Abstract
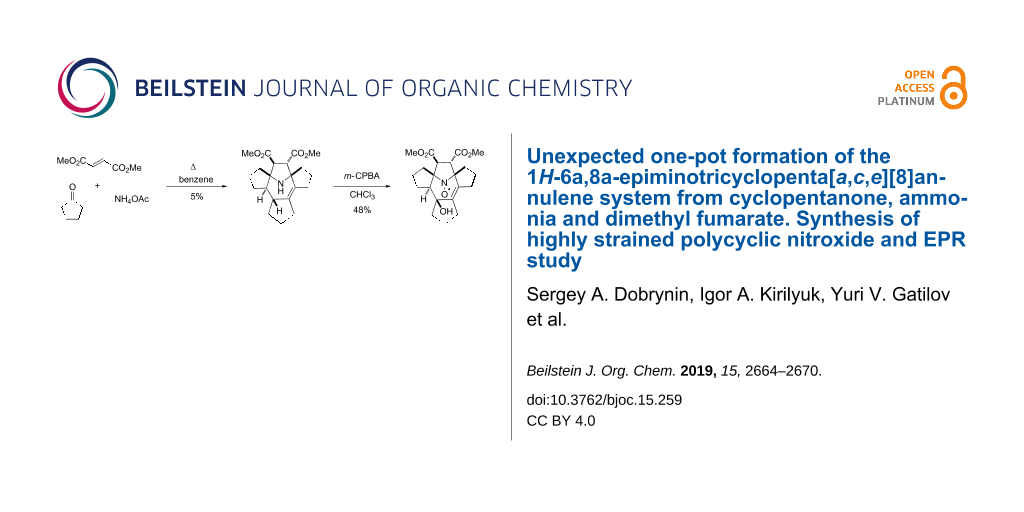
The unexpected formation of a highly strained polycyclic amine was observed in a one-pot synthesis from cyclopentanone, dimethyl fumarate and ammonium acetate. This multistep reaction includes 1,3-dipolar cycloaddition of dimethyl fumarate to the cyclic azomethine ylide formed in situ from cyclopentanone and ammonia. The polycyclic amine product was easily converted into a sterically shielded polycyclic nitroxide. The EPR spectra and spin relaxation behavior of the nitroxide were studied in solution. The spin relaxation seems well suited for the use as a biological spin label and are comparable with those of cyclic nitroxides with two spirocyclic moieties adjacent to the N–O· group.

Graphical Abstract
Introduction
Domino reactions have attracted much attention as an approach for the synthesis of complex molecules in a few steps [1]. The utility of multicomponent reactions involving amines, activated olefins and carbonyl compounds for the synthesis of heterocyclic compounds has been repeatedly demonstrated [2,3]. We recently used a domino reaction of amino acid, ketone and dimethyl fumarate for the one-pot synthesis of a substituted pyrrolidine, which then was converted into a reduction-resistant pyrrolidine nitroxide [4]. Here we report the unexpected formation of a highly strained polycyclic amine from cyclopentanone, dimethyl fumarate and ammonium acetate. This multistep reaction obviously includes the 1,3-dipolar cycloaddition of dimethyl fumarate with cyclic azomethine ylide formed in situ from cyclopentanone and ammonia. The polycyclic amine product was then converted into a sterically shielded polycyclic nitroxide.
Sterically hindered nitroxides have high chemical stability [5] and can be used as spin labels to study biopolymers in cells [6]. The introduction of spirocyclic moieties has a smaller effect on the reduction rates of nitroxides than does the introduction of linear alkyl substituents. However, spirocyclic nitroxides may have much longer spin relaxation times at 70–150 K which make them attractive agents for spin labeling [7-9]. Sterically hindered nitroxides can be used as spin labels for measurements at room temperature [10]. In this paper we examined the properties of this new nitroxide, in particular, the electron spin relaxation at different temperatures in water/glycerol solution.
