Abstract
After prosperous domino reactions towards benzopyrans, the products were used as the starting material in Lewis acid catalyzed and organocatalytic Diels–Alder reactions to build up a tricyclic system. Herein, an asymmetric induction up to 96% enantiomeric excess was obtained by the use of imidazolidinone catalysts. This approach can be utilized to construct the tricyclic system in numerous natural products, in particular the scaffold of tetrahydrocannabinol (THC) being the most representative one. Compared with other published methods, condensation with a preexisting cyclohexane moiety in the precursor is needed to gain the heterogenic tricycle systems, whereas we present a novel strategy towards cannabinoid derivatives based on a flexible modular synthesis.

Graphical Abstract
Introduction
The Diels–Alder reaction is one of the most important processes for carbon–carbon-bond formation in organic chemistry [1,2]. Especially in the synthesis of natural products it is a widely used method [3-7]. Some examples are shown in Figure 1. The first application was the total synthesis of the steroid Cortisone (1) in 1952 by Woodward et al. [8]. Another example, indicating the importance of the well-known [4 + 2] cycloaddition in natural-product synthesis, is the first published total synthesis of Taxol (2) by Nicolaou. Two different [4 + 2]-cycloaddition reactions were applied to set up each of the two six-membered rings of the target molecule [9,10]. As a final example, Dynemicin A (3) should be mentioned, which is an enediyne consisting of a complex heterocyclic skeleton and a network of sensitive functional groups, exhibiting antitumor and antibiotic activity [11]. Three independent research groups (Schreiber, Myers and Danishefsky) successfully applied [4 + 2]-cycloaddition reactions in elegant and divergent strategies to reach the target molecule [12-14].
![[1860-5397-8-160-1]](/bjoc/content/figures/1860-5397-8-160-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: An assortment of natural products synthesized by Diels–Alder reactions.
Figure 1: An assortment of natural products synthesized by Diels–Alder reactions.
Given the importance of the Diels–Alder reaction, considerable efforts have been directed towards increasing the reaction rate and enantioselectivity. In the past century, catalysts that were employed for the enantioselective synthesis of organic compounds, such as pharmaceuticals, agrochemicals, or fine chemicals, were either transition-metal complexes or enzymes. In the past few years, however, organocatalysis has emerged as an alternative approach for the catalytic production of enantiomerically pure organic compounds [15,16]. These organocatalysts have several important advantages. They are metal-free, usually nontoxic, stable, moisture-insensitive, and often easy to obtain. Because of their inertness towards oxygen and moisture, the use of absolute solvents, inert atmosphere, low temperature, etc., is, in many instances, not required. Furthermore, due to the absence of transition metals, organocatalytic methods are especially attractive for the preparation of compounds that do not tolerate metal contamination, e.g., active pharmaceutical ingredients.
MacMillan’s imidazolidinone-based organocatalysts are general catalysts for a variety of asymmetric transformations. The first highly enantioselective organocatalytic Diels–Alder reaction was reported by MacMillan in his pioneering work in 2000 [17]. The activated iminium ion, formed through condensation of imidazolidinone and an α,β-unsaturated aldehyde, underwent reactions with various dienes to yield [4 + 2]-cycloadducts in excellent yields and enantioselectivities.
Results and Discussion
On our way towards a metal-free total synthesis of (−)-Δ9-tetrahydrocannabinol (4, Figure 2) we developed a convenient organocatalytic Diels–Alder route to generate the desired tricycle 5 of a model-system that also represents an alternative approach to key-intermediate 6 in Danishefsky’s total synthesis of Dynemicin A (3) [14].
![[1860-5397-8-160-2]](/bjoc/content/figures/1860-5397-8-160-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Intermediates towards the total synthesis of (−)-Δ9-tetrahydrocannabinol (4).
Figure 2: Intermediates towards the total synthesis of (−)-Δ9-tetrahydrocannabinol (4).
Our strategy, using the [4 + 2]-cycloaddition to obtain the cannabinoid tricycle system, is able to employ a variety of dienophiles and, hence, provides a versatile entry to this significant group of naturally occurring compounds [18], whereas the achieved Diels–Alder products can be further modified to gain a variety of heterocycles. Most published strategies are based on the use of a preexisting cyclohexane moiety in the starting material to form the heterogenic tricycle [19-28].
In former experiments of our group [29-31] it became apparent that without a catalyst no conversion towards the desired product occurred. Only the homodimer of the diene 10 could be isolated. Metal-based Lewis acids (e.g., catechol boronates) proved to be inefficient or too reactive. Due to the fact that other groups have used thiourea-derivatives as successful catalysts [32], we decided to test, on a model system, whether these catalysts could increase the reaction rate based on specific hydrogen bonds. In our initial screening we used the proven Schreiner catalyst 1,3-bis(3,5-bis(trifluoromethyl)phenyl)thiourea, which gave a satisfying 60% yield. In order to optimize the yield and to introduce chiral elements, we screened a number of analogues. These thiourea catalysts 9a–l were easy to obtain from the corresponding isothiocyanates 7a,b and chiral amines 8a–f in a one-step synthesis (Scheme 1) [33]. The yields were good to excellent in all procedures (Table 1).
![[1860-5397-8-160-i1]](/bjoc/content/inline/1860-5397-8-160-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of thiourea catalysts 9a–l.
Scheme 1: Synthesis of thiourea catalysts 9a–l.
