used for logP prediction, MarvinSketch 20.11, ChemAxon (https://www.chemaxon.com).
Demethylated cinchona squaramide 6
The demethylated cinchona squaramide was prepared according to the literature procedure [39]. To the best of our knowledge, the NMR assignment has not been reported yet. 1H NMR (methanol-d4, 600 MHz, 295 K) δ 8.68 (d, 3JH,H = 4.7 Hz, 1H), 8.01 (bs, 2H), 7.94 (d, 3JH,H = 9.1 Hz, 1H), 7.72 (bs, 1H), 7.55 (m, 2H), 7.40 (dd, 3JH,H = 9.1 Hz, 4JH,H = 2.5 Hz, 1H), 6.15 (bs, 1H), 5.91 (m, 1H), 5.07 (bd, 2JH,H = 17.2 Hz, 1H), 5.02 (bd, 2JH,H = 10.4 Hz, 1H), 3.49 (m, 1H), 3.48 (m, 1H), 3.33 (m, 1H), 2.86 (m, 1H), 2.82 (m, 1H), 2.41 (bs, 1H), 1.69 (m, 1H), 1.68 (m, 2H), 1.54 (m, 1H), 0.85 (m, 1H); 13C NMR (methanol-d4, 150 MHz, 295 K) δ 185.7, 182.1, 170.4, 164.8, 158.3, 147.6, 145.3, 144.6, 142.5, 142.3, 133.9 (q, 2JC,F = 33.4 Hz), 131.7, 129.7, 124.5 (q, 1JC,F = 272.0 Hz), 123.9, 120.0, 119.3 (q, 3JC,F = 3.0 Hz), 116.7 (m), 115.2, 105.4, 61.9, 56.9, 54.7, 41.8, 40.7, 28.8, 28.3, 26.9; for the full 1H and 13C assignment of demethylated cinchona squaramide structure, see Supporting Information File 1.
Cinchona squaramide organocatalyst with linker, 7
![[Graphic 22]](/bjoc/content/inline/1860-5397-19-133-i26.svg?max-width=637&scale=1.18182)
The demethylated cinchona squaramide 6 (732 mg, 1.19 mmol) was dissolved in DMF (15 mL), and caesium carbonate (1.75 g, 5.37 mmol, 4.5 equiv) was added to the solution at 0 °C. The reaction mixture was stirred for 20 min at this temperature. Then, the DMF (10 mL) solution of tosylated N-Boc-protected ethanolamine (955 mg, 3.03 mmol, 2.5 equiv, Scheme 2) was added dropwise. The reaction mixture was stirred at 50 °C for 8 hours. The volatile components were removed under reduced pressure. To the resulting orange oil, water (60 mL) was added, and the product was extracted with dichloromethane (3 × 60 mL). The combined organic phase was washed with water (2 × 60 mL), dried over MgSO4, and concentrated in vacuo. The intermediate (1.24 g) was used in the next reaction without purification. The crude, N-Boc-protected intermediate was dissolved in dichloromethane (25 mL) and the solution cooled to 0 °C with an ice bath. Next, trifluoroacetic acid (5.74 mL, 75 mmol, 63 equiv) was added dropwise. The reaction mixture was stirred at room temperature for 1 h. Then, it was cooled to 0 °C, and a 40% NaOH(aq) solution was added to set the pH to 13. To this mixture, water (60 mL) was added, and it was extracted with DCM/MeOH 20:1 (60 mL). After the separation of the phases, the aqueous phase was extracted again with DCM/MeOH 20:1 (3 × 40 mL). The combined organic phase was dried over MgSO4 and concentrated in vacuo. The obtained residue (609 mg, 78% for 2 steps) was a dark orange solid, and we used this product in the next step without further purifications. TLC (SiO2; DCM/MeOH/25% NH4OH(aq) 10:1:0.01, Rf 0.22); mp 158–160 °C; ![[Graphic 23]](/bjoc/content/inline/1860-5397-19-133-i27.svg?max-width=637&scale=1.18182) −51.8 (c 1.00, DMSO); IR (cm−1) νmax: 2957, 2923, 2853, 1664, 1620, 1609, 1560, 1508, 1437, 1378, 1331, 1277, 1201, 1175, 1126, 1021, 930, 884, 832, 799, 719, 700, 679, 620, 550, 521, 414; 1H NMR (500 MHz, MeOH-d4, 298 K) δ 8.77 (d, J = 4.7 Hz, 1H), 8.03–7.96 (m, 3H), 7.94 (bs, 1H), 7.69 (d, J = 4.7 Hz, 1H), 7.51 (dd, J = 2.57 Hz, 9.27 Hz, overlapped, 1H) 7.49 (bs, overlapped, 1H), 6.28 (d, J = 8.9 Hz, 1H), 5.95 (ddd, J = 17.5, 10.3, 7.7 Hz, 1H), 5.08 (bd, J = 17.5 Hz, 1H), 5.03 (bd, J = 10.3 Hz, 1H), 4.37 (m, 2H), 3.69–3.59 (m, 1H), 3.56–3.47 (m, 1H), 3.30–3.27 (m, overlapped, 1H), 2.99 (s, 1H), 2.93–2.86 (m, 1H), 2.86 (s, 1H), 2.84–2.76 (m, 1H), 2.43–2.37 (m, 1H), 1.70–1.61 (m, overlapped, 4H), 1.27 (m, 1H), 0.73–0.66 (m, 1H) ppm; 13C NMR (126 MHz, MeOH-d4, 298 K) δ 186.4, 182.8, 170.9, 165.9, 159.7, 149.3, 146.1, 146.0, 143.5, 143.0, 134.2 (q, J = 33.1 Hz), 132.4, 129.9, 125.1 (q, J = 272.1 Hz), 124.9, 120.3, 117.0, 115.8, 103.7, 69.6, 61.4, 57.5, 55.4, 42.2, 41.5, 41.2, 37.4, 32.1, 29.4, 29.0, 28.0 ppm.; HRESI(+)-MS (m/z): [M + H+] calcd for C33H32F6N5O3, 660.2409; found, 660.2424.