Results and Discussion
Synthesis
Polycyclic amine
A mixture cyclopentanone, dimethyl fumarate and ammonium acetate was refluxed in benzene in a Dean–Stark apparatus. The reaction mixture underwent a strong tarring and after standard extraction with aqueous sulfuric acid, the main product 1 was isolated in a low yield (ca. 5%, see Scheme 1). In another experiment a much higher yield was obtained (ca. 15%), however, we failed to reproduce this result and in subsequent experiments the yields were close to the 5% limit. The 1H NMR spectra of 1 show signals of two ester groups and multiple methylene and methyne protons with an overall intensity of 22 protons. The 13C NMR spectrum showed signals from 21 carbon atoms, including an isolated, fully substituted C=C moiety at 133.5 and 137.5 ppm, methoxy groups at 51.5 and 51.6 ppm and carboxylate groups at 172.7 and 175.3 ppm, respectively. There are also four signals originating from CH groups at 45.9, 55.1, 59.9 and 60.4 ppm. The two downfield CH groups belong to isolated spin systems in the 1H NMR spectra at 3.19 and 3.44 ppm with hfs constants of 3 Hz. In the HMBC spectrum, the latter protons show cross peaks with the carboxylate carbons and with nodal carbons at 76.08 and 76.12 ppm. The above data imply the presence of a 2,2,5,5-tetrasubstituted-3,4-bis(methoxycarbonyl)pyrrolidine ring. The 1H signal at 3.19 ppm shows a cross peak with an atom of a fully substituted C=C moiety, while another lower field signal of a CH group at 3.44 ppm interacts with a CH-group carbon at 55.1 ppm. The hydrogen of the latter group shows cross peaks with the carbon of the 3aCH group at 45.9 ppm and with the carbon of the 11bC=C moiety at 137.5 ppm. These interactions clearly indicate a 9-azabicyclo[4.2.1]non-2-ene system in the structure of 1. The remaining assignments were made on the basis of HSQC, COSY 1H,1H and 1H,13C NMR spectra (see Table 1). The structure of the compound was finally confirmed by X-ray analysis and elemental analysis data (Figure 1). A possible mechanism for the formation of 1 is presented in Scheme 2.
![[1860-5397-15-259-i1]](/bjoc/content/inline/1860-5397-15-259-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 1: NMR Assignment of 1 based on the HSQC, COSY 1H,1H and 1H,13C spectra.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-259-i5.svg?max-width=637&scale=1.0)
|
|||||
| 13C (ppm) | 1H (ppm) | 13C (ppm) | 1H (ppm) | ||
| 10CH2 | 21.0 | 1.71; 2.00 | 3aCH | 45.9 | 2.45 |
| 2CH2 | 24.1 | 1.43; 1.68 | CH3 | 51.5; 51.6 | 3.57; 3.66 |
| 5CH2 | 25.3 | 1.51; 1.63 | 3bCH | 55.1 | 2.39 |
| 11CH2 | 30.1 | 1.93; 2.24 | 7CH | 59.9 | 3.44 |
| 1CH2 | 32.4 | 2.01; 2.15 | 8CH | 60.4 | 3.19 |
| 4CH2 | 33.9 | 1.41; 1.94 | 6aC, 8aC | 76.08; 76.12 | – |
| 3CH2 | 34.4 | 1.11; 1.87 | 11aC | 133.5 | |
| 6CH2 | 36.4 | 1.45; 1.63 | 11bC | 137.5 | |
| 9CH2 | 40.9 | 1.96; 2.01 | CO2 | 172.7; 175.3 | |
![[1860-5397-15-259-1]](/bjoc/content/figures/1860-5397-15-259-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray structure of compound 1 (one of the two enantiomers present in the crystal).
Figure 1: X-ray structure of compound 1 (one of the two enantiomers present in the crystal).
![[1860-5397-15-259-i2]](/bjoc/content/inline/1860-5397-15-259-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Possible mechanism for the formation of 1.