Table 1: Yields of thiourea syntheses.
| Entry | R1 | R2 | Product | Yield [%] |
|---|---|---|---|---|
| 1 | H | Cy | (R)-9a [34] | 99 |
| 2 | H | Cy | (S)-9a [34] | 95 |
| 3 | H | Ph | (R)-9b | 99 |
| 4 | H | Ph | (S)-9b [34] | 99 |
| 5 | H | p-BrC6H4 | (R)-9c | 99 |
| 6 | H | p-BrC6H4 | (S)-9c | 99 |
| 7 | H | cyclopropyl | (R)-9d | 99 |
| 8 | H | m-ClC6H4 | (S)-9e | 99 |
| 9 | H | m-BrC6H4 | (S)-9f | 99 |
| 10 | CF3 | Cy | (R)-9g | 69 |
| 11 | CF3 | Cy | (S)-9g | 62 |
| 12 | CF3 | Ph | (R)-9h | 89 |
| 13 | CF3 | Ph | (S)-9h [35] | 99 |
| 14 | CF3 | p-BrC6H4 | (R)-9i | 99 |
| 15 | CF3 | p-BrC6H4 | (S)-9i | 99 |
| 16 | CF3 | cyclopropyl | (R)-9j | 91 |
| 17 | CF3 | m-ClC6H4 | (S)-9k | 99 |
| 18 | CF3 | m-BrC6H4 | (S)-9l | 76 |
These thioureas were used in an intermolecular Diels–Alder reaction of diene 10 [36] with acrolein (11) to obtain cannabinoid tricycle 5 shown in Scheme 2.
![[1860-5397-8-160-i2]](/bjoc/content/inline/1860-5397-8-160-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Organocatalytic Diels–Alder reaction with thiourea-catalysis.
Scheme 2: Organocatalytic Diels–Alder reaction with thiourea-catalysis.
In all cases we only achieved one cis-diastereomer and the carbonyl function was always in the 10-position. The reaction was carried out at low temperature and high dilution to avoid the dimer formation of diene 10. An increase of the temperature to 40 °C resulted in a higher conversion but unfortunately no enantioselectivity was observed. We obtained good to excellent yields from 68% to 99% depending on the substitution of the thiourea-catalyst 9a–l (Table 2).
Table 2: Summarized results of the Diels–Alder reaction.
| Entry | Catalyst | Yield [%]a |
|---|---|---|
| 1 | (R)-9a | 97 |
| 2 | (S)-9a | 91 |
| 3 | (R)-9b | 99 |
| 4 | (S)-9b | 85 |
| 5 | (R)-9c | 93 |
| 6 | (S)-9c | 92 |
| 7 | (R)-9d | 74 |
| 8 | (S)-9e | 82 |
| 9 | (S)-9f | 77 |
| 10 | (R)-9g | 68 |
| 11 | (S)-9g | 78 |
| 12 | (R)-9h | 73 |
| 13 | (S)-9h | 88 |
| 14 | (R)-9i | 76 |
| 15 | (S)-9i | 85 |
| 16 | (R)-9j | 87 |
| 17 | (S)-9k | 93 |
| 18 | (S)-9l | 83 |
aThe conversion is quantitative with respect to 10, the byproduct is the uncatalyzed dimer of compound 10.
When the substitution of the various thioureas 9a–l is further compared with the obtained yields in the Diels–Alder reaction (Table 2), the following tendencies are also observed. A noticeable fact is that, in contrast to the thioureas with a cyclopropyl- and m-halophenyl-moiety (Table 2, entries 7–9), the corresponding bis(trifluoromethyl)thioureas (Table 2, entries 16–18) provide a higher conversion.
Next to the previously described thiourea catalysts, we also analyzed iminium-ion catalysts according to MacMillan on various model systems (Figure 3) [37]. The effect of these catalysts is based on the formation of iminium ions by condensation of the dienophile bearing a carbonyl group, with the sterically hindered imidazole catalyst. In this way, one side is shielded and only the other side can be attacked by the diene. Hence, reaction rate and asymmetric induction are increased.
![[1860-5397-8-160-3]](/bjoc/content/figures/1860-5397-8-160-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis of imidazolidinone catalysts [38] is the premise for the construction of the demanding amide 13, which was afforded by the reaction of (S)-phenylalanine methyl ester hydrochloride (12) with methylamine (Scheme 3). In the second step of the synthesis, amide 13 was cyclized with different aldehydes 14 and addition of catalytic amounts of FeCl3 at high temperatures into (2S,5S)- or (2R,5S)-imidazolidinone 15 [39,40].
![[1860-5397-8-160-i3]](/bjoc/content/inline/1860-5397-8-160-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of electron poor imidazolidinone catalysts.
Scheme 3: Synthesis of electron poor imidazolidinone catalysts.
To gain electron-poor catalysts for the degradation of the dienophilic LUMO in the Diels–Alder reaction and to consequently increase the reaction rate, we used aldehydes with electron-withdrawing groups in ortho-, meta- and para-positions (Scheme 3, Table 3). Reaction with p-nitrobenzaldehyde gave compound 16 in 66% yield (Figure 4).