−51.8 (c 1.00, DMSO); IR (cm−1) νmax: 2957, 2923, 2853, 1664, 1620, 1609, 1560, 1508, 1437, 1378, 1331, 1277, 1201, 1175, 1126, 1021, 930, 884, 832, 799, 719, 700, 679, 620, 550, 521, 414; 1H NMR (500 MHz, MeOH-d4, 298 K) δ 8.77 (d, J = 4.7 Hz, 1H), 8.03–7.96 (m, 3H), 7.94 (bs, 1H), 7.69 (d, J = 4.7 Hz, 1H), 7.51 (dd, J = 2.57 Hz, 9.27 Hz, overlapped, 1H) 7.49 (bs, overlapped, 1H), 6.28 (d, J = 8.9 Hz, 1H), 5.95 (ddd, J = 17.5, 10.3, 7.7 Hz, 1H), 5.08 (bd, J = 17.5 Hz, 1H), 5.03 (bd, J = 10.3 Hz, 1H), 4.37 (m, 2H), 3.69–3.59 (m, 1H), 3.56–3.47 (m, 1H), 3.30–3.27 (m, overlapped, 1H), 2.99 (s, 1H), 2.93–2.86 (m, 1H), 2.86 (s, 1H), 2.84–2.76 (m, 1H), 2.43–2.37 (m, 1H), 1.70–1.61 (m, overlapped, 4H), 1.27 (m, 1H), 0.73–0.66 (m, 1H) ppm; 13C NMR (126 MHz, MeOH-d4, 298 K) δ 186.4, 182.8, 170.9, 165.9, 159.7, 149.3, 146.1, 146.0, 143.5, 143.0, 134.2 (q, J = 33.1 Hz), 132.4, 129.9, 125.1 (q, J = 272.1 Hz), 124.9, 120.3, 117.0, 115.8, 103.7, 69.6, 61.4, 57.5, 55.4, 42.2, 41.5, 41.2, 37.4, 32.1, 29.4, 29.0, 28.0 ppm.; HRESI(+)-MS (m/z): [M + H+] calcd for C33H32F6N5O3, 660.2409; found, 660.2424.
Methyl 3,4,5-tris(octadecyloxy)benzoate (9)
![[Graphic 24]](/bjoc/content/inline/1860-5397-19-133-i28.svg?max-width=637&scale=1.18182)
Methyl gallate (8, 15 g, 0.082 mol) was dissolved in DMF (150 mL), and 1-bromooctadecane (87.3 g, 0.262 mol) and potassium carbonate (67.5 g, 0.489 mol) were added. To the resulting reaction mixture, further DMF (150 mL) was added. After stirring for 16 hours at 80 °C, the reaction mixture was cooled, and diluted with toluene, water, and chloroform to precipitate the product. The crude product was filtered and recrystallized using chloroform/methanol mixed solvents. For a typical recrystallization of 1 gram crude product, 30 mL chloroform and 90 mL methanol were applied. The recrystallized solid was filtered to obtain product 9 as a white solid (49.2 g, 64%). The products had the same spectroscopic data than those reported in the literature [32].
Lipophilic organocatalyst 2
![[Graphic 25]](/bjoc/content/inline/1860-5397-19-133-i29.svg?max-width=637&scale=1.18182)
The cinchona squaramide with the aminoethylene linker 7 (534 mg, 0.81 mmol, 1.05 equiv) and triethylamine (1.09 mL, 8.26 mmol, 10.2 equiv) were dissolved in dichloromethane (8.8 mL). Then, a solution of 3,4,5-tris(octadecyloxy)benzoyl chloride (11, 729.3 mg, 0.77 mmol) in dichloromethane (22 mL) was added. To the resulting yellow solution further dichloromethane (12.6 mL) was added, and the mixture was stirred for 24 hours. Next, the reaction mixture was washed with water (2 × 30 mL), and the organic phase was dried over MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, DCM/MeOH/NH3(aq) 20:1:0.01 → DCM/MeOH/NH3 10:1:0.01). The product was dissolved in a small amount of dichloromethane (1 mL) and added dropwise to acetonitrile (100 mL) to precipitate the product as an off-white solid (698 mg, 58%). TLC (SiO2, DCM/MeOH 20:1, Rf 0.35); mp 68–69 °C; ![[Graphic 26]](/bjoc/content/inline/1860-5397-19-133-i30.svg?max-width=637&scale=1.18182) +14.1 (c 1.00, CHCl3); IR (cm−1) νmax: 3267, 2916, 2850, 1792, 1689, 1622, 1604, 1587, 1551, 1466, 1437, 1378, 1335, 1277, 1226, 1181, 1133, 1117, 1047, 1000, 930, 879, 851, 821, 764, 721, 701, 680, 619, 526; 1H NMR (500 MHz, CDCl3, 318 K) δ 8.73 (d, J = 3.6 Hz, 1H), 8.03 (m, 1H), 7.90–7.62 (m, 2H), 7.57–7.42 (m, overlapped, 1H), 7.40–7.33 (m, overlapped, 3H), 7.28 (m, 1H), 7.08 (br s, 2H), 6.30 (br s, 1H), 5.70 (m, 1H), 5.21–4.90 (m, 2H), 4.45 (m, 2H), 4.06–3.88 (m, overlapped, 10H), 3.76–3.69 (m, 2H), 3.61–3.19 (m, 2H), 3.09–2.65 (m, 2H), 1.93–1.61 (m, overlapped, 7H), 1.52–1.11 (m, overlapped, 94H), 0.88 (t, J = 7.0 Hz, 9H,) ppm; 13C NMR (75 MHz, CDCl3, 318 K) δ 185.4, 180.3, 175.4, 168.3, 165.1, 157.8, 153.3, 153.0, 147.9, 145.1, 141.6, 141.4, 140.3, 132.7 (q, 2JC,F = 34.2 Hz), 132.3, 130.3, 129.0, 127.6, 122,9 (q, 1JC,F = 272.0 Hz), 118.4, 116.5, 116.1, 108.0, 105.9, 101.3, 73.7, 73.6, 69.5, 69.2, 66.7, 60.4, 53.5, 41.2, 39.1, 32.1, 30.5, 29.9, 29.8, 29.6, 29.5, 26.3, 26,2 22.8, 14.2 ppm; 19F NMR −63.1 ppm; HRESI(+)-MS (m/z): [M + H+] calcd for C94H144F6N5O7, 1569.0970; found, 1569.957.