Scheme 2: Possible mechanism for the formation of 1.
It is well-known that cyclopentanone is prone to self-condensation. In the presence of ammonia these reactions may lead to heterocycle formation [11]. Presumably, a prototropic shift in the enamine-imine intermediate 4 is followed by electrocyclization to the cyclic azomethine ylide, which then reacts with dimethyl fumarate in a 1,3-dipolar cycloaddition. The suggested mechanism accounts for the trans-position of the methyne hydrogens in the azepine ring: electrocyclization proceeds via a conrotatory mechanism due to the antisymmetry of the HOMO (Figure 2).
![[1860-5397-15-259-2]](/bjoc/content/figures/1860-5397-15-259-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A possible mechanism for the trans-position of the methyne hydrogens in the azepine ring: the electrocyclization proceeds via a conrotatory mechanism due to the antisymmetry of the HOMO.
Figure 2: A possible mechanism for the trans-position of the methyne hydrogens in the azepine ring: the elect...
The selective formation of a single diastereomer in the 1,3-dipolar cycloaddition reaction is likely a result from secondary interactions of orbitals of the π-systems of the dipole and dipolarophile (Figure 3).
![[1860-5397-15-259-3]](/bjoc/content/figures/1860-5397-15-259-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selective formation of a single diastereomer in the 1,3-dipolar cycloaddition reaction.
Figure 3: Selective formation of a single diastereomer in the 1,3-dipolar cycloaddition reaction.
Nitroxide
Oxidation of 1 with m-CPBA afforded the nitroxide 6 with 48% yield (Scheme 3). It is noteworthy that the oxidation of the amino group is accompanied by the stereospecific hydroxylation at position 4 of the 2,3,4,7-tetrahydroazepine ring. The structure assignment was based on the single-crystal X-ray analysis (Figure 4) and a possible mechanism for this hydroxylation is shown in Scheme 4. Oxidation of amines with peracids is known to proceed through oxoammonium cation formation [12]. The close proximity of this reactive group to the allyl hydrogen results in hydride abstraction with the formation of carbocation 9 and subsequent cyclization to the bicyclic alkoxyamine 10. The resulting isoxazolidine ring is then opened with m-CPBA in the usual way, retaining the configuration of the asymmetric center at 3aC-OH [13] (Scheme 4).
![[1860-5397-15-259-i3]](/bjoc/content/inline/1860-5397-15-259-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-259-4]](/bjoc/content/figures/1860-5397-15-259-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray structure of compound 6 (one of the two enantiomers present in the crystal).
Figure 4: X-ray structure of compound 6 (one of the two enantiomers present in the crystal).
![[1860-5397-15-259-i4]](/bjoc/content/inline/1860-5397-15-259-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: A proposed mechanism for nitroxide 6 synthesis.
Scheme 4: A proposed mechanism for nitroxide 6 synthesis.
The steric strain in molecules 1 and 6 is characterized by an elongation of the bonds C6A–C7 1.579(2), C8–C8A 1.573(2) Å in 1 and C3B–C6A 1.580(2), C8–C8A 1.573(2) Å in 6. The conformation of the azepane ring in 1 is close to a distorted boat with a kink at the C3A–N1 line, while in 6 the conformation of this ring is close to a distorted half-chair. Presumably, this difference is due to the formation of an intramolecular hydrogen bond O6–H…O5 (H…O 1.95(2) Å, O–H…O 154(2)°) in molecule 6. In the crystal of 1 the amino group participates in intermolecular hydrogen bonding N1–H…O3 (H…O 2.39(2) Å, N–H…O 167(1)°) forming chains of molecules along the a-axis.
