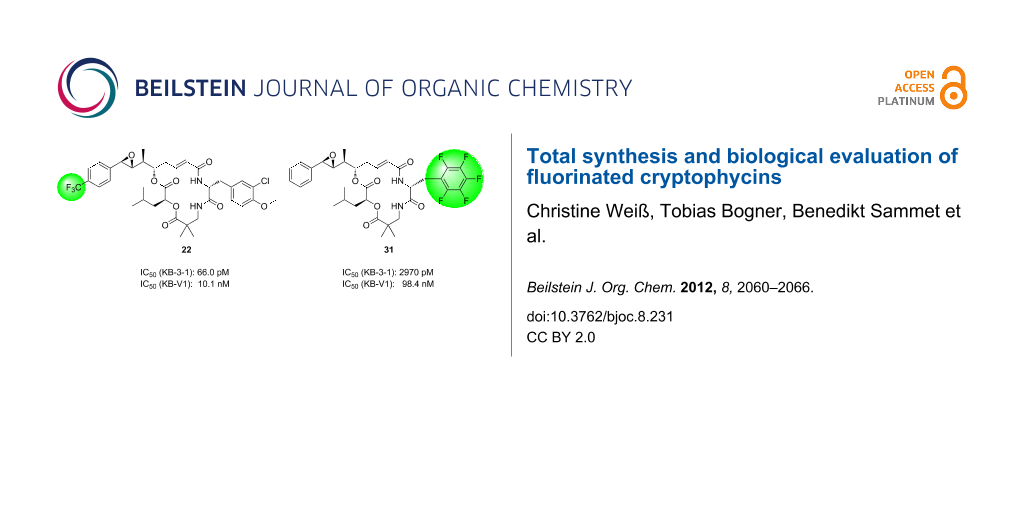
Abstract
Cryptophycins are cytotoxic natural products that exhibit considerable activities even against multi-drug-resistant tumor cell lines. As fluorinated pharmaceuticals have become more and more important during the past decades, fluorine-functionalized cryptophycins were synthesized and evaluated in cell-based cytotoxicity assays. The unit A trifluoromethyl-modified cryptophycin proved to be highly active against KB-3-1 cells and exhibited an IC50 value in the low picomolar range. However, the replacement of the 3-chloro-4-methoxyphenyl-substituent in unit B by a pentafluorophenyl moiety resulted in a significant loss of activity.

Graphical Abstract
Introduction
Cryptophycins form a class of cytotoxic sixteen-membered macrocyclic depsipeptides. Cryptophycin-1 (1) was isolated for the first time in 1990 from cyanobacteria Nostoc sp. ATCC 53789 [1] (Figure 1). Moore et al. isolated cryptophycin-1 from the related Nostoc strain GSV 224, investigated the stereochemistry, and described the cytotoxicity [2]. At the same time Kobayashi et al. succeeded in a full structural analysis and described the first total synthesis of another member of the cryptophycin family [3,4]. Twenty-eight naturally occurring cryptophycins have been isolated up to this day [5-7], while numerous synthetic analogues have been synthesized in the frame of structure–activity-relationship studies [8,9]. Cryptophycins display remarkable biological activity against multi-drug-resistant (MDR) tumor cells. Such tumor cells express a P-glycoprotein, a drug efflux pump that transports xenobiotics out of the cell. A synthetic analogue, cryptohycin-52 (2, LY355703), has been investigated in clinical trials. However, this development was discontinued because of neurotoxic side effects and lacking efficacy in vivo [10,11].
![[1860-5397-8-231-1]](/bjoc/content/figures/1860-5397-8-231-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of cryptophycin-1 (1) and -52 (2).
Figure 1: Structures of cryptophycin-1 (1) and -52 (2).
Fluorinated drugs are gaining increasing importance, and currently about 20% of all pharmaceuticals on the world market contain fluorine substituents [12,13]. Fluorination is supposed to enhance bioavailability and receptor selectivity. The van der Waals-radius of a fluorine substituent (1.47 Å) lies between the value of a hydrogen substituent (1.20 Å) and an oxygen substituent (1.52 Å). However, despite this similarity in size, a fluorine substituent exerts considerable electronic effects due to the high electronegativity. A trifluoromethyl substituted analogue of epothilone, another important tubulin-binding cytotoxic drug, was shown to retain the cytotoxic activity of the parent compound. At the same time nonspecific side effects due to oxidative degradation were prevented by the introduction of the CF3 group [14,15]. Likewise, partially fluorinated taxoids, analogues of paclitaxel and docetaxel, displayed biological activity even exceeding that of the parent nonfluorinated compounds [16]. The interesting biological profile of fluorinated cytotoxic agents prompted us to synthesize partially fluorinated analogues of cryptophycins.