+14.1 (c 1.00, CHCl3); IR (cm−1) νmax: 3267, 2916, 2850, 1792, 1689, 1622, 1604, 1587, 1551, 1466, 1437, 1378, 1335, 1277, 1226, 1181, 1133, 1117, 1047, 1000, 930, 879, 851, 821, 764, 721, 701, 680, 619, 526; 1H NMR (500 MHz, CDCl3, 318 K) δ 8.73 (d, J = 3.6 Hz, 1H), 8.03 (m, 1H), 7.90–7.62 (m, 2H), 7.57–7.42 (m, overlapped, 1H), 7.40–7.33 (m, overlapped, 3H), 7.28 (m, 1H), 7.08 (br s, 2H), 6.30 (br s, 1H), 5.70 (m, 1H), 5.21–4.90 (m, 2H), 4.45 (m, 2H), 4.06–3.88 (m, overlapped, 10H), 3.76–3.69 (m, 2H), 3.61–3.19 (m, 2H), 3.09–2.65 (m, 2H), 1.93–1.61 (m, overlapped, 7H), 1.52–1.11 (m, overlapped, 94H), 0.88 (t, J = 7.0 Hz, 9H,) ppm; 13C NMR (75 MHz, CDCl3, 318 K) δ 185.4, 180.3, 175.4, 168.3, 165.1, 157.8, 153.3, 153.0, 147.9, 145.1, 141.6, 141.4, 140.3, 132.7 (q, 2JC,F = 34.2 Hz), 132.3, 130.3, 129.0, 127.6, 122,9 (q, 1JC,F = 272.0 Hz), 118.4, 116.5, 116.1, 108.0, 105.9, 101.3, 73.7, 73.6, 69.5, 69.2, 66.7, 60.4, 53.5, 41.2, 39.1, 32.1, 30.5, 29.9, 29.8, 29.6, 29.5, 26.3, 26,2 22.8, 14.2 ppm; 19F NMR −63.1 ppm; HRESI(+)-MS (m/z): [M + H+] calcd for C94H144F6N5O7, 1569.0970; found, 1569.957.
General procedure for the solvent screening of stereoselective Michael addition of acetylacetone to trans-β-nitrostyrene
![[Graphic 27]](/bjoc/content/inline/1860-5397-19-133-i31.svg?max-width=637&scale=1.18182)
First, trans-β-nitrostyrene (12, 11.9 mg, 0.08 mmol) and the lipophilic organocatalyst 2 (6.3 mg, 0.004 mmol, 5 mol %) were dissolved in the appropriate solvent (500 µL). Then, acetylacetone (13, 21.1 μL, 0.205 mmol) was added. The reaction mixture was stirred for 24 hours at room temperature. After the reaction was completed, the solvent was evaporated, and the crude product was purified by preparative thin-layer chromatography (SiO2, hexane/ethyl acetate 2:1, Rf 0.36) to obtain the product (S)-14 as pale-yellow crystals. The products had the same spectroscopic data than those of reported (the absolute configuration was determined by the optical rotation of the products) [26]. HPLC: Phenomenex Lux Cellulose-3 column (3 mm, 250 × 4.6 mm), eluent CH3CN/H2O (0.1% formic acid) 40:60, isocratic mode; 0.6 mL min−1; UV detector 222 nm, 30 °C. The retention time for the (S)-enantiomer was 12.3 min, for the (R)-enantiomer 14.7 min.
General procedure for recycling of the lipophilic organocatalyst in the stereoselective Michael addition of acetylacetone to trans-β-nitrostyrene
trans-β-Nitrostyrene (12, 57 mg, 0.382 mmol) and the lipophilic organocatalyst 2 (30 mg, 0.0191 mmol, 5 mol %) were dissolved in toluene (2.0 mL), and acetylacetone (13, 99 μL, 0.962 mmol) was added. The reaction mixture was stirred for 24 hours at room temperature. Then, the volatile components were evaporated, and acetonitrile (2 mL) was added. The dissolution of the product and the suspension of the catalyst were aided by using an ultrasonic bath. The reaction mixture was transferred to Eppendorf tubes, and the insoluble components were separated by centrifugation (8 min, 13500 rpm). After the separation, the precipitated catalyst was similarly washed twice with acetonitrile (2 × 2 mL). The combined acetonitrile phase was evaporated, and the crude product was purified by using preparative thin-layer chromatography (SiO2, hexane/ethyl acetate 2:1, Rf 0.36) to obtain the product (S)-14. The catalyst was dried in vacuo and reused in the following reaction cycle.