EPR measurements
Figure 4 demonstrates the X-band CW EPR spectra of nitroxide 6 in water/glycerol solution at 180 K and at room temperature with simulations (red) using the parameters listed in the caption. The electron spin relaxation of nitroxides with different bulky substituents has been studied in water/glycerol solutions at low temperatures in numerous papers [9,14-16]. Previously, we have investigated the electron spin relaxation of a series of nitroxides with different bulky substituents in water solution and in trehalose [10]. Because 6 is a new class of a hindered nitroxide, we investigated its electron spin relaxation properties in water/glycerol solution which is the solvent of choice for biomolecular distance measurements by PELDOR/DEER. If the rotation of the radical is prevented, the primary relaxation mechanisms are (i) modulation of the 14N hyperfine interaction (hfi) anisotropy of the NO group by librational motion, and (ii) modulation of the hfi with other nuclei in the radical by rotation of the groups containing those nuclei (e.g., rotation of methyl groups). The temperature dependence of spin relaxation reveals the relevant mechanism. For comparison, relaxation of a spirocyclohexane-substituted nitroxide [10] is also shown in Figure 5. The Tm vs T dependence generally shows consistent trends in frozen solutions. For 2,5-tetramethyl-substituted pyrrolidine and piperidine nitroxides, a local maximum appears in their phase relaxation (1/Tm) at T > 100 K as thermally activated rotation of their methyl groups becomes rapid. Above 140 K this rotation is rapid enough to average the hfi anisotropy and to cause some decrease in the phase relaxation rates. Finally, at T > 220 K the librational motion of the NO group dominates phase relaxation and causes the relaxation rate to increase with temperature in soft matter [14,17]. Consequently, the common tetramethyl-substituted nitroxides are characterized by a local bell-shaped maximum in their phase relaxation at T > 100 K, that is absent in more hindered nitroxides. In contrast, no bell-like shape is observed in the phase relaxation of nitroxides with two spirocyclohexane moieties adjacent to the N-O• group [8-10] or for nitroxide 6. The temperature dependence of 1/Tm is similar for both radicals in Figure 5 with some divergence above 180 K.
![[1860-5397-15-259-5]](/bjoc/content/figures/1860-5397-15-259-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: A, B) Temperature dependence of the electron spin relaxation times in water/glycerol at X-band frequency. The solid red dots are for nitroxide 6; the open black squares are for spirocyclohexane-substituted nitroxide. C, D) CW EPR spectra of nitroxide 6 at 180 K (C) and at room temperature (D) in water/glycerol. Black lines are experimental spectra and the red lines are simulations for hfi, g-factor and correlation times of A(N) = [0.71; 0.71; 3.71] mT, g = [2.0087; 2.0055; 2.0019], tcorr = 0.53 ns (at 298 K).
Figure 5: A, B) Temperature dependence of the electron spin relaxation times in water/glycerol at X-band freq...
Conclusion
An unexpected formation of a highly strained polycyclic amine from cyclopentanone, dimethyl fumarate and ammonium acetate was observed, and this amine was converted to a sterically-shielded polycyclic nitroxide. The temperature dependence of the electron spin relaxation of the nitroxide in water/glycerol solution is very similar to that of the previously studied nitroxides with two spirocyclohexane moieties adjacent to the N-O• group despite their very different structures.
Experimental
1H NMR spectra were recorded at 400 or 600 MHz, and 13C NMR spectra were recorded at 100 or 150 MHz, respectively. 1H and 13C NMR chemical shifts (δ) were internally referenced to the residual solvent peak. IR spectra were acquired on an FTIR spectrometer in KBr and are reported in wavenumbers (cm−1). Reactions were monitored by TLC using UV light 254 nm, 1% aqueous permanganate and Dragendorff reagent as visualizing agents. Column chromatography was performed on silica gel 60 (70–230 mesh). X-ray diffraction data were obtained with a Bruker KAPPA APEX II diffractometer using ϕ, ω scans with Mo Kα radiation (λ = 0.71073 Å) and a graphite monochromator. CCDC 1947797 (for 1) and 1947798 (for 6) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre at http://www.ccdc.cam.ac.uk.