Table 3: Results of the conversion of various aldehydes 14 with amide 13.
| Entry | (Aldehyde 14) R = | Catalyst 15 | Yield [%] |
|---|---|---|---|
|
1
2 |
t-Bu |
(2R,5S)-15a
(2S,5S)-15a |
35 [41]
25 [40] |
|
3
4 |
o-NO2-C6H4 |
(2R,5S)-15b
(2S,5S)-15b |
42
52 |
|
5
6 |
m-F-C6H4 |
(2R,5S)-15c
(2S,5S)-15c |
28
39 |
|
7
8 |
p-F-C6H4 |
(2R,5S)-15d
(2S,5S)-15d |
32
43 |
|
9
10 |
p-Br-C6H4 |
(2R,5S)-15e
(2S,5S)-15e |
39
45 |
|
11
12 |
p-CN-C6H4 |
(2R,5S)-15f
(2S,5S)-15f |
38
46 |
![[1860-5397-8-160-4]](/bjoc/content/figures/1860-5397-8-160-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Crystal structure of the side product from the reaction of 13.
Figure 4: Crystal structure of the side product from the reaction of 13.
The organocatalytic species 15 were synthesized in moderate yields. A conspicuous feature is that the yields of the (2S,5S)-derivatives, with the exception of the tert-butyl substituted catalyst 15a (Table 3, entries 1–2), are always higher than those of the corresponding (2R,5S)-components (Table 3, entries 3–12). The configuration of the afforded imidazolidinone catalysts 15 could be confirmed by NOESY experiments and from their X-ray structures (Figure 5).
![[1860-5397-8-160-5]](/bjoc/content/figures/1860-5397-8-160-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Confirmation of the relative configuration with NOESY experiments and X-ray crystal structures of two imidazolidinones 15d and 15f.
Figure 5: Confirmation of the relative configuration with NOESY experiments and X-ray crystal structures of t...
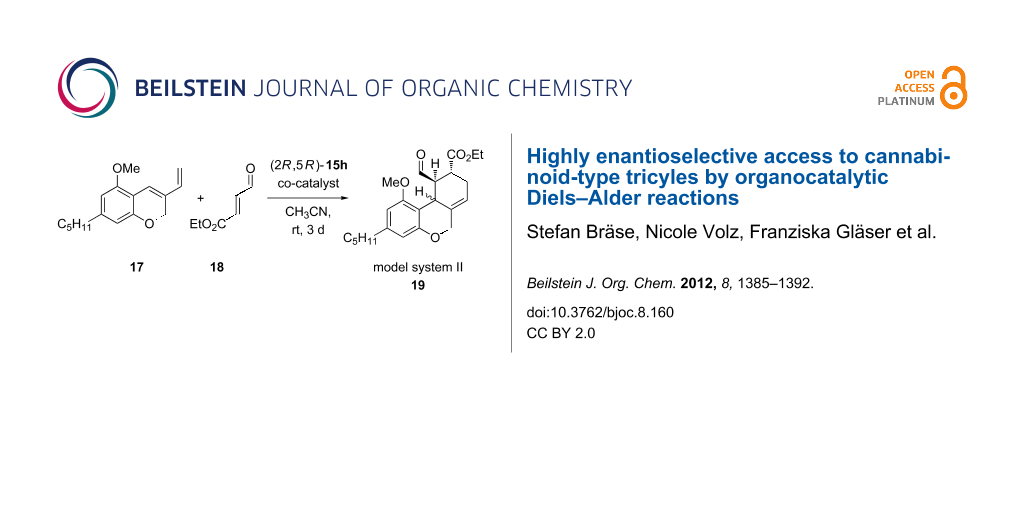
Before testing the catalysts in the Diels–Alder reaction, we analyzed a few co-catalysts with the commercially available imidazolidinone catalyst 15h. A pentyl-substituted tricycle was used as a model system (model system II) 19 (Scheme 4, Table 4).
![[1860-5397-8-160-i4]](/bjoc/content/inline/1860-5397-8-160-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 4: Summary of the screening of co-catalysts.
| Entry | Co-catalyst | cis-19 | trans-19 | ||
|---|---|---|---|---|---|
| Yield [%] | ee [%] | Yield [%] | ee [%] | ||
| 1 | HCl (1 M) | 51 | 96 | 12 | 91 |
| 2 | HClO4 (60%) | 6 | 95 | 10 | n.d. |
| 3 | TFA | 31 | 96 | 9 | 77 |
| 4 | p-TSA·H2O | 50 | n.d. | 15 | 90 |
| 5 | TfOH | — | — | — | — |
n.d.: not determined.
Hydrochloric acid has been proven to be the best co-catalyst (Table 4, entry 1), providing cis-19 in good yield and with an enantiomeric excess of 96%. Even perchloric acid and trifluoroacetic acid gave high enantiomeric excesses but in much poorer yields (Table 4, entries 2 and 3). Using para-toluenesulfonic acid also afforded cis-19 in good yield, but its enantiomeric excess could not be determined (Table 4, entry 4). Trifluoromethanesulfonic acid as co-catalyst only led to decomposition (Table 4, entry 5).
Under these optimized reaction conditions, we performed a catalyst screening with the previously synthesized imidazolidinone catalysts 15 to study their asymmetric induction (Scheme 5). As starting material we used compound 17 with the THC-typical pentyl side chain. The results are summarized in Table 5.
![[1860-5397-8-160-i5]](/bjoc/content/inline/1860-5397-8-160-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Screening of imidazolidinone catalysts 15.
Scheme 5: Screening of imidazolidinone catalysts 15.