The depsipeptidic character of the cryptophycins suggests four different fragments to be assembled in the total synthesis, named unit A–D (Figure 1). Unit A is an α,β-unsaturated δ-hydroxy acid that usually also contains a benzylic epoxide or a benzylic double bond. Unit B represents a chlorinated O-methyl-D-tyrosine derivative, while unit C is a β2-amino acid, usually β2-homoalanine. Finally, unit D is leucic acid, the hydroxy analogue of leucine. Numerous synthetic analogues have been obtained in the frame of structure–activity-relationship studies (SAR-studies), as reviewed in [17,18].
Unit A para-alkoxymethyl derivatives of cryptophycin-52 have been synthesized and were shown to retain cytotoxicity even against MDR tumor cell lines [19]. The introduction of a fluorine substituent in the same position also provides a cytotoxic analogue, albeit with decreased biological activity by a factor of 5 [8].
In unit B the chlorine and the methoxy substituents at the D-tyrosine residue were crucial for high antimitotic activity [17,18]. Moore et al. patented the synthesis of fluorinated analogues of cryptophycin-1 and cryptophycin-52 [20]. In particular, derivative 3 was shown to retain biological activity (IC50 = 39 pM) and was active against the tumor cell line KB-3-1 [21] (Figure 2). The chlorohydrin derived from 4 that also contained a fluorine substituent in the para-position of the unit A phenyl ring was patented as a promising candidate [22].
![[1860-5397-8-231-2]](/bjoc/content/figures/1860-5397-8-231-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Fluorinated derivatives of cryptophycin-1 and -52 [20-22].
Figure 2: Fluorinated derivatives of cryptophycin-1 and -52 [20-22].
In the frame of our on-going SAR studies on cryptophycins [19,23-30], we envisaged the synthesis of analogues of cryptophycin-52 with a para-trifluoromethyl substituent at the unit A aryl ring. In addition, we targeted the replacement of the unit B by a D-pentafluorophenylalanine residue.
Results and Discussion
Cryptophycin-52 with a para-trifluoromethyl substituted unit A
The synthesis of the para-trifluoromethyl substituted unit A started with a modified Knoevenagel condensation [23,31]. The required aldehyde 9 was obtained by DIBAL-H reduction of the corresponding methyl ester 8 and was found to decompose upon chromatographic purification (Scheme 1). However, it can usually be employed in the Knoevenagel condensation without purification. Reaction of 9 with malonic acid in the presence of piperidine/acetic acid gave the β,γ-unsaturated carboxylic acid 10. The latter compound was transformed into the methyl ester by treatment with SOCl2 in methanol. The resulting ester 11 could then be directly employed without purification in the asymmetric dihydroxylation with osmium tetroxide and (DHQD)2PHAL, in close analogy to a previously published procedure [23]. The initially formed vicinal diol cyclizes under the reaction conditions to give lactone 12 in enantiomerically pure form (chiral HPLC: Chiralpak OD®). Deprotonation of 12 with 2.5 equiv of LDA, followed by treatment with iodomethane furnished the α-methyl substituted lactone 13. Treatment of this compound with acetone dimethyl acetal in methanol in the presence of an acidic ion exchanger resulted in acetonide protection of the vicinal diol, accompanied by methyl ester formation. The methyl ester 14 was subsequently reduced with DIBAL-H to give the aldehyde 15. In order to avoid epimerisation, this aldehyde was not purified, but filtered through Celite only and then reacted with allyl-tri-n-butyltin to give the homoallyl alcohol 16. The magnesium bromide diethyl etherate mediated allylation proceeded under substrate control and with complete diastereoselectivity [23,32].