Synthesis of dimethyl 2,3,3a,3b,4,5,6,7,8,9,10,11-dodecahydro-1H-6a,8a-epiminotricyclopenta[a,c,e][8]annulene-7,8-dicarboxylate (1)
A mixture of ammonium acetate (616 mg, 8 mmol), cyclopentanone (1.3 mL, 15 mmol), dimethyl fumarate (576 mg, 4 mmol) and benzene (10 mL) was placed in a Dean–Stark apparatus and stirred under reflux for 48 h. The solvent was distilled off in vacuum and the residue was dissolved in ethyl acetate. The organic layer was washed with 5% sodium hydrogen carbonate solution and then extracted with 5% sulfuric acid. Acidic extracts were basified with Na2CO3 and extracted with ethyl acetate. The extract was dried with Na2CO3 and the solvent was distilled off in vacuum to give a dark oil, which was purified using column chromatography on silica gel (hexane/ethyl acetate 4:1). 1: 70 mg (5%); colorless crystals; mp 146.8–148.4 °C; IR (KBr): 3298 (N-H), 1734, 1718 (C=O); 1H NMR (400 MHz, CDCl3, δ) 1.08–1.16 (m, 1H), 1.35–1.48 (m, 3H), 1.49–1.57 (m, 1H), 1.60–1.69 (m, 3H), 1.70–1.76 (m, 1H), 1.84–1.90 (m,1H), 1.91–1.98 (m, 2H), 1.99–2.08 (m, 3H), 2.14–2.20 (m, 1H), 2.20–2.29 (m, 1H), 2.36–2.41 (m, 1H), 2.42–2.49 (m, 1H), 2.58–2.80 (br, 1H), 3.19 (d Jd = 3.1 Hz, 1H), 3.44 (d Jd = 3.1 Hz, 1H), 3.57 (s, 3H), 3.66 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3, δ) 21.0, 24.0, 25.3, 30.0, 32.3, 33.8, 34.3, 36.3, 40.8, 45.9, 51.4, 51.5, 55.0, 59.8, 60.4, 76.0, 76.1, 133.5, 137.4, 172.7, 175.3; Anal. calcd for С21H29NO4: C, 70.17; H, 8.13; N, 3.90; found: C, 69.84; H, 7.89; N, 3.96. X-ray: triclinic system, P-1, a = 6.2467(4), b = 9.2039(8), c = 16.7575(15) Å, α = 98.858(4), β = 94.176(3), γ = 95.362(3)º, V = 944.07(13) Å3, Z = 2, 2θmax = 27.271°, 4168 independent reflections (Rint 0.0429), R1 0.0602, wR2 0.1696, S 1.030 [for 3234 I > 2σ(I)].
Synthesis of 3a-hydroxy-7,8-bis(methoxycarbonyl)-2,3,3a,3b,4,5,6,7,8,9,10,11-dodecahydro-1H-6a,8a-epiminotricyclopenta[a,c,e][8]annulene N-oxyl (6)
A 70% m-CPBA solution (200 mg, 0.9 mmol) was added to a solution of amine 1 (108 mg, 0.3 mmol) in chloroform (2 mL) and stirred at room temperature for 30 min. The reaction mixture was washed 3 times with a 5% solution of sodium bicarbonate. The extract was dried with Na2CO3 and the solvent was distilled off in vacuum to give a red oil, which was purified using column chromatography on silica gel (hexane/ethyl acetate 4:1). 2: 56 mg (48%); red crystals; mp 127.0 °C dec, IR (KBr): 3438 (O-H) 1730 (C=O); X-ray: triclinic system, P-1, a = 8.2240(5), b = 10.9423(6), c = 11.4515(8) Å, α = 83.440(3), β = 75.611(3), γ = 83.119(2)º, V = 987.17(11) Å3, Z = 2, 2θmax = 27.292°, 4388 independent reflections (Rint 0.0375), R1 0.0439, wR2 0.1215, S 1.011 [for 3451 I > 2σ(I)].