General procedure for the solvent screening of stereoselective Michael addition of the cyclohexyl derivative of Meldrum’s acid to 4-chloro-trans-β-nitrostyrene
![[Graphic 28]](/bjoc/content/inline/1860-5397-19-133-i32.svg?max-width=637&scale=1.18182)
Cyclohexyl derivative of Meldrum’s acid 15 (11.7 mg, 0.064 mmol), 4-chloro-trans-β-nitrostyrene (16, 17.5 mg, 0.096 mmol), and the lipophilic organocatalyst 2 (1, 5, or 10 mol %) were dissolved in the appropriate solvent (470 µL), and stirred for 3 hours at room temperature. Then, the solvent was evaporated, and the crude product was purified by preparative thin-layer chromatography (SiO2, hexane/ethyl acetate/AcOH 2:1:0.01, Rf 0.34) to obtain the product as a pale-yellow foam. To the best of our knowledge, the synthesis of (S)-17 has not been reported so far. TLC (SiO2, hexane/acetone 1:3, Rf 0.85); mp 61–69 °C; ![[Graphic 29]](/bjoc/content/inline/1860-5397-19-133-i33.svg?max-width=637&scale=1.18182) −5.7 (c 1.00, CHCl3); IR (cm−1) νmax 2941, 2860, 1780, 1744, 1709, 1637, 1553, 1493, 1448, 1433, 1416, 1367, 1339, 1299, 1272, 1223, 1184, 1133, 1091, 1068, 1036, 1014, 998, 976, 956, 912, 49, 825, 783, 743, 718, 679, 654, 530, 425; 1H NMR (500 MHz, CDCl3, 298 K) δ 7.33–7.28 (m, 4H), 5.35 (dd, J = 13.9, 8.7 Hz, 1H), 5.00 (dd, J = 13.9, 6.7 Hz, 1H), 4.63 (ddd, J = 8.8, 6.7, 3.1 Hz, 1H), 4.02 (d, J = 3.2 Hz, 1H), 1.91 (t, J = 6.2 Hz, 2H), 1.67 (m, overlapped, 2H), 1.48–1.42 (m, 2H), 1.30–1.26 (m, overlapped, 4H) ppm; 13C NMR (75 MHz, CDCl3, 298 K) δ 164.4, 163.9, 135.1, 133.7, 130.7, 129.5, 106.9, 76.1, 49.0, 41.4, 36.8, 36.7, 29.8, 21.8 ppm; HRESI(+)-MS (m/z): [M + Na+] calcd for C17H18ClNO6Na, 390.0720; found, 390.0682; HPLC: Phenomenex Lux Cellulose-3 column (3 mm, 250 × 4.6 mm), eluent CH3CN/H2O (0.2% formic acid) 40:60, isocratic mode; 1 mL min−1; UV detector 265 nm, 35 °C. The retention time for the (S)-enantiomer was 27.2 min, for the (R)-enantiomer 29.1 min.
−5.7 (c 1.00, CHCl3); IR (cm−1) νmax 2941, 2860, 1780, 1744, 1709, 1637, 1553, 1493, 1448, 1433, 1416, 1367, 1339, 1299, 1272, 1223, 1184, 1133, 1091, 1068, 1036, 1014, 998, 976, 956, 912, 49, 825, 783, 743, 718, 679, 654, 530, 425; 1H NMR (500 MHz, CDCl3, 298 K) δ 7.33–7.28 (m, 4H), 5.35 (dd, J = 13.9, 8.7 Hz, 1H), 5.00 (dd, J = 13.9, 6.7 Hz, 1H), 4.63 (ddd, J = 8.8, 6.7, 3.1 Hz, 1H), 4.02 (d, J = 3.2 Hz, 1H), 1.91 (t, J = 6.2 Hz, 2H), 1.67 (m, overlapped, 2H), 1.48–1.42 (m, 2H), 1.30–1.26 (m, overlapped, 4H) ppm; 13C NMR (75 MHz, CDCl3, 298 K) δ 164.4, 163.9, 135.1, 133.7, 130.7, 129.5, 106.9, 76.1, 49.0, 41.4, 36.8, 36.7, 29.8, 21.8 ppm; HRESI(+)-MS (m/z): [M + Na+] calcd for C17H18ClNO6Na, 390.0720; found, 390.0682; HPLC: Phenomenex Lux Cellulose-3 column (3 mm, 250 × 4.6 mm), eluent CH3CN/H2O (0.2% formic acid) 40:60, isocratic mode; 1 mL min−1; UV detector 265 nm, 35 °C. The retention time for the (S)-enantiomer was 27.2 min, for the (R)-enantiomer 29.1 min.
General procedure for recycling of the lipophilic organocatalyst in the stereoselective Michael addition of the cyclohexyl derivative of Meldrum’s acid to 4-chloro-trans-β-nitrostyrene
Cyclohexyl derivative of Meldrum’s acid 15 (35.2 mg, 0.19 mmol), 4-chloro-trans-β-nitrostyrene (16, 52.7 mg, 0.29 mmol), and the lipophilic organocatalyst 2 (30 mg, 0.0019 mmol, 10 mol %) were dissolved in toluene or anisole (1.4 mL), and stirred for 3 hours at room temperature. After the reaction was completed, the solvent was evaporated, and acetonitrile (1.5 mL) was added. The dissolution of the product and the suspension of the catalyst were aided by using an ultrasonic bath. The reaction mixture was transferred to Eppendorf tubes, and the insoluble components were separated by centrifugation (8 min, 13500 rpm). After the separation, the precipitated catalyst was similarly washed twice with acetonitrile (2 × 1.5 mL). The combined acetonitrile phase was evaporated, and the crude product was purified by using preparative thin-layer chromatography (SiO2, hexane/ethyl acetate/AcOH 2:1:0.01, Rf 0.34) to obtain the product (S)-17. The catalyst was dried in vacuo and reused in the following reaction cycle.