Table 5: Catalyst screening towards model system II 19.
| Entry | Catalyst 15 | cis-19 | ||
|---|---|---|---|---|
| R = | Yield [%] | ee [%] | ||
| 1 | t-Bu | (2R,5R)-15h | 51 | 96 |
|
2
3 |
t-Bu |
(2R,5S)-15a
(2S,5S)-15a |
47
66 |
−24
−98 |
|
4
5 |
o-NO2-C6H4 |
(2R,5S)-15b
(2S,5S)-15b |
43
50 |
−89
−29 |
|
6
7 |
m-F-C6H4 |
(2R,5S)-15c
(2S,5S)-15c |
47
55 |
−78
−38 |
|
8
9 |
p-F-C6H4 |
(2R,5S)-15d
(2S,5S)-15d |
48
54 |
−79
−37 |
|
10
11 |
p-CN-C6H4 |
(2R,5S)-15f
(2S,5S)-15f |
52
54 |
−83
−43 |
The used organocatalysts, having a (S)-configured stereocenter in the 5-position, should prefer the formation of opposite enantiomers, compared to the commercially available (2R,5R)-15h. This is indicated by negative enantiomeric excess (Table 5). Due to low yields of diastereomer trans-19 and the less successful determination of their enantiomeric excess, Table 5 only contains results for cis-19. Except for in the case of 15a, we observe tendencies such as the achievement of higher yields through the use of (2S,5S)-configurated imidazolidinones 15 compared to the yields afforded by (2R,5S)-configurated catalysts 15. There is an opposite trend for the enantiomeric excess, which is also related to the steric hindrance of the phenyl substituent, i.e., the smaller the substituent, the lower the enantiomeric excess. Application of the known tert-butylimidazolidinone catalyst 15a provides the highest yield (66%) and enantiomeric excess (98% ee) with its (2S,5S)-derivate (Table 5, entry 3).
Conclusion
In conclusion, we have demonstrated that the Diels–Alder reaction of 3-vinyl-2H-chromene with acrolein can be accelerated with different thioureas, obtained in a one-step synthesis (in some cases with quantitative conversion). Application of imidazolidinone catalysts, inspired by the work of MacMillan, achieved good yields up to 66% and enantiomeric excesses up to 98%.
Experimental
Crystal structure determinations
The single-crystal X-ray diffraction study was carried out on a Nonius Kappa-CCD (9a, 16) or Bruker-Nonius APEXII diffractometer (9c, 9k, 9l, 15d, 15f) at 123(2) K, by using Mo Kα radiation (λ = 0.71073 Å). Direct methods (SHELXS-97) [42] were used for structure solution, and refinement was carried out with SHELXL-97 [42] (full-matrix least-squares on F2). Hydrogen atoms were localized by difference electron density determination and refined by using a riding model (H(N) free). The absolute configurations of 9a, 9c, 9k, 9l were determined by refinement of Flack’s x-parameter [43] and by using Bayesian statistics on Bijvoet differences (Hooft’s y-parameter) [44].
The absolute configuration of 15d and 15f could not be determined reliably by refinement of Flack’s x-parameter [43], nor by using Bayesian statistics on Bijvoet differences (Hooft’s y-parameter) [44]. The enantiomer was assigned by reference to an unchanging chiral center in the synthetic procedure.
Semi-empirical absorption corrections were applied for 9c and 9l. In 9l one CF3-group is disordered.
9a: colorless, C15H22N2S, M = 262.41, crystal size 0.30 × 0.10 × 0.05 mm, monoclinic, space group P21 (no. 4): a = 8.5864(5) Å, b = 22.2641(17) Å, c = 8.6811(6) Å, β = 117.350(4)°, V = 1474.04(17) Å3, Z = 4, ρ(calc) = 1.182 Mg m−3, F(000) = 568, μ = 0.206 mm−1, 8575 reflections (2θmax = 50°), 4687 unique [Rint = 0.057], 337 parameters, 5 restraints, R1 (I > 2σ(I)) = 0.048, wR2 (all data) = 0.119, GOOF = 1.09, largest diff. peak and hole 0.235/−0.290 e Å−3, x = −0.01(8), y = −0.05(5).
9c: colorless, C15H15BrN2S, M = 335.26, crystal size 0.25 × 0.10 × 0.05 mm, monoclinic, space group P21 (no. 4): a = 16.2276(4) Å, b = 5.5562(2) Å, c = 17.3953(6) Å, β = 108.894(2)°, V = 1483.92(8) Å3, Z = 4, ρ(calc) = 1.501 Mg m−3, F(000) = 680, μ = 2.899 mm−1, 11045 reflections (2θmax = 50°), 4958 unique [Rint = 0.037], 355 parameters, 5 restraints, R1 (I > 2σ(I)) = 0.030, wR2 (all data) = 0.072, GOOF = 1.06, largest diff. peak and hole 0.387/−0.504 e Å−3, x = −0.018(7), y = −0.012(2).
9k: colorless, C17H13ClF6N2S, M = 426.80, crystal size 0.20 × 0.10 × 0.05 mm, monoclinic, space group P21 (no. 4): a = 8.1359(6) Å, b =16.2464(13) Å, c = 13.9980(7) Å, β = 92.246(4)°, V = 1848.8(2) Å3, Z = 4, ρ(calc) = 1.533 Mg m−3, F(000) = 864, μ = 0.381 mm−1, 9231 reflections (2θmax = 50°), 5469 unique [Rint = 0.039], 499 parameters, 5 restraints, R1 (I > 2σ(I)) = 0.055, wR2 (all data) = 0.116, GOOF = 1.16, largest diff. peak and hole 0.388/−0.252 e Å−3, x = 0.07(9), y = 0.07(4).