EPR measurements
Continuous wave (CW) EPR measurements were carried out using a commercial Bruker Elexsys E540 X-band spectrometer. The CW EPR spectra were simulated using EasySpin [18]. Pulse EPR experiments were carried out using a commercial Bruker Elexsys E580 X/Q-band spectrometer equipped with an Oxford helium flow cryostat and temperature control system. An ER 4118X-MD5W resonator was used for X-band measurements. Tm was measured using a two-pulse electron spin echo (ESE) sequence; T1 was measured using the inversion-recovery technique with a π-pulse for inversion and a two-pulse ESE sequence for detection. The π-pulse lengths were nominally 20 ns.
Supporting Information
| Supporting Information File 1: Copies of the IR and 1H, 13C, HMBC, HSQC NMR spectra and X-ray analysis data. | ||
| Format: PDF | Size: 510.2 KB | Download |
References
-
Tietze, L. F., Ed. Domino reactions: concepts for efficient organic synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527671304
Return to citation in text: [1] -
Hill, M. D. Chem. – Eur. J. 2010, 16, 12052–12062. doi:10.1002/chem.201001100
Return to citation in text: [1] -
Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2014, 43, 4633–4657. doi:10.1039/c3cs60015g
Return to citation in text: [1] -
Dobrynin, S. A.; Glazachev, Y. I.; Gatilov, Y. V.; Chernyak, E. I.; Salnikov, G. E.; Kirilyuk, I. A. J. Org. Chem. 2018, 83, 5392–5397. doi:10.1021/acs.joc.8b00085
Return to citation in text: [1] -
Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; Ouari, O.; Mileo, E. Angew. Chem., Int. Ed. 2018, 57, 1366–1370. doi:10.1002/anie.201710184
Return to citation in text: [1] -
Bleicken, S.; Assafa, T. E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. ChemistryOpen 2019, 8, 1057–1065. doi:10.1002/open.201900119
Return to citation in text: [1] -
Meyer, V.; Swanson, M. A.; Clouston, L. J.; Boratyński, P. J.; Stein, R. A.; Mchaourab, H. S.; Rajca, A.; Eaton, S. S.; Eaton, G. R. Biophys. J. 2015, 108, 1213–1219. doi:10.1016/j.bpj.2015.01.015
Return to citation in text: [1] -
Rajca, A.; Kathirvelu, V.; Roy, S. K.; Pink, M.; Rajca, S.; Sarkar, S.; Eaton, S. S.; Eaton, G. R. Chem. – Eur. J. 2010, 16, 5778–5782. doi:10.1002/chem.200903102
Return to citation in text: [1] [2] -
Kathirvelu, V.; Smith, C.; Parks, C.; Mannan, M. A.; Miura, Y.; Takeshita, K.; Eaton, S. S.; Eaton, G. R. Chem. Commun. 2009, 454–456. doi:10.1039/b817758a
Return to citation in text: [1] [2] [3] -
Kuzhelev, A. A.; Strizhakov, R. K.; Krumkacheva, O. A.; Polienko, Y. F.; Morozov, D. A.; Shevelev, G. Y.; Pyshnyi, D. V.; Kirilyuk, I. A.; Fedin, M. V.; Bagryanskaya, E. G. J. Magn. Reson. 2016, 266, 1–7. doi:10.1016/j.jmr.2016.02.014
Return to citation in text: [1] [2] [3] [4] -