Gram-scale synthesis of baclofen precursor 17
Cyclohexyl derivative of Meldrum’s acid 15 (938.4 mg, 5.1 mmol), 4-chloro-trans-β-nitrostyrene (16, 1404.2 mg, 7.6 mmol), and the lipophilic organocatalyst 2 (80 mg, 0.05 mmol, 1 mol %) were dissolved in toluene or anisole (37.6 mL), and stirred for 5 hours at room temperature. After the reaction was completed, the solvent was evaporated, and acetonitrile (30 mL) was added. The dissolution of the product and the suspension of the catalyst were aided by using an ultrasonic bath. The resulting solid was filtrated, and washed with acetonitrile (3 × 10 mL). The combined acetonitrile phase was evaporated, and the crude product was purified by using column chromatography (SiO2, heptane/ethyl acetate/AcOH 4:1:0.01) to obtain the product ((S)-17, 1.675 g, 89%).
Synthesis of nitro acid (S)-18
![[Graphic 30]](/bjoc/content/inline/1860-5397-19-133-i34.svg?max-width=637&scale=1.18182)
In a 150 mL pressure flask (Synthware), the baclofen precursor (S)-17 (0.5 g, 1.36 mmol) was dissolved in THF (71 mL), and 1 M HCl (aq. solution, 36 mL) was added. The reaction mixture was heated in a 100 °C oil bath for 5 hours. Then, the volatile components were evaporated, and the aqueous phase was extracted with EtOAc (3 × 10 mL). The combined organic phase was dried over MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, heptane/ethyl acetate/AcOH 4:1:0.01 → heptane/ethyl acetate/AcOH 1:1:0.01) to gain the product (S)-18 as a white solid (232 mg, 70%). TLC (SiO2, heptane/ethyl acetate/AcOH 1:1:0.01, Rf 0.52, visualized by bromocresol green); mp 82–84 °C (lit. [40] 78–80 °C); ![[Graphic 31]](/bjoc/content/inline/1860-5397-19-133-i35.svg?max-width=637&scale=1.18182) −10.5 (c 2.00, MeOH), (lit. [38]:
−10.5 (c 2.00, MeOH), (lit. [38]: ![[Graphic 32]](/bjoc/content/inline/1860-5397-19-133-i36.svg?max-width=637&scale=1.18182) −10.1 (c 2.00, MeOH)). The products had the same spectroscopic data than those reported in the literature [41].
−10.1 (c 2.00, MeOH)). The products had the same spectroscopic data than those reported in the literature [41].
Supporting Information
| Supporting Information File 1: Experimental part, NMR, IR, and chiral HPLC spectra. | ||
| Format: PDF | Size: 2.3 MB | Download |
Acknowledgements
The authors are grateful to Péter Bagi for his help with the chiral HPLC measurements.
Funding
This research was funded by the National Research, Development, and Innovation Office (grant number FK138037), the Richter Gedeon Excellence PhD Scholarship of the Richter Gedeon Talentum Foundation, Richter Gedeon Plc. (G. D.). Further support was provided by the UNKP-22-3-I-BME-125 New National Excellence Program of the Ministry for Culture and Innovation sourced from the National Research, Development and Innovation Fund. Project no. RRF-2.3.1-21-2022-00015 has been implemented with the support provided by the European Union.
References
-
García Mancheño, O.; Waser, M. Eur. J. Org. Chem. 2023, 26, e202200950. doi:10.1002/ejoc.202200950
Return to citation in text: [1] -
Xiang, S.-H.; Tan, B. Nat. Commun. 2020, 11, 3786. doi:10.1038/s41467-020-17580-z
Return to citation in text: [1] -
da Gama Oliveira, V.; do Carmo Cardoso, M. F.; da Silva Magalhães Forezi, L. Catalysts 2018, 8, 605. doi:10.3390/catal8120605
Return to citation in text: [1] -
Krištofíková, D.; Modrocká, V.; Mečiarová, M.; Šebesta, R. ChemSusChem 2020, 13, 2828–2858. doi:10.1002/cssc.202000137
Return to citation in text: [1] -
Castelvecchi, D.; Stoye, E. Nature 2021, 598, 247–248. doi:10.1038/d41586-021-02704-2
Return to citation in text: [1] -
Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Chem. Soc. Rev. 2021, 50, 1522–1586. doi:10.1039/d0cs00196a
Return to citation in text: [1] [2] [3] -
Burke, A. J. Expert Opin. Drug Discovery 2023, 18, 37–46. doi:10.1080/17460441.2023.2160437
Return to citation in text: [1] -
Reyes, E.; Prieto, L.; Milelli, A. Molecules 2022, 28, 271. doi:10.3390/molecules28010271