9l: colorless, C17H13BrF6N2S, M = 471.26, crystal size 0.35 × 0.20 × 0.10 mm, monoclinic, space group P21 (no. 4): a = 8.2910(7) Å, b = 16.1565(8) Å, c = 14.0204(10) Å, β = 92.073(3)°, V = 1876.9(2) Å3, Z = 4, ρ(calc) = 1.668 Mg m−3, F(000) = 936, μ = 2.362 mm−1, 10315 reflections (2θmax = 50°), 5936 unique [Rint = 0.046], 494 parameters, 320 restraints, R1 (I > 2σ(I)) = 0.056, wR2 (all data) = 0.125, GOOF = 1.09, largest diff. peak and hole 1.033/−0.634 e Å−3, x = −0.014(11), y = −0.009(6).
15d: colorless, C17H17FN2O, M = 284.33, crystal size 0.45 × 0.25 × 0.10 mm, orthorhombic, space group P212121 (no. 19): a = 8.4559(3) Å, b = 10.5000(4) Å, c = 16.6177(5) Å, V = 1475.43(9) Å3, Z = 4, ρ(calc) = 1.280 Mg m−3, F(000) = 600, μ = 0.089 mm−1, 11771 reflections (2θmax = 50°), 2602 unique [Rint = 0.045], 194 parameters, 1 restraint, R1 (I > 2σ(I)) = 0.038, wR2 (all data) = 0.084, GOOF = 1.11, largest diff. peak and hole 0.135/−0.173 e Å−3, x = −0.8(10), y = 0.3(5).
15f: colorless, C18H17N3O, M = 291.35, crystal size 0.50 × 0.40 × 0.30 mm, monoclinic, space group P21 (no. 4): a = 6.0140(1) Å, b =15.7002(4) Å, c = 8.5232(2) Å, β = 95.211(2)°, V = 801.44(3) Å3, Z = 2, ρ(calc) = 1.207 Mg m−3, F(000) = 308, μ = 0.077 mm−1, 13805 reflections (2θmax = 55°), 3628 unique [Rint = 0.027], 203 parameters, 2 restraints, R1 (I > 2σ(I)) = 0.033, wR2 (all data) = 0.077, GOOF = 1.08, largest diff. peak and hole 0.181/−0.152 e Å−3, x = −0.2(11), y = 0.8(5).
16: colorless, C17H17N3O3, M = 311.34, crystal size 0.30 × 0.05 × 0.05 mm, monoclinic, space group P21/c (no. 14): a = 18.265(5) Å, b =10.935(2) Å, c = 7.857(2) Å, β = 101.60(1)°, V = 1537.2(6) Å3, Z = 4, ρ(calc) = 1.345 Mg m−3, F(000) = 656, μ = 0.094 mm−1, 4930 reflections (2θmax = 50°), 2685 unique [Rint = 0.128], 212 parameters, 1 restraint, R1 (I > 2σ(I)) = 0.071, wR2 (all data) = 0.185, GOOF = 0.99, largest diff. peak and hole 0.227/−0.247 e Å−3.
Crystallographic data (excluding structure factors) for the structures reported in this work have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications no.’s CCDC-865479 (9a), CCDC-865480 (9c), CCDC-865481 (9k), CCDC-865482 (9l), CCDC-965483 (15d), CCDC-865484 (15f), and CCDC-865485 (16). Copies of the data can be obtained free of charge on application to The Director, CCDC, 12 Union Road, Cambridge DB2 1EZ, UK (Fax: int. code +(1223)336-033; email: deposit@ccdc.cam.ac.uk).
Supporting Information
Supporting Information File:
| Supporting Information File 1: Experimental data for all new compounds. | ||
| Format: PDF | Size: 678.1 KB | Download |
References
-
Brieger, G.; Bennett, J. N. Chem. Rev. 1980, 80, 63–97. doi:10.1021/cr60323a004
Return to citation in text: [1] -
Stocking, E. M.; Williams, R. M. Angew. Chem., Int. Ed. 2003, 42, 3078–3115. doi:10.1002/anie.200200534
Return to citation in text: [1] -
Mergott, D. J.; Zuend, S. J.; Jacobsen, E. N. Org. Lett. 2008, 10, 745–748. doi:10.1021/ol702781q
Return to citation in text: [1] -
Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2002, 41, 1668–1698. doi:10.1002/1521-3773(20020517)41:10<1668::AID-ANIE1668>3.0.CO;2-Z
Return to citation in text: [1] -
Takao, K.-I.; Munakata, R.; Tadano, K.-I. Chem. Rev. 2005, 105, 4779–4807. doi:10.1021/cr040632u
Return to citation in text: [1] -
Friedrich, A.; Jainta, M.; Nising, C. F.; Bräse, S. Synlett 2008, 589–591. doi:10.1055/s-2008-1032085
Return to citation in text: [1] -
Tietze, L.-F.; von Kiedrowski, G.; Harms, K.; Clegg, W.; Sheldrick, G. Angew. Chem. 1980, 92, 130–131. doi:10.1002/ange.19800920217
Return to citation in text: [1] -
Woodward, R. B.; Sondheimer, F.; Taub, D.; Heusler, K.; McLamore, W. M. J. Am. Chem. Soc. 1952, 74, 4223–4251. doi:10.1021/ja01137a001
Return to citation in text: [1] -
Nicolaou, K. C.; Yang, Z.; Liu, J.-J.; Ueno, H.; Nantermet, P. G.; Guy, R. K.; Claiborne, C. F.; Renaud, J.; Couladouros, E. A.; Paulvannan, K.; Sorensen, E. J. Nature 1994, 367, 630–634. doi:10.1038/367630a0
Return to citation in text: [1] -
Nicolaou, K. C.; Natermet, P. G.; Ueno, H.; Guy, R. K.; Couladouros, E. A.; Sorensen, E. J. J. Am. Chem. Soc. 1995, 117, 634–644. doi:10.1021/ja00107a007
Return to citation in text: [1] -
Konishi, M.; Ohkuma, H.; Matsumoto, K.; Tsuno, T.; Kamei, H.; Miyaki, T.; Oki, T.; Kawaguchi, H.; Van Duyne, G. D.; Clardy, J. J. Antibiot. 1989, 42, 1449–1452. doi:10.7164/antibiotics.42.1449
Return to citation in text: [1] -
Taunton, J.; Wood, J. L.; Schreiber, S. L. J. Am. Chem. Soc. 1993, 115, 10378–10379. doi:10.1021/ja00075a071
Return to citation in text: [1] -
Myers, A. G.; Fraley, M. E.; Tom, N. J.; Cohen, S. B.; Madr, D. J. Chem. Biol. 1995, 2, 33–43. doi:10.1016/1074-5521(95)90078-0
Return to citation in text: [1] -
Shair, M. D.; Yoon, T. Y.; Mosny, K. K.; Chou, T. C.; Danishefsky, S. J. J. Am. Chem. Soc. 1996, 118, 9509–9525. doi:10.1021/ja960040w
Return to citation in text: [1] [2] -
Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650
Return to citation in text: [1] -
Rouhi, A. M. Chem. Eng. News 2004, 82, 37–41.