Edgar, O. B.; Johnson, D. H. J. Chem. Soc. 1958, 3925–3944. doi:10.1039/jr9580003925
Return to citation in text: [1] -
Sen', V. D.; Golubev, V. A.; Efremova, N. N. Russ. Chem. Bull. 1982, 31, 53–63. doi:10.1007/bf00954410
Return to citation in text: [1] -
Ali, S. A.; Wazeer, M. I. M. Tetrahedron 1993, 49, 4339–4354. doi:10.1016/s0040-4020(01)85751-3
Return to citation in text: [1] -
Biller, J. R.; Elajaili, H.; Meyer, V.; Rosen, G. M.; Eaton, S. S.; Eaton, G. R. J. Magn. Reson. 2013, 236, 47–56. doi:10.1016/j.jmr.2013.08.006
Return to citation in text: [1] [2] -
Kirilyuk, I. A.; Polienko, Y. F.; Krumkacheva, O. A.; Strizhakov, R. K.; Gatilov, Y. V.; Grigor’ev, I. A.; Bagryanskaya, E. G. J. Org. Chem. 2012, 77, 8016–8027. doi:10.1021/jo301235j
Return to citation in text: [1] -
Kirilina, E. P.; Dzuba, S. A.; Maryasov, A. G.; Tsvetkov, Y. D. Appl. Magn. Reson. 2001, 21, 203–221. doi:10.1007/bf03162452
Return to citation in text: [1] -
Sato, H.; Kathirvelu, V.; Fielding, A.; Blinco, J. P.; Micallef, A. S.; Bottle, S. E.; Eaton, S. S.; Eaton, G. R. Mol. Phys. 2007, 105, 2137–2151. doi:10.1080/00268970701724966
Return to citation in text: [1] -
Stoll, S.; Schweiger, A. J. Magn. Reson. 2006, 178, 42–55. doi:10.1016/j.jmr.2005.08.013
Return to citation in text: [1]
| 1. | Tietze, L. F., Ed. Domino reactions: concepts for efficient organic synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527671304 |
| 6. | Bleicken, S.; Assafa, T. E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. ChemistryOpen 2019, 8, 1057–1065. doi:10.1002/open.201900119 |
| 8. | Rajca, A.; Kathirvelu, V.; Roy, S. K.; Pink, M.; Rajca, S.; Sarkar, S.; Eaton, S. S.; Eaton, G. R. Chem. – Eur. J. 2010, 16, 5778–5782. doi:10.1002/chem.200903102 |
| 9. | Kathirvelu, V.; Smith, C.; Parks, C.; Mannan, M. A.; Miura, Y.; Takeshita, K.; Eaton, S. S.; Eaton, G. R. Chem. Commun. 2009, 454–456. doi:10.1039/b817758a |
| 10. | Kuzhelev, A. A.; Strizhakov, R. K.; Krumkacheva, O. A.; Polienko, Y. F.; Morozov, D. A.; Shevelev, G. Y.; Pyshnyi, D. V.; Kirilyuk, I. A.; Fedin, M. V.; Bagryanskaya, E. G. J. Magn. Reson. 2016, 266, 1–7. doi:10.1016/j.jmr.2016.02.014 |
| 5. | Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; Ouari, O.; Mileo, E. Angew. Chem., Int. Ed. 2018, 57, 1366–1370. doi:10.1002/anie.201710184 |
| 18. | Stoll, S.; Schweiger, A. J. Magn. Reson. 2006, 178, 42–55. doi:10.1016/j.jmr.2005.08.013 |
| 4. | Dobrynin, S. A.; Glazachev, Y. I.; Gatilov, Y. V.; Chernyak, E. I.; Salnikov, G. E.; Kirilyuk, I. A. J. Org. Chem. 2018, 83, 5392–5397. doi:10.1021/acs.joc.8b00085 |
| 10. | Kuzhelev, A. A.; Strizhakov, R. K.; Krumkacheva, O. A.; Polienko, Y. F.; Morozov, D. A.; Shevelev, G. Y.; Pyshnyi, D. V.; Kirilyuk, I. A.; Fedin, M. V.; Bagryanskaya, E. G. J. Magn. Reson. 2016, 266, 1–7. doi:10.1016/j.jmr.2016.02.014 |