![[1860-5397-8-231-i1]](/bjoc/content/inline/1860-5397-8-231-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Access to the trifluoromethyl substituted unit A-building block 16. Reagents and conditions: (a) SOCl2, MeOH, 0 °C → rt, 16 h; (b) DIBAL-H, CH2Cl2, −78 °C, 3.5 h; (c) malonic acid, piperidine, AcOH, DMSO, 65 °C, 1.5 h; (d) SOCl2, MeOH, 0 °C → rt, 1 h; (e) K2CO3, K2OsO4∙2H2O, K3[Fe(CN)6], (DHQD)2-PHAL, CH3SO2NH2, t-BuOH/H2O, 0 °C, 42 h; (f) LDA, MeI, THF, −78 °C, 3 d; (g) (CH3)2C(OCH3)2, MeOH, Amberlyst-15®, rt, 8 d; (h) DIBAL-H, CH2Cl2, −78 °C, 4.5 h; (i) AllylSnBu3, MgBr2∙Et2O, CH2Cl2, −78 °C, 15 h.
Scheme 1: Access to the trifluoromethyl substituted unit A-building block 16. Reagents and conditions: (a) SO...
Cross-metathesis of homoallyl alcohol 16 with the unit B derived acrylamide 17 provided the α,β-unsaturated δ-hydroxy carboxamide 18 (Scheme 2). In order to bring about complete metathesis of 16, the acrylamide 17 had to be employed in 1.2-fold excess, which resulted in a contamination of the cross-metathesis product 18 with minor amounts of the homo-coupling product 23. The latter could not be separated by flash chromatography on this stage, but did not interfere with the subsequent Yamaguchi esterification of 18 with the unit C–D segment 19 and was removed on this stage [33]. Fmoc cleavage of the seco-depsipeptide 20 liberated the free amino group of unit C, which under the reaction conditions displaced the trichloroethylester of unit B resulting in macrocyclization according to Moher et al. [34]. In the final steps the dioxolane ring of 21 was cleaved with trifluoroacetic acid in the presence of water. The resulting vicinal diol was not purified, but reacted with a large excess of trimethyl orthoformate. The cyclic orthoester resulting from this transformation was directly subjected to reaction with acetyl bromide to form a bromohydrin formate. This was then treated with a potassium carbonate/ethylene glycol/dimethoxyethane-emulsion to bring about cleavage of the formyl ester accompanied by epoxide formation as previously described by us [19]. The trifluoromethyl substituted cryptohycin-52 analogue 22 was obtained in a yield of 39% over the final four steps. It was purified by column chromatography, followed by lyophilization.
![[1860-5397-8-231-i2]](/bjoc/content/inline/1860-5397-8-231-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Assembly of units A–D and macrocyclization, followed by diol-epoxide transformation to give the trifluoromethyl substituted analogue 22 of cryptophycin-52. Reagents and conditions: (a) Grubbs II catalyst, CH2Cl2, reflux, 16 h; (b) 19, DMAP, NEt3, 2,4,6-trichlorobenzoylchloride, THF, 0 °C, 1 h; (c) piperidine, DMF, rt, 16 h; (d) TFA, CH2Cl2, H2O, 0 °C, 3 h; (e) (CH3O)3CH, PPTS, CH2Cl2, rt, 2 h; (f) AcBr, CH2Cl2, rt, 4 h; (g) K2CO3, DME, ethylene glycol, rt, 3 min.
Scheme 2: Assembly of units A–D and macrocyclization, followed by diol-epoxide transformation to give the tri...
Cryptophycin-52 with D-pentafluorophenylalanine as unit B
The N-acryloyl derivative 26 of D-pentafluorophenylalanine was obtained by carbodiimide esterification of commercially available Boc-D-pentafluorophenylalanine (24) with trichloroethanol, followed by cleavage of Boc and reaction with acryloylchloride in the presence of base [19] (Scheme 3).
![[1860-5397-8-231-i3]](/bjoc/content/inline/1860-5397-8-231-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the pentafluorophenylalanine building block 26. Reagents and conditions: (a) pyridine, trichloroethanol, DCC, CH2Cl2, 0 °C, 20 h; (b) 1. TFA, rt, 2 h; 2. NEt3, acryloylchloride, CH2Cl2, 0 °C, 7 h.
Scheme 3: Synthesis of the pentafluorophenylalanine building block 26. Reagents and conditions: (a) pyridine,...
The cryptophycin analogue with D-pentafluorophenylalanine as unit B was synthesized by the same convergent route as described for derivative 22. Homoallyl alcohol 27 [23] was reacted with the D-pentafluorophenylalanine derivative 26 in a cross-metathesis reaction in the presence of Grubbs II catalyst (Scheme 4). The resulting α,β-unsaturated δ-hydroxy carboxamide 28, representing units A and B was then esterified with 19 under Yamaguchi conditions with 2,4,6-trichlorobenzoylchloride and triethylamine in the presence of catalytic amounts of DMAP. Macrocyclization was brought about by cleavage of the Fmoc protecting group from the unit C amino group, which concomitantly displaced the trichloroethylester at unit B to result in the macrocyclic product 30 [34]. Cleavage of the dioxolane liberated the vicinal diol, which was then subjected to the final diol-epoxide transformation to provide the cryptophycin-52 analogue 31 in a yield of 14% over the final four steps.