Return to citation in text: [1] -
Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s
Return to citation in text: [1] -
Kapeller, D. C.; Bräse, S. ACS Comb. Sci. 2011, 13, 554–561. doi:10.1021/co200107s
Return to citation in text: [1] -
Teske, J. A.; Deiters, A. Org. Lett. 2008, 10, 2195–2198. doi:10.1021/ol800589e
Return to citation in text: [1] -
Mechoulam, R.; Gaoni, Y. J. Am. Chem. Soc. 1965, 87, 3273–3275. doi:10.1021/ja01092a065
Return to citation in text: [1] -
Evans, D. A.; Shaughnessy, E. A.; Barnes, D. M. Tetrahedron Lett. 1997, 38, 3193–3194. doi:10.1016/S0040-4039(97)00609-6
Return to citation in text: [1] -
Trost, B. M.; Dogra, K. Org. Lett. 2007, 9, 861–863. doi:10.1021/ol063022k
Return to citation in text: [1] -
Chu, C.; Ramamurthy, A.; Makriyannis, A.; Tius, M. A. J. Org. Chem. 2003, 68, 55–61. doi:10.1021/jo0264978
Return to citation in text: [1] -
Healy, M. P.; Parsons, A. F.; Rawlinson, J. G. T. Synlett 2008, 329–332. doi:10.1055/s-2008-1032049
Return to citation in text: [1] -
Tius, M. A.; Kannangara, G. S. K. Tetrahedron 1992, 48, 9173–9186. doi:10.1016/S0040-4020(01)85608-8
Return to citation in text: [1] -
Huffman, J. W.; Zhang, X.; Wu, M. J.; Joyner, H. H.; Pennington, W. T. J. Org. Chem. 1991, 56, 1481–1489. doi:10.1021/jo00004a027
Return to citation in text: [1] -
Nilsson, J. L. G.; Nilsson, I. M.; Agurell, S.; Ben-Zvi, Z.; Mechoulam, R. Acta Pharm. Suec. 1972, 9, 215–220.
Return to citation in text: [1] -
Majumdar, N.; Korthals, K. A.; Wulff, W. D. J. Am. Chem. Soc. 2012, 134, 1357–1362. doi:10.1021/ja210655g
Return to citation in text: [1] -
Lesch, B.; Toräng, J.; Nieger, M.; Bräse, S. Synthesis 2005, 1888–1900. doi:10.1055/s-2005-870008
Return to citation in text: [1] -
Metje, C. Synthese von THC-Derivaten durch Lewissäure-katalysierte Diels–Alder-Reaktionen. Diploma Thesis, Universität Clausthal-Zellerfeld, 2005.
Return to citation in text: [1] -
Bröhmer, M. C.; Volz, N.; Bräse, S. Synlett 2009, 1383–1386. doi:10.1055/s-0029-1217179
Return to citation in text: [1] -
Schreiner, P. R. Chem. Soc. Rev. 2003, 32, 289–296. doi:10.1039/b107298f
Return to citation in text: [1] -
Jiang, L.; Zheng, H.-T.; Liu, T.-Y.; Yue, L.; Chen, Y.-C. Tetrahedron 2007, 63, 5123–5128. doi:10.1016/j.tet.2007.04.011
Return to citation in text: [1] -
Karame, I.; Lorraine, T. M.; Lemaire, M. J. Mol. Catal. A: Chem. 2003, 196, 137–143. doi:10.1016/S1381-1169(02)00643-X
(9a, 9b).
Return to citation in text: [1] [2] [3] -
Sibi, M. P.; Itoh, K. J. Am. Chem. Soc. 2007, 129, 8064–8065. doi:10.1021/ja071739c
(9h).
Return to citation in text: [1] -
Volz, N. Sauerstoff-Heterocyclen als neue, selektive Liganden für die Cannabinoid-Rezeptoren. Ph.D. Thesis, Karlsruher Institut für Technologie, 2010.
Return to citation in text: [1] -
Northrup, A. B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 2458–2460. doi:10.1021/ja017641u
Return to citation in text: [1] -
Samulis, L.; Tomkinson, N. C. O. Tetrahedron 2011, 67, 4263–4267. doi:10.1016/j.tet.2011.04.009
Return to citation in text: [1] -
Feenstra, R. W.; Stokkingreef, E. H. M.; Reichwein, A. M.; Lousberg, W. B. H.; Ottenheijm, H. C. J.; Kamphuis, J.; Boesten, W. H. J.; Schoemaker, H. E.; Meijer, E. M. Tetrahedron 1990, 46, 1745–1756. doi:10.1016/S0040-4020(01)81979-7
Return to citation in text: [1] -
Wilson, R. M.; Jen, W. S.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 11616–11617. doi:10.1021/ja054008q
Return to citation in text: [1] [2] -
MacMillan, D. W. C. Enantioselective transformations of α,β-unsaturated aldehydes by a wide variety of reactions using imidazolidinone enantiomers as chiral organic catalysts. Patent WO 2003002491, Jan 9, 2003.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1] [2] -
Flack, H. D. Acta Crystallogr., Sect. A: Found. Crystallogr. 1983, A39, 876–881. doi:10.1107/S0108767383001762
Return to citation in text: [1] [2] -
Hooft, R. W. W.; Straver, L. H.; Spek, A. L. J. Appl. Crystallogr. 2008, 41, 96–103. doi:10.1107/S0021889807059870
Return to citation in text: [1] [2]
| 44. | Hooft, R. W. W.; Straver, L. H.; Spek, A. L. J. Appl. Crystallogr. 2008, 41, 96–103. doi:10.1107/S0021889807059870 |
| 43. | Flack, H. D. Acta Crystallogr., Sect. A: Found. Crystallogr. 1983, A39, 876–881. doi:10.1107/S0108767383001762 |
| 44. | Hooft, R. W. W.; Straver, L. H.; Spek, A. L. J. Appl. Crystallogr. 2008, 41, 96–103. doi:10.1107/S0021889807059870 |
| 1. | Brieger, G.; Bennett, J. N. Chem. Rev. 1980, 80, 63–97. doi:10.1021/cr60323a004 |
| 2. | Stocking, E. M.; Williams, R. M. Angew. Chem., Int. Ed. 2003, 42, 3078–3115. doi:10.1002/anie.200200534 |
| 11. | Konishi, M.; Ohkuma, H.; Matsumoto, K.; Tsuno, T.; Kamei, H.; Miyaki, T.; Oki, T.; Kawaguchi, H.; Van Duyne, G. D.; Clardy, J. J. Antibiot. 1989, 42, 1449–1452. doi:10.7164/antibiotics.42.1449 |
| 34. |
Karame, I.; Lorraine, T. M.; Lemaire, M. J. Mol. Catal. A: Chem. 2003, 196, 137–143. doi:10.1016/S1381-1169(02)00643-X
(9a, 9b). |
| 9. | Nicolaou, K. C.; Yang, Z.; Liu, J.-J.; Ueno, H.; Nantermet, P. G.; Guy, R. K.; Claiborne, C. F.; Renaud, J.; Couladouros, E. A.; Paulvannan, K.; Sorensen, E. J. Nature 1994, 367, 630–634. doi:10.1038/367630a0 |
| 10. | Nicolaou, K. C.; Natermet, P. G.; Ueno, H.; Guy, R. K.; Couladouros, E. A.; Sorensen, E. J. J. Am. Chem. Soc. 1995, 117, 634–644. doi:10.1021/ja00107a007 |
| 34. |
Karame, I.; Lorraine, T. M.; Lemaire, M. J. Mol. Catal. A: Chem. 2003, 196, 137–143. doi:10.1016/S1381-1169(02)00643-X
(9a, 9b). |
| 8. | Woodward, R. B.; Sondheimer, F.; Taub, D.; Heusler, K.; McLamore, W. M. J. Am. Chem. Soc. 1952, 74, 4223–4251. doi:10.1021/ja01137a001 |
| 3. | Mergott, D. J.; Zuend, S. J.; Jacobsen, E. N. Org. Lett. 2008, 10, 745–748. doi:10.1021/ol702781q |
| 4. | Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2002, 41, 1668–1698. doi:10.1002/1521-3773(20020517)41:10<1668::AID-ANIE1668>3.0.CO;2-Z |
| 5. | Takao, K.-I.; Munakata, R.; Tadano, K.-I. Chem. Rev. 2005, 105, 4779–4807. doi:10.1021/cr040632u |
| 6. | Friedrich, A.; Jainta, M.; Nising, C. F.; Bräse, S. Synlett 2008, 589–591. doi:10.1055/s-2008-1032085 |
| 7. | Tietze, L.-F.; von Kiedrowski, G.; Harms, K.; Clegg, W.; Sheldrick, G. Angew. Chem. 1980, 92, 130–131. doi:10.1002/ange.19800920217 |
| 33. | Jiang, L.; Zheng, H.-T.; Liu, T.-Y.; Yue, L.; Chen, Y.-C. Tetrahedron 2007, 63, 5123–5128. doi:10.1016/j.tet.2007.04.011 |
| 14. | Shair, M. D.; Yoon, T. Y.; Mosny, K. K.; Chou, T. C.; Danishefsky, S. J. J. Am. Chem. Soc. 1996, 118, 9509–9525. doi:10.1021/ja960040w |
| 19. | Teske, J. A.; Deiters, A. Org. Lett. 2008, 10, 2195–2198. doi:10.1021/ol800589e |
| 20. | Mechoulam, R.; Gaoni, Y. J. Am. Chem. Soc. 1965, 87, 3273–3275. doi:10.1021/ja01092a065 |
| 21. | Evans, D. A.; Shaughnessy, E. A.; Barnes, D. M. Tetrahedron Lett. 1997, 38, 3193–3194. doi:10.1016/S0040-4039(97)00609-6 |
| 22. | Trost, B. M.; Dogra, K. Org. Lett. 2007, 9, 861–863. doi:10.1021/ol063022k |
| 23. | Chu, C.; Ramamurthy, A.; Makriyannis, A.; Tius, M. A. J. Org. Chem. 2003, 68, 55–61. doi:10.1021/jo0264978 |
| 24. | Healy, M. P.; Parsons, A. F.; Rawlinson, J. G. T. Synlett 2008, 329–332. doi:10.1055/s-2008-1032049 |
| 25. | Tius, M. A.; Kannangara, G. S. K. Tetrahedron 1992, 48, 9173–9186. doi:10.1016/S0040-4020(01)85608-8 |
| 26. | Huffman, J. W.; Zhang, X.; Wu, M. J.; Joyner, H. H.; Pennington, W. T. J. Org. Chem. 1991, 56, 1481–1489. doi:10.1021/jo00004a027 |
| 27. | Nilsson, J. L. G.; Nilsson, I. M.; Agurell, S.; Ben-Zvi, Z.; Mechoulam, R. Acta Pharm. Suec. 1972, 9, 215–220. |
| 28. | Majumdar, N.; Korthals, K. A.; Wulff, W. D. J. Am. Chem. Soc. 2012, 134, 1357–1362. doi:10.1021/ja210655g |
| 17. | Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s |
| 29. | Lesch, B.; Toräng, J.; Nieger, M.; Bräse, S. Synthesis 2005, 1888–1900. doi:10.1055/s-2005-870008 |
| 30. | Metje, C. Synthese von THC-Derivaten durch Lewissäure-katalysierte Diels–Alder-Reaktionen. Diploma Thesis, Universität Clausthal-Zellerfeld, 2005. |
| 31. | Bröhmer, M. C.; Volz, N.; Bräse, S. Synlett 2009, 1383–1386. doi:10.1055/s-0029-1217179 |
| 15. | Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650 |
| 16. | Rouhi, A. M. Chem. Eng. News 2004, 82, 37–41. |
| 12. | Taunton, J.; Wood, J. L.; Schreiber, S. L. J. Am. Chem. Soc. 1993, 115, 10378–10379. doi:10.1021/ja00075a071 |
| 13. | Myers, A. G.; Fraley, M. E.; Tom, N. J.; Cohen, S. B.; Madr, D. J. Chem. Biol. 1995, 2, 33–43. doi:10.1016/1074-5521(95)90078-0 |
| 14. | Shair, M. D.; Yoon, T. Y.; Mosny, K. K.; Chou, T. C.; Danishefsky, S. J. J. Am. Chem. Soc. 1996, 118, 9509–9525. doi:10.1021/ja960040w |
| 18. | Kapeller, D. C.; Bräse, S. ACS Comb. Sci. 2011, 13, 554–561. doi:10.1021/co200107s |
| 36. | Volz, N. Sauerstoff-Heterocyclen als neue, selektive Liganden für die Cannabinoid-Rezeptoren. Ph.D. Thesis, Karlsruher Institut für Technologie, 2010. |
| 34. |
Karame, I.; Lorraine, T. M.; Lemaire, M. J. Mol. Catal. A: Chem. 2003, 196, 137–143. doi:10.1016/S1381-1169(02)00643-X
(9a, 9b). |
| 35. |
Sibi, M. P.; Itoh, K. J. Am. Chem. Soc. 2007, 129, 8064–8065. doi:10.1021/ja071739c
(9h). |
| 42. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112–122. doi:10.1107/S0108767307043930 |
| 43. | Flack, H. D. Acta Crystallogr., Sect. A: Found. Crystallogr. 1983, A39, 876–881. doi:10.1107/S0108767383001762 |
| 40. | Wilson, R. M.; Jen, W. S.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 11616–11617. doi:10.1021/ja054008q |
| 42. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112–122. doi:10.1107/S0108767307043930 |
| 39. | Feenstra, R. W.; Stokkingreef, E. H. M.; Reichwein, A. M.; Lousberg, W. B. H.; Ottenheijm, H. C. J.; Kamphuis, J.; Boesten, W. H. J.; Schoemaker, H. E.; Meijer, E. M. Tetrahedron 1990, 46, 1745–1756. doi:10.1016/S0040-4020(01)81979-7 |
| 40. | Wilson, R. M.; Jen, W. S.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 11616–11617. doi:10.1021/ja054008q |
| 41. | MacMillan, D. W. C. Enantioselective transformations of α,β-unsaturated aldehydes by a wide variety of reactions using imidazolidinone enantiomers as chiral organic catalysts. Patent WO 2003002491, Jan 9, 2003. |
| 37. | Northrup, A. B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 2458–2460. doi:10.1021/ja017641u |
| 38. | Samulis, L.; Tomkinson, N. C. O. Tetrahedron 2011, 67, 4263–4267. doi:10.1016/j.tet.2011.04.009 |
© 2012 Bräse et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)