Return to citation in text: [1] -
Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450–5453. doi:10.1021/ol102294g
Return to citation in text: [1] [2] -
Bagheri, I.; Mohammadi, L.; Zadsirjan, V.; Heravi, M. M. ChemistrySelect 2021, 6, 1008–1066. doi:10.1002/slct.202003034
Return to citation in text: [1] -
Marqués‐López, E.; Merino, P.; Tejero, T.; Herrera, R. P. Eur. J. Org. Chem. 2009, 2401–2420. doi:10.1002/ejoc.200801097
Return to citation in text: [1] -
Merino, P.; Marqués-López, E.; Tejero, T.; Herrera, R. P. Synthesis 2010, 1–26. doi:10.1055/s-0029-1217130
Return to citation in text: [1] -
Dargó, G.; Nagy, S.; Kis, D.; Bagi, P.; Mátravölgyi, B.; Tóth, B.; Huszthy, P.; Drahos, L.; Kupai, J. Synthesis 2022, 54, 3823–3830. doi:10.1055/s-0040-1719886
Return to citation in text: [1] -
Fulgheri, T.; Della Penna, F.; Baschieri, A.; Carlone, A. Curr. Opin. Green Sustainable Chem. 2020, 25, 100387. doi:10.1016/j.cogsc.2020.100387
Return to citation in text: [1] -
Rodríguez-Escrich, C.; Pericàs, M. A. Chem. Rec. 2019, 19, 1872–1890. doi:10.1002/tcr.201800097
Return to citation in text: [1] -
Corma, A.; Garcia, H. Adv. Synth. Catal. 2006, 348, 1391–1412. doi:10.1002/adsc.200606192
Return to citation in text: [1] -
Rostamnia, S.; Doustkhah, E. RSC Adv. 2014, 4, 28238–28248. doi:10.1039/c4ra03773a
Return to citation in text: [1] -
Ferré, M.; Pleixats, R.; Wong Chi Man, M.; Cattoën, X. Green Chem. 2016, 18, 881–922. doi:10.1039/c5gc02579f
Return to citation in text: [1] -
Benaglia, M.; Puglisi, A.; Cozzi, F. Chem. Rev. 2003, 103, 3401–3430. doi:10.1021/cr010440o
Return to citation in text: [1] -
Nagy, S.; Fehér, Z.; Kárpáti, L.; Bagi, P.; Kisszékelyi, P.; Koczka, B.; Huszthy, P.; Pukánszky, B.; Kupai, J. Chem. – Eur. J. 2020, 26, 13513–13522. doi:10.1002/chem.202001993
Return to citation in text: [1] [2] -
Rodríguez-Rodríguez, M.; Maestro, A.; Andrés, J. M.; Pedrosa, R. Adv. Synth. Catal. 2020, 362, 2744–2754. doi:10.1002/adsc.202000238
Return to citation in text: [1] -
Gawande, M. B.; Branco, P. S.; Varma, R. S. Chem. Soc. Rev. 2013, 42, 3371–3393. doi:10.1039/c3cs35480f
Return to citation in text: [1] -
Dalpozzo, R. Green Chem. 2015, 17, 3671–3686. doi:10.1039/c5gc00386e
Return to citation in text: [1] -
O’Neal, E. J.; Lee, C. H.; Brathwaite, J.; Jensen, K. F. ACS Catal. 2015, 5, 2615–2622. doi:10.1021/acscatal.5b00149
Return to citation in text: [1] -
Kisszékelyi, P.; Fehér, Z.; Nagy, S.; Bagi, P.; Kozma, P.; Garádi, Z.; Dékány, M.; Huszthy, P.; Mátravölgyi, B.; Kupai, J. Symmetry 2021, 13, 521. doi:10.3390/sym13030521
Return to citation in text: [1] [2] -
Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. ACS Catal. 2018, 8, 7430–7438. doi:10.1021/acscatal.8b01706
Return to citation in text: [1] [2] [3] [4] -
Benaglia, M., Ed. Recoverable and Recyclable Catalysts; John Wiley & Sons: New York, NY, USA, 2009. doi:10.1002/9780470682005
Return to citation in text: [1] [2] -
Jichu, T.; Inokuma, T.; Aihara, K.; Kohiki, T.; Nishida, K.; Shigenaga, A.; Yamada, K.-i.; Otaka, A. ChemCatChem 2018, 10, 3402–3405. doi:10.1002/cctc.201800714
Return to citation in text: [1] -
Kisszekelyi, P.; Alammar, A.; Kupai, J.; Huszthy, P.; Barabas, J.; Holtzl, T.; Szente, L.; Bawn, C.; Adams, R.; Szekely, G. J. Catal. 2019, 371, 255–261. doi:10.1016/j.jcat.2019.01.041
Return to citation in text: [1] -
Olpe, H.-R.; Demiéville, H.; Baltzer, V.; Bencze, W. L.; Koella, W. P.; Wolf, P.; Haas, H. L. Eur. J. Pharmacol. 1978, 52, 133–136. doi:10.1016/0014-2999(78)90032-8
Return to citation in text: [1] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s
Return to citation in text: [1] -
Kim, S.; Matsumoto, M.; Chiba, K. Chem. – Eur. J. 2013, 19, 8615–8620. doi:10.1002/chem.201300655
Return to citation in text: [1] [2] -
Dargo, G.; Kis, D.; Gede, M.; Kumar, S.; Kupai, J.; Szekely, G. Chem. Eng. J. 2023, 471, 144365. doi:10.1016/j.cej.2023.144365
Return to citation in text: [1] [2] -