| 2. | Hill, M. D. Chem. – Eur. J. 2010, 16, 12052–12062. doi:10.1002/chem.201001100 |
| 3. | Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2014, 43, 4633–4657. doi:10.1039/c3cs60015g |
| 14. | Biller, J. R.; Elajaili, H.; Meyer, V.; Rosen, G. M.; Eaton, S. S.; Eaton, G. R. J. Magn. Reson. 2013, 236, 47–56. doi:10.1016/j.jmr.2013.08.006 |
| 17. | Sato, H.; Kathirvelu, V.; Fielding, A.; Blinco, J. P.; Micallef, A. S.; Bottle, S. E.; Eaton, S. S.; Eaton, G. R. Mol. Phys. 2007, 105, 2137–2151. doi:10.1080/00268970701724966 |
| 12. | Sen', V. D.; Golubev, V. A.; Efremova, N. N. Russ. Chem. Bull. 1982, 31, 53–63. doi:10.1007/bf00954410 |
| 9. | Kathirvelu, V.; Smith, C.; Parks, C.; Mannan, M. A.; Miura, Y.; Takeshita, K.; Eaton, S. S.; Eaton, G. R. Chem. Commun. 2009, 454–456. doi:10.1039/b817758a |
| 14. | Biller, J. R.; Elajaili, H.; Meyer, V.; Rosen, G. M.; Eaton, S. S.; Eaton, G. R. J. Magn. Reson. 2013, 236, 47–56. doi:10.1016/j.jmr.2013.08.006 |
| 15. | Kirilyuk, I. A.; Polienko, Y. F.; Krumkacheva, O. A.; Strizhakov, R. K.; Gatilov, Y. V.; Grigor’ev, I. A.; Bagryanskaya, E. G. J. Org. Chem. 2012, 77, 8016–8027. doi:10.1021/jo301235j |
| 16. | Kirilina, E. P.; Dzuba, S. A.; Maryasov, A. G.; Tsvetkov, Y. D. Appl. Magn. Reson. 2001, 21, 203–221. doi:10.1007/bf03162452 |
| 11. | Edgar, O. B.; Johnson, D. H. J. Chem. Soc. 1958, 3925–3944. doi:10.1039/jr9580003925 |
| 10. | Kuzhelev, A. A.; Strizhakov, R. K.; Krumkacheva, O. A.; Polienko, Y. F.; Morozov, D. A.; Shevelev, G. Y.; Pyshnyi, D. V.; Kirilyuk, I. A.; Fedin, M. V.; Bagryanskaya, E. G. J. Magn. Reson. 2016, 266, 1–7. doi:10.1016/j.jmr.2016.02.014 |
| 10. | Kuzhelev, A. A.; Strizhakov, R. K.; Krumkacheva, O. A.; Polienko, Y. F.; Morozov, D. A.; Shevelev, G. Y.; Pyshnyi, D. V.; Kirilyuk, I. A.; Fedin, M. V.; Bagryanskaya, E. G. J. Magn. Reson. 2016, 266, 1–7. doi:10.1016/j.jmr.2016.02.014 |
| 7. | Meyer, V.; Swanson, M. A.; Clouston, L. J.; Boratyński, P. J.; Stein, R. A.; Mchaourab, H. S.; Rajca, A.; Eaton, S. S.; Eaton, G. R. Biophys. J. 2015, 108, 1213–1219. doi:10.1016/j.bpj.2015.01.015 |
| 8. | Rajca, A.; Kathirvelu, V.; Roy, S. K.; Pink, M.; Rajca, S.; Sarkar, S.; Eaton, S. S.; Eaton, G. R. Chem. – Eur. J. 2010, 16, 5778–5782. doi:10.1002/chem.200903102 |
| 9. | Kathirvelu, V.; Smith, C.; Parks, C.; Mannan, M. A.; Miura, Y.; Takeshita, K.; Eaton, S. S.; Eaton, G. R. Chem. Commun. 2009, 454–456. doi:10.1039/b817758a |
| 13. | Ali, S. A.; Wazeer, M. I. M. Tetrahedron 1993, 49, 4339–4354. doi:10.1016/s0040-4020(01)85751-3 |
© 2019 Dobrynin et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)