![[1860-5397-8-231-i4]](/bjoc/content/inline/1860-5397-8-231-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Convergent synthesis of the pentafluorinated cryptophycin 31. Reagents and conditions: (a) Grubbs II catalyst, CH2Cl2, reflux, 16 h; (b) 19, DMAP, NEt3, 2,4,6-trichlorobenzoylchloride, THF, 0 °C, 1 h; (c) piperidine, DMF, rt, 16 h; (d) TFA, CH2Cl2, H2O, 0 °C, 3 h; (e) (CH3O)3CH, PPTS, CH2Cl2, rt, 2 h; (f) AcBr, CH2Cl2, rt, 4 h; (g) K2CO3, DME, ethylene glycol, rt, 3 min.
Scheme 4: Convergent synthesis of the pentafluorinated cryptophycin 31. Reagents and conditions: (a) Grubbs I...
The biological activities of the fluorine-functionalized cryptophycin analogues were determined in a resazurin assay with the tumor cell line KB-3-1 and its MDR correlate KB-V1. The IC50 values of the fluorinated cryptophycins 22 and 31 were compared to cryptophycin-52 in Table 1 [17]. While the cytotoxicity of the unit A-modified analogue 22 against the tumor cell line KB-3-1 was only by about a factor of 4 decreased compared to cryptophycin-52, the pentafluorophenylalanine-containing derivative 31 was much less active. A significant loss in activity of both analogues against the MDR cell line KB-V1 was observed. The degree of activity against MDR tumor cells can be described by the resistance factor FR, which is defined as the ratio of the IC50 value for the MDR cell line and the value for the nonresistant cell line. A high FR means a high loss of activity due to the cellular resistance mechanisms. Analogue 22 exhibited a high FR value whereas compound 31 showed a lower loss of activity.
Conclusion
The synthesis of selectively fluorinated cryptophycin-52 analogues succeeded and both target compounds could be obtained. The two analogues were less active, both against the tumor cell line KB-3-1 and its MDR subclone KB-V1. This fact was quite surprising because the fluorinated cryptophycins were expected to display higher lipophilicity compared to the parent compound cryptophycin-52 and, therefore, exhibit equal or even higher activities. In contrast, more amphiphilic or polar compounds are usually good substrates for the P-glycoprotein efflux pump resulting in a decreased bioactivity.
Supporting Information
| Supporting Information File 1: Full experimental procedures and detailed analytical data for the synthesis of all compounds. | ||
| Format: PDF | Size: 515.4 KB | Download |
References
-
Schwartz, R. E.; Hirsch, C. F.; Sesin, D. F.; Flor, J. E.; Chartrain, M.; Fromtling, R. E.; Harris, G. H.; Salvatore, M. J.; Liesch, J. M.; Yudin, K. J. Ind. Microbiol. Biotechnol. 1990, 5, 113–123. doi:10.1007/BF01573860
Return to citation in text: [1] -
Trimurtulu, G.; Ohtani, I.; Patterson, G. M. L.; Moore, R. E.; Corbett, T. H.; Valeriote, F. A.; Demchik, L. J. Am. Chem. Soc. 1994, 116, 4729–4737. doi:10.1021/ja00090a020
Return to citation in text: [1] -
Kobayashi, M.; Kurosu, M.; Ohyabu, N.; Wang, W.; Fujii, S.; Kitagawa, I. Chem. Pharm. Bull. 1994, 42, 2196–2198. doi:10.1248/cpb.42.2196
Return to citation in text: [1] -
Kobayashi, M.; Aoki, S.; Ohyabu, N.; Kurosu, M.; Wang, W.; Kitagawa, I. Tetrahedron Lett. 1994, 35, 7969–7972. doi:10.1016/S0040-4039(00)78398-5
Return to citation in text: [1] -
Golakoti, T.; Ogino, J.; Heltzel, C. E.; Le Husebo, T.; Jensen, C. M.; Larsen, L. K.; Patterson, G. M. L.; Moore, R. E.; Mooberry, S. L. J. Am