Alder, C. M.; Hayler, J. D.; Henderson, R. K.; Redman, A. M.; Shukla, L.; Shuster, L. E.; Sneddon, H. F. Green Chem. 2016, 18, 3879–3890. doi:10.1039/c6gc00611f
Return to citation in text: [1] [2] -
Hayashi, Y. Chem. Sci. 2016, 7, 866–880. doi:10.1039/c5sc02913a
Return to citation in text: [1] -
Kimmel, K. L.; Weaver, J. D.; Ellman, J. A. Chem. Sci. 2012, 3, 121–125. doi:10.1039/c1sc00441g
Return to citation in text: [1] [2] -
Jiang, H.; Zhang, J.-M.; Du, W.-Q.; Zhu, S.-Z. Chin. J. Chem. 2007, 25, 86–89. doi:10.1002/cjoc.200790023
Return to citation in text: [1] -
Camps, P.; Muñoz-Torrero, D.; Sánchez, L. Tetrahedron: Asymmetry 2004, 15, 2039–2044. doi:10.1016/j.tetasy.2004.05.021
Return to citation in text: [1] [2] -
Hajra, S.; Jana, B. Org. Lett. 2017, 19, 4778–4781. doi:10.1021/acs.orglett.7b02150
Return to citation in text: [1] -
Felluga, F.; Gombac, V.; Pitacco, G.; Valentin, E. Tetrahedron: Asymmetry 2005, 16, 1341–1345. doi:10.1016/j.tetasy.2005.02.019
Return to citation in text: [1] -
Zu, L.; Xie, H.; Li, H.; Wang, J.; Wang, W. Adv. Synth. Catal. 2007, 349, 2660–2664. doi:10.1002/adsc.200700353
Return to citation in text: [1]
| 6. | Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Chem. Soc. Rev. 2021, 50, 1522–1586. doi:10.1039/d0cs00196a |
| 35. | Hayashi, Y. Chem. Sci. 2016, 7, 866–880. doi:10.1039/c5sc02913a |
| 36. | Kimmel, K. L.; Weaver, J. D.; Ellman, J. A. Chem. Sci. 2012, 3, 121–125. doi:10.1039/c1sc00441g |
| 36. | Kimmel, K. L.; Weaver, J. D.; Ellman, J. A. Chem. Sci. 2012, 3, 121–125. doi:10.1039/c1sc00441g |
| 1. | García Mancheño, O.; Waser, M. Eur. J. Org. Chem. 2023, 26, e202200950. doi:10.1002/ejoc.202200950 |
| 2. | Xiang, S.-H.; Tan, B. Nat. Commun. 2020, 11, 3786. doi:10.1038/s41467-020-17580-z |
| 3. | da Gama Oliveira, V.; do Carmo Cardoso, M. F.; da Silva Magalhães Forezi, L. Catalysts 2018, 8, 605. doi:10.3390/catal8120605 |
| 4. | Krištofíková, D.; Modrocká, V.; Mečiarová, M.; Šebesta, R. ChemSusChem 2020, 13, 2828–2858. doi:10.1002/cssc.202000137 |
| 27. | Benaglia, M., Ed. Recoverable and Recyclable Catalysts; John Wiley & Sons: New York, NY, USA, 2009. doi:10.1002/9780470682005 |
| 38. | Camps, P.; Muñoz-Torrero, D.; Sánchez, L. Tetrahedron: Asymmetry 2004, 15, 2039–2044. doi:10.1016/j.tetasy.2004.05.021 |
| 6. | Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Chem. Soc. Rev. 2021, 50, 1522–1586. doi:10.1039/d0cs00196a |
| 28. | Jichu, T.; Inokuma, T.; Aihara, K.; Kohiki, T.; Nishida, K.; Shigenaga, A.; Yamada, K.-i.; Otaka, A. ChemCatChem 2018, 10, 3402–3405. doi:10.1002/cctc.201800714 |
| 41. | Zu, L.; Xie, H.; Li, H.; Wang, J.; Wang, W. Adv. Synth. Catal. 2007, 349, 2660–2664. doi:10.1002/adsc.200700353 |
| 6. | Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Chem. Soc. Rev. 2021, 50, 1522–1586. doi:10.1039/d0cs00196a |
| 7. | Burke, A. J. Expert Opin. Drug Discovery 2023, 18, 37–46. doi:10.1080/17460441.2023.2160437 |
| 8. | Reyes, E.; Prieto, L.; Milelli, A. Molecules 2022, 28, 271. doi:10.3390/molecules28010271 |
| 22. | Gawande, M. B.; Branco, P. S.; Varma, R. S. Chem. Soc. Rev. 2013, 42, 3371–3393. doi:10.1039/c3cs35480f |
| 23. | Dalpozzo, R. Green Chem. 2015, 17, 3671–3686. doi:10.1039/c5gc00386e |
| 26. | Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. ACS Catal. 2018, 8, 7430–7438. doi:10.1021/acscatal.8b01706 |
| 5. | Castelvecchi, D.; Stoye, E. Nature 2021, 598, 247–248. doi:10.1038/d41586-021-02704-2 |
| 24. | O’Neal, E. J.; Lee, C. H.; Brathwaite, J.; Jensen, K. F. ACS Catal. 2015, 5, 2615–2622. doi:10.1021/acscatal.5b00149 |
| 25. | Kisszékelyi, P.; Fehér, Z.; Nagy, S.; Bagi, P.; Kozma, P.; Garádi, Z.; Dékány, M.; Huszthy, P.; Mátravölgyi, B.; Kupai, J. Symmetry 2021, 13, 521. doi:10.3390/sym13030521 |
| 26. | Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. ACS Catal. 2018, 8, 7430–7438. doi:10.1021/acscatal.8b01706 |
| 40. | Felluga, F.; Gombac, V.; Pitacco, G.; Valentin, E. Tetrahedron: Asymmetry 2005, 16, 1341–1345. doi:10.1016/j.tetasy.2005.02.019 |
| 14. | Fulgheri, T.; Della Penna, F.; Baschieri, A.; Carlone, A. Curr. Opin. Green Sustainable Chem. 2020, 25, 100387. doi:10.1016/j.cogsc.2020.100387 |
| 16. | Corma, A.; Garcia, H. Adv. Synth. Catal. 2006, 348, 1391–1412. doi:10.1002/adsc.200606192 |
| 17. | Rostamnia, S.; Doustkhah, E. RSC Adv. 2014, 4, 28238–28248. doi:10.1039/c4ra03773a |
| 18. | Ferré, M.; Pleixats, R.; Wong Chi Man, M.; Cattoën, X. Green Chem. 2016, 18, 881–922. doi:10.1039/c5gc02579f |
| 39. | Hajra, S.; Jana, B. Org. Lett. 2017, 19, 4778–4781. doi:10.1021/acs.orglett.7b02150 |
| 12. | Merino, P.; Marqués-López, E.; Tejero, T.; Herrera, R. P. Synthesis 2010, 1–26. doi:10.1055/s-0029-1217130 |
| 13. | Dargó, G.; Nagy, S.; Kis, D.; Bagi, P.; Mátravölgyi, B.; Tóth, B.; Huszthy, P.; Drahos, L.; Kupai, J. Synthesis 2022, 54, 3823–3830. doi:10.1055/s-0040-1719886 |
| 19. | Benaglia, M.; Puglisi, A.; Cozzi, F. Chem. Rev. 2003, 103, 3401–3430. doi:10.1021/cr010440o |
| 20. | Nagy, S.; Fehér, Z.; Kárpáti, L.; Bagi, P.; Kisszékelyi, P.; Koczka, B.; Huszthy, P.; Pukánszky, B.; Kupai, J. Chem. – Eur. J. 2020, 26, 13513–13522. doi:10.1002/chem.202001993 |
| 21. | Rodríguez-Rodríguez, M.; Maestro, A.; Andrés, J. M.; Pedrosa, R. Adv. Synth. Catal. 2020, 362, 2744–2754. doi:10.1002/adsc.202000238 |
| 32. | Kim, S.; Matsumoto, M.; Chiba, K. Chem. – Eur. J. 2013, 19, 8615–8620. doi:10.1002/chem.201300655 |
| 11. | Marqués‐López, E.; Merino, P.; Tejero, T.; Herrera, R. P. Eur. J. Org. Chem. 2009, 2401–2420. doi:10.1002/ejoc.200801097 |
| 37. | Jiang, H.; Zhang, J.-M.; Du, W.-Q.; Zhu, S.-Z. Chin. J. Chem. 2007, 25, 86–89. doi:10.1002/cjoc.200790023 |
| 10. | Bagheri, I.; Mohammadi, L.; Zadsirjan, V.; Heravi, M. M. ChemistrySelect 2021, 6, 1008–1066. doi:10.1002/slct.202003034 |
| 15. | Rodríguez-Escrich, C.; Pericàs, M. A. Chem. Rec. 2019, 19, 1872–1890. doi:10.1002/tcr.201800097 |
| 38. | Camps, P.; Muñoz-Torrero, D.; Sánchez, L. Tetrahedron: Asymmetry 2004, 15, 2039–2044. doi:10.1016/j.tetasy.2004.05.021 |
| 26. | Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. ACS Catal. 2018, 8, 7430–7438. doi:10.1021/acscatal.8b01706 |
| 20. | Nagy, S.; Fehér, Z.; Kárpáti, L.; Bagi, P.; Kisszékelyi, P.; Koczka, B.; Huszthy, P.; Pukánszky, B.; Kupai, J. Chem. – Eur. J. 2020, 26, 13513–13522. doi:10.1002/chem.202001993 |
| 25. | Kisszékelyi, P.; Fehér, Z.; Nagy, S.; Bagi, P.; Kozma, P.; Garádi, Z.; Dékány, M.; Huszthy, P.; Mátravölgyi, B.; Kupai, J. Symmetry 2021, 13, 521. doi:10.3390/sym13030521 |
| 26. | Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. ACS Catal. 2018, 8, 7430–7438. doi:10.1021/acscatal.8b01706 |
| 29. | Kisszekelyi, P.; Alammar, A.; Kupai, J.; Huszthy, P.; Barabas, J.; Holtzl, T.; Szente, L.; Bawn, C.; Adams, R.; Szekely, G. J. Catal. 2019, 371, 255–261. doi:10.1016/j.jcat.2019.01.041 |
| 30. | Olpe, H.-R.; Demiéville, H.; Baltzer, V.; Bencze, W. L.; Koella, W. P.; Wolf, P.; Haas, H. L. Eur. J. Pharmacol. 1978, 52, 133–136. doi:10.1016/0014-2999(78)90032-8 |
| 34. | Alder, C. M.; Hayler, J. D.; Henderson, R. K.; Redman, A. M.; Shukla, L.; Shuster, L. E.; Sneddon, H. F. Green Chem. 2016, 18, 3879–3890. doi:10.1039/c6gc00611f |
| 34. | Alder, C. M.; Hayler, J. D.; Henderson, R. K.; Redman, A. M.; Shukla, L.; Shuster, L. E.; Sneddon, H. F. Green Chem. 2016, 18, 3879–3890. doi:10.1039/c6gc00611f |
| 33. | Dargo, G.; Kis, D.; Gede, M.; Kumar, S.; Kupai, J.; Szekely, G. Chem. Eng. J. 2023, 471, 144365. doi:10.1016/j.cej.2023.144365 |
| 33. | Dargo, G.; Kis, D.; Gede, M.; Kumar, S.; Kupai, J.; Szekely, G. Chem. Eng. J. 2023, 471, 144365. doi:10.1016/j.cej.2023.144365 |
| 32. | Kim, S.; Matsumoto, M.; Chiba, K. Chem. – Eur. J. 2013, 19, 8615–8620. doi:10.1002/chem.201300655 |
| 27. | Benaglia, M., Ed. Recoverable and Recyclable Catalysts; John Wiley & Sons: New York, NY, USA, 2009. doi:10.1002/9780470682005 |
| 31. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
© 2023 Dargó et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.