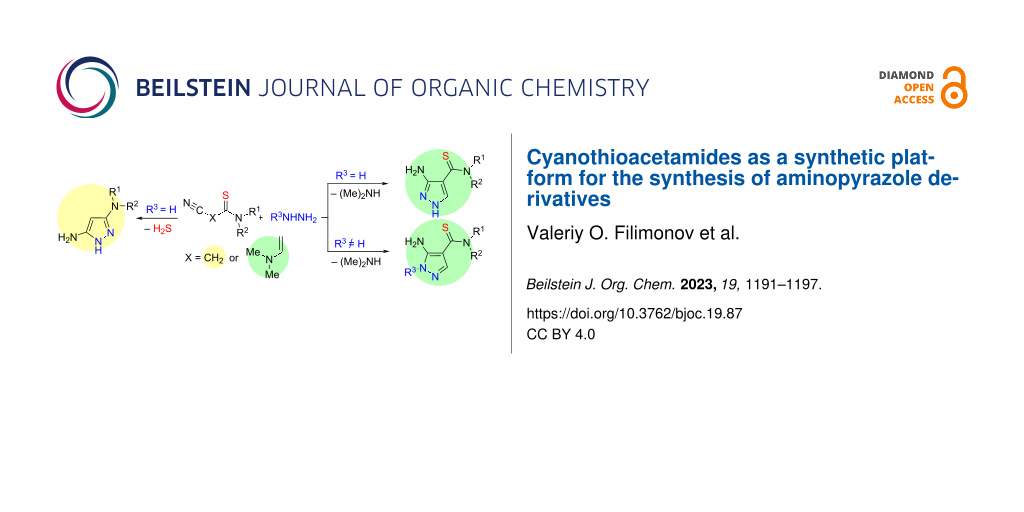
Abstract
It was shown that the reaction of 2-cyanothioacetamides with hydrazine involves both cyano- and thioamide groups, and 3,5-diaminopyrazoles are formed. In the reaction of 2-cyano-3-(dimethylamino)-N,N-dimethylprop-2-enethioamides with hydrazine and its derivatives, the interaction proceeds with the participation of cyano- and enamine groups, not affecting the thiocarbamoyl group, and leads to the formation of 4-thiocarbamoylpyrazoles. A synthesis method has been developed and a series of 1-substituted-4-thiocarbamoyl pyrazoles has been thus synthesized. The structure of the reaction products was studied using NMR spectroscopy and mass spectrometry and confirmed by X-ray diffraction analysis.

Graphical Abstract
Introduction
Compounds containing a pyrazole cycle exhibit a variety of biological activity. Their application is known in pharmacy [1-4] and agro-industry [5-8]. Especially such compounds are used to protect plants from insects and weeds (Figure 1).
![[1860-5397-19-87-1]](/bjoc/content/figures/1860-5397-19-87-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of bioactive pyrazoles for the protection of cultivated plants and drugs containing a thiocarbonyl group.
Figure 1: Examples of bioactive pyrazoles for the protection of cultivated plants and drugs containing a thio...
Thus, pinoxaden is a commercially available inhibitor of acetyl-CoA carboxylase and affects the biosynthesis of fatty acids in plants, which leads to herbicidal activity [9]. Pyraclonil is used as a protoporphyrinogen oxidase inhibitor for weed control. Such inhibitors not only block the production of chlorophyll and gem in plant pests, but also lead to the formation of highly reactive molecules that attack and destroy lipids and protein membranes of weed cells [10]. Fipronil is an inhibitor of GABA receptors and affects the nervous system of insects as a broad-spectrum insecticide [11,12].
On the other hand, thioamides are an important class of organic compounds. 6-Thioguanine and mercaptopurine (Figure 1), containing an intracyclic thioamide group, are antagonists of purine bases and are well known as cytostatic drugs [13]. The thioamide group as an amide isostere is used in medical chemistry to increase thermal and proteolytic stability and improve the pharmacokinetic properties of biologically active substances containing amide groups [14].
The presence of a pyrazole core and a thioamide group within the hybrid molecules that we are planning to obtain, allows us to expect both an increase in their activity and the emergence of other types of biological activity, and also high synthetic potential of such compounds.
The most common method for obtaining 3,5-disubstituted pyrazoles is the cyclocondensation of 1,3-bielectrophilic reagents with hydrazine, which acts as a 1,2-dinucleophile [15,16]. It is worthy to note that a small number of methods for the synthesis of 3,5-diaminopyrazoles are presented in the literature. These syntheses are multistage [17] or are essentially a transformation of the structure of the previously obtained pyrazoles [18] (Scheme 1A).
![[1860-5397-19-87-i1]](/bjoc/content/inline/1860-5397-19-87-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Syntheses of 4,5-diamino- and 4-thiocarbamoyl-5-aminopyrazoles.
Scheme 1: Syntheses of 4,5-diamino- and 4-thiocarbamoyl-5-aminopyrazoles.
Methods for the synthesis of pyrazoles containing thioamide and amino groups in a molecule are even less developed than methods for obtaining 3,5-diaminopyrazoles, and presented in the literature by two examples only [19-21] (Scheme 1B).
Thus, the development of effective methods for the production of 3,5-diaminopyrazoles and 3,5-diaminothiocarbamoylpyrazoles is an actual task which was chosen the aim of current study. Here, we first present the formation of 3,5-diaminopyrazoles by reaction of 2-cyanothioacetamides with hydrazine (Scheme 1C). Our paper also contains the data on our study of reaction of 3-amino-2-cyanoprop-2-enethioamides with phenyl- and tosylhydrazines leading to novel 3-amino-4-thiocarbamoylpyrazoles (Scheme 1D).
Results and Discussion
Considering that the construction of the pyrazole cycle can be carried out by the interaction of hydrazine with 1,3-bielectrophilic reagents, we paid attention to the structure of 2-cyanothioacetamides 1 and 3-amino-2-cyanoprop-2-enethioamides 2 [22] which combine in one molecule cyano- and thioamide groups, as well as a fragment of enamine, each in principle being capable of interaction with hydrazine (3a) (Figure 2).
![[1860-5397-19-87-2]](/bjoc/content/figures/1860-5397-19-87-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of starting materials.
Figure 2: Structures of starting materials.
First, we have studied the reaction of thioamides 1a–c with hydrazine (3a). It was found that the reaction proceeds smoothly in ethanol at 80 °C to form 3,5-diaminopyrazoles 4a–c (Scheme 2).
![[1860-5397-19-87-i2]](/bjoc/content/inline/1860-5397-19-87-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 3,5-diaminopyrazoles 4а–с and thiocarbamoyl-NH-pyrazoles 5a–e. Reaction conditions: 1a–c or 2a–e (1 mmol), 3a (80% aq solution) (1.1 mmol), EtOH, 80 °С, 4‒6 h.
Scheme 2: Synthesis of 3,5-diaminopyrazoles 4а–с and thiocarbamoyl-NH-pyrazoles 5a–e. Reaction conditions: 1a–...
The structures of compounds 4a–c were confirmed by 1H and 13C NMR spectroscopy data, as well as high-resolution mass spectrometry (HRMS). Compound 4a was previously obtained by another method [18]. The spectral characteristics of diaminopyrazole 4a reported in [18] correspond to the data given in the current work.
The formation of 3,5-diaminopyrazoles 4a–c occurs, presumably, as a result of a sequential attack of electrophilic carbon atoms of the cyano- and thioamide groups of thioamides 1a–c by nucleophilic nitrogen atoms of hydrazine (3a) and is accompanied by the elimination of hydrogen sulfide (Scheme 2).
Thus, we have shown that when 2-cyanothioacetamides 1a–c react with hydrazine hydrate (3a) in ethanol, both groups (thioamide and cyano) interact with hydrazine with the elimination of hydrogen sulfide and the formation of 3,5-diaminopyrazoles 4a–c (Scheme 2). It should be noted that such a reaction has not been described in the literature so far.
On the contrary, the reaction of thioamides 2a–e with hydrazine (3a) does not affect the thioamide group, and only the enamine and cyano groups participate in the construction of the pyrazole cycle. Thus, the desired 4(3)-thiocarbamoyl-NH-pyrazoles 5a–e were obtained with a yield of 68–78% (Scheme 2).
These experiments allowed us to conclude that in the compounds 2a–e under study, the enamine group has a higher reactivity towards hydrazine than the thioamide one, which leads to the preservation of the thioamide group during the reaction.
The structures of compounds 5a–e were confirmed by 1H and 13C NMR spectroscopy and HRMS, as well as X-ray diffraction analysis of a single crystal of compound 5b.
The involvement of arylhydrazines 3b,c in the reaction with enamines 2a–d similarly leads to the formation of 1-aryl-5-amino-4-thiocarbamoyl pyrazoles 6a–f with yields of 63–86% (Scheme 3).
![[1860-5397-19-87-i3]](/bjoc/content/inline/1860-5397-19-87-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1-aryl-5-amino-4-thiocarbamoylpyrazoles 6a–f. Reaction conditions: 2a–d (1 mmol), 3b,c (1.1 mmol), aq HCl (1.1 mmol), EtOH, 80 °С, 12‒16 h.
Scheme 3: Synthesis of 1-aryl-5-amino-4-thiocarbamoylpyrazoles 6a–f. Reaction conditions: 2a–d (1 mmol), 3b,c...
However, we have found that when replacing hydrazine hydrate with arylhydrazines and arylsulfonylhydrazines, the reaction in ethanol was not detected, neither at room temperature nor at reflux. The progress was achieved only upon addition of hydrochloric acid. This is probably due to the protonation of the dimethylamino moiety or/and that dimethylamine hydrochloride is a better leaving group than the free base.
The structures of compounds 6a–f were confirmed by 1H and 13C NMR spectroscopy and HRMS, as well as X-ray diffraction analysis of a single crystal of compound 6e.
It is interesting to note that the replacement of hydrazine (3a) with arylsulfonylhydrazines 3d–g and benzoylhydrazine (3h) in the reaction with enamines 2b,d leads to the formation of 1-sulfonylpyrazoles 7a–j and 1-benzoylpyrazole 7k with a significantly higher yield (72–94%, Scheme 4).
![[1860-5397-19-87-i4]](/bjoc/content/inline/1860-5397-19-87-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 1-sulfonylpyrazoles 7a–j and 1-benzoylpyrazole 7k. Conditions for 7a–j: 2b,d,e (1 mmol), 3d–g (1.1 mmol), aq HCl (1.1 mmol), EtOH, rt, 12‒16 h. Conditions for 7k: 2b (1 mmol), 3h (1.1 mmol), aq HCl (1.1 mmol), EtOH, 60 °C, 5 h. Conditions for 5b: 7b (1 mmol), aq HCl (1 mmol), EtOH, 80 °C, 24 h.
Scheme 4: Synthesis of 1-sulfonylpyrazoles 7a–j and 1-benzoylpyrazole 7k. Conditions for 7a–j: 2b,d,e (1 mmol...
We have noticed that 1-sulfonylpyrazole 7b is unstable when heated in ethanol in the presence of hydrochloric acid and converts into 3-aminopyrazole-4-carbothioamide 5b. It is worth noting that the two-stage method of obtaining 3-aminopyrazole-4-carbothioamide 5b described above is more preferable (Scheme 4) than the one-stage method using hydrazine 3a (Scheme 2), (yields 89 × 97 = 86% and 75%, respectively).
Conclusion
In order to develop an effective method for the synthesis of functional derivatives of pyrazoles, the reactions of 2-cyanothioacetamides and their derivatives, 3-amino-2-cyanoprop-2-enethioamides, with hydrazine, (substituted)phenyl- and (substituted)phenylsulfonylhydrazines were studied. The reaction between these compounds proceeds smoothly in ethanol to form 3,4-diaminopyrazoles, 5-amino-4-thiocarbamoylpyrazoles, 1-(substituted)phenyl- or 1-(substituted)phenylsulfonyl-4-thiocarbamoylpyrazoles in moderate to high yields. It was concluded that in 2-cyanothioacetamides, cyano and enamino groups are more active in the reaction with hydrazines than the thiocarbamoyl function.
Experimental
X-ray structure determination of 5b, 6a, 7a
5b: Crystal data for C8H12N4S (M = 196.27 g/mol): monoclinic, space group P21/c (no. 14), a = 10.449(2) Å, b = 9.6365(17) Å, c = 9.406(2) Å, β = 97.10(2)°, V = 939.8(3) Å3, Z = 4, μ(Mo Kα) = 0.302 mm−1, Dcalc = 1.387 g/cm3, 4629 reflections measured (5.736° ≤ 2Θ ≤ 58.142°), 2229 unique (Rint = 0.0315, Rsigma = 0.0388) which were used in all calculations. The final R1 = 0.0403, wR2 = 0.0956 (I > 2σ(I)) and R1 = 0.0510, wR2 = 0.1042 (all data). Largest diff. peak/hole 0.281/−0.285 ēÅ−3.
6e: Crystal data for C15H15F3N4OS (M = 356.37 g/mol): triclinic, space group P-1 (no. 2), a = 6.1592(10) Å, b = 12.0568(18) Å, c = 12.1206(19) Å, α = 109.032 (14)°, β = 101.540 (13)°, γ = 100.648 (13), V = 802.7(2) Å3, Z = 2, μ(Mo Kα) = 0.244 mm−1, Dcalc = 1.474 g/cm3, 5870 reflections measured (6.154° ≤ 2Θ ≤ 58.47°), 3676 unique (Rint = 0.0326, Rsigma = 0.0537) were used in all calculations. The final R1 = 0.0513, wR2 = 0.1241 (I > 2σ(I)) and R1 = 0.0685, wR2 = 0.1431 (all data). Largest diff. peak/hole 0.256/−0.392 ēÅ−3.
7a: Crystal data for C14H16N4O2S2 (M = 336.43 g/mol): monoclinic, space group P21/c (no. 14), a = 15.431(4) Å, b = 10.307(3) Å, c = 9.946(2) Å, β = 103.45(2)°, V = 1538.5(7) Å3, Z = 4, μ(Mo Kα) = 0.358 mm−1, Dcalc = 1.452 g/cm3, 7847 reflections measured (4.794° ≤ 2Θ ≤ 58.972°), 3663 unique (Rint = 0.0461, Rsigma = 0.0641) were used in all calculations. The final R1 = 0.0580, wR2 = 0.1295 (I > 2σ(I)) and R1 = 0.0993, wR2 = 0.1673(all data). Largest diff. peak/hole 0.244/−0.450 ēÅ−3.
The experiments were accomplished on the automated X-ray diffractometer «Xcalibur 3» with CCD detector following standard procedures (Mo Kα-irradiation, graphite monochromator, ω-scans with 1o step at T = 295(2) K). Empirical absorption correction was applied. The structure was solved using the intrinsic phases in ShelXT program [23] and refined by ShelXL [24] using full-matrix least-squared method for non-hydrogen atoms. The H-atoms were placed in the calculated positions and were refined in isotropic approximation. The solution and refinement of the structures were accomplished with the Olex program package [25].
CCDC 2250448 (5b), CCDC 2250451 (6e) and CCDC 2250453 (7a) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
References
-
Dadiboyena, S.; Nefzi, A. Eur. J. Med. Chem. 2011, 46, 5258–5275. doi:10.1016/j.ejmech.2011.09.016
Return to citation in text: [1] -
Mahesh, P.; Akshinthala, P.; Katari, N. K.; Gupta, L. K.; Panwar, D.; Sharma, M. K.; Jonnalagadda, S. B.; Gundla, R. ACS Omega 2023, 8, 25698–25709. doi:10.1021/acsomega.2c07539
Return to citation in text: [1] -
Sabnis, R. W. ACS Med. Chem. Lett. 2023. doi:10.1021/acsmedchemlett.3c00271
Return to citation in text: [1] -
Mykhailiuk, P. K. Chem. Rev. 2021, 121, 1670–1715. doi:10.1021/acs.chemrev.0c01015
Return to citation in text: [1] -
Kang, J.; Yue, X. L.; Chen, C. S.; Li, J. H.; Ma, H. J. Molecules 2016, 21, 39. doi:10.3390/molecules21010039
Return to citation in text: [1] -
Dong, L.; Chang, W.; Yang, W.; Xu, Z.; Cheng, J.; Shao, X.; Xu, X.; Li, Z. J. Agric. Food Chem. 2023. doi:10.1021/acs.jafc.3c03193
Return to citation in text: [1] -
Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. New J. Chem. 2017, 41, 16–41. doi:10.1039/c6nj03181a
Return to citation in text: [1] -
Ebenezer, O.; Shapi, M.; Tuszynski, J. A. Biomedicines 2022, 10, 1124. doi:10.3390/biomedicines10051124
Return to citation in text: [1] -
Takano, H. K.; Ovejero, R. F. L.; Belchior, G. G.; Maymone, G. P. L.; Dayan, F. E. Sci. Agric. (Piracicaba, Braz.) 2021, 78, e20190102. doi:10.1590/1678-992x-2019-0102
Return to citation in text: [1] -
Deng, W.; Yang, Q.; Chen, Y.; Yang, M.; Xia, Z.; Zhu, J.; Chen, Y.; Cai, J.; Yuan, S. J. Agric. Food Chem. 2020, 68, 2623–2630. doi:10.1021/acs.jafc.9b07342
Return to citation in text: [1] -
Das, P. C.; Cao, Y.; Cherrington, N.; Hodgson, E.; Rose, R. L. Chem.-Biol. Interact. 2006, 164, 200–214. doi:10.1016/j.cbi.2006.09.013
Return to citation in text: [1] -
Narahashi, T.; Zhao, X.; Ikeda, T.; Salgado, V. L.; Yeh, J. Z. Pestic. Biochem. Physiol. 2010, 97, 149–152. doi:10.1016/j.pestbp.2009.07.008
Return to citation in text: [1] -
Crouwel, F.; Buiter, H. J. C.; de Boer, N. K. J. Crohn's Colitis 2022, 16, 1177–1183. doi:10.1093/ecco-jcc/jjac004
Return to citation in text: [1] -
Dyachenko, V. D.; Dyachenko, I. V.; Nenajdenko, V. G. Russ. Chem. Rev. 2018, 87, 1–27. doi:10.1070/rcr4760
Return to citation in text: [1] -
Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459
Return to citation in text: [1] -
Fustero, S.; Simón-Fuentes, A.; Sanz-Cervera, J. F. Org. Prep. Proced. Int. 2009, 41, 253–290. doi:10.1080/00304940903077832
Return to citation in text: [1] -
Gopalsamy, A.; Ciszewski, G.; Shi, M.; Berger, D.; Hu, Y.; Lee, F.; Feldberg, L.; Frommer, E.; Kim, S.; Collins, K.; Wojciechowicz, D.; Mallon, R. Bioorg. Med. Chem. Lett. 2009, 19, 6890–6892. doi:10.1016/j.bmcl.2009.10.074
Return to citation in text: [1] -
Lim, F. P. L.; Tan, K. C.; Luna, G.; Tiekink, E. R. T.; Dolzhenko, A. V. Tetrahedron 2019, 75, 2314–2321. doi:10.1016/j.tet.2019.03.003
Return to citation in text: [1] [2] [3] -
Bovara, R.; Largaiolli, R.; Meroni, G. J. Labelled Compd. 1971, 7, 357–359. doi:10.1002/jlcr.2590070325
Return to citation in text: [1] -
Khosropour, A. R.; Noei, J.; Mirjafari, A. J. Iran. Chem. Soc. 2010, 7, 752–758. doi:10.1007/bf03246065
Return to citation in text: [1] -
Naito, Y.; Akahoshi, F.; Takeda, S.; Okada, T.; Kajii, M.; Nishimura, H.; Sugiura, M.; Fukaya, C.; Kagitani, Y. J. Med. Chem. 1996, 39, 3019–3029. doi:10.1021/jm9507993
Return to citation in text: [1] -
Lugovik, K. I.; Eltyshev, A. K.; Benassi, E.; Belskaya, N. P. Chem. – Asian J. 2017, 12, 2410–2425. doi:10.1002/asia.201700721
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1]
| 25. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 1. | Dadiboyena, S.; Nefzi, A. Eur. J. Med. Chem. 2011, 46, 5258–5275. doi:10.1016/j.ejmech.2011.09.016 |
| 2. | Mahesh, P.; Akshinthala, P.; Katari, N. K.; Gupta, L. K.; Panwar, D.; Sharma, M. K.; Jonnalagadda, S. B.; Gundla, R. ACS Omega 2023, 8, 25698–25709. doi:10.1021/acsomega.2c07539 |
| 3. | Sabnis, R. W. ACS Med. Chem. Lett. 2023. doi:10.1021/acsmedchemlett.3c00271 |
| 4. | Mykhailiuk, P. K. Chem. Rev. 2021, 121, 1670–1715. doi:10.1021/acs.chemrev.0c01015 |
| 11. | Das, P. C.; Cao, Y.; Cherrington, N.; Hodgson, E.; Rose, R. L. Chem.-Biol. Interact. 2006, 164, 200–214. doi:10.1016/j.cbi.2006.09.013 |
| 12. | Narahashi, T.; Zhao, X.; Ikeda, T.; Salgado, V. L.; Yeh, J. Z. Pestic. Biochem. Physiol. 2010, 97, 149–152. doi:10.1016/j.pestbp.2009.07.008 |
| 23. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 10. | Deng, W.; Yang, Q.; Chen, Y.; Yang, M.; Xia, Z.; Zhu, J.; Chen, Y.; Cai, J.; Yuan, S. J. Agric. Food Chem. 2020, 68, 2623–2630. doi:10.1021/acs.jafc.9b07342 |
| 24. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 9. | Takano, H. K.; Ovejero, R. F. L.; Belchior, G. G.; Maymone, G. P. L.; Dayan, F. E. Sci. Agric. (Piracicaba, Braz.) 2021, 78, e20190102. doi:10.1590/1678-992x-2019-0102 |
| 18. | Lim, F. P. L.; Tan, K. C.; Luna, G.; Tiekink, E. R. T.; Dolzhenko, A. V. Tetrahedron 2019, 75, 2314–2321. doi:10.1016/j.tet.2019.03.003 |
| 5. | Kang, J.; Yue, X. L.; Chen, C. S.; Li, J. H.; Ma, H. J. Molecules 2016, 21, 39. doi:10.3390/molecules21010039 |
| 6. | Dong, L.; Chang, W.; Yang, W.; Xu, Z.; Cheng, J.; Shao, X.; Xu, X.; Li, Z. J. Agric. Food Chem. 2023. doi:10.1021/acs.jafc.3c03193 |
| 7. | Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. New J. Chem. 2017, 41, 16–41. doi:10.1039/c6nj03181a |
| 8. | Ebenezer, O.; Shapi, M.; Tuszynski, J. A. Biomedicines 2022, 10, 1124. doi:10.3390/biomedicines10051124 |
| 18. | Lim, F. P. L.; Tan, K. C.; Luna, G.; Tiekink, E. R. T.; Dolzhenko, A. V. Tetrahedron 2019, 75, 2314–2321. doi:10.1016/j.tet.2019.03.003 |
| 17. | Gopalsamy, A.; Ciszewski, G.; Shi, M.; Berger, D.; Hu, Y.; Lee, F.; Feldberg, L.; Frommer, E.; Kim, S.; Collins, K.; Wojciechowicz, D.; Mallon, R. Bioorg. Med. Chem. Lett. 2009, 19, 6890–6892. doi:10.1016/j.bmcl.2009.10.074 |
| 19. | Bovara, R.; Largaiolli, R.; Meroni, G. J. Labelled Compd. 1971, 7, 357–359. doi:10.1002/jlcr.2590070325 |
| 20. | Khosropour, A. R.; Noei, J.; Mirjafari, A. J. Iran. Chem. Soc. 2010, 7, 752–758. doi:10.1007/bf03246065 |
| 21. | Naito, Y.; Akahoshi, F.; Takeda, S.; Okada, T.; Kajii, M.; Nishimura, H.; Sugiura, M.; Fukaya, C.; Kagitani, Y. J. Med. Chem. 1996, 39, 3019–3029. doi:10.1021/jm9507993 |
| 15. | Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459 |
| 16. | Fustero, S.; Simón-Fuentes, A.; Sanz-Cervera, J. F. Org. Prep. Proced. Int. 2009, 41, 253–290. doi:10.1080/00304940903077832 |
| 22. | Lugovik, K. I.; Eltyshev, A. K.; Benassi, E.; Belskaya, N. P. Chem. – Asian J. 2017, 12, 2410–2425. doi:10.1002/asia.201700721 |
| 14. | Dyachenko, V. D.; Dyachenko, I. V.; Nenajdenko, V. G. Russ. Chem. Rev. 2018, 87, 1–27. doi:10.1070/rcr4760 |
| 13. | Crouwel, F.; Buiter, H. J. C.; de Boer, N. K. J. Crohn's Colitis 2022, 16, 1177–1183. doi:10.1093/ecco-jcc/jjac004 |
| 18. | Lim, F. P. L.; Tan, K. C.; Luna, G.; Tiekink, E. R. T.; Dolzhenko, A. V. Tetrahedron 2019, 75, 2314–2321. doi:10.1016/j.tet.2019.03.003 |
© 2023 Filimonov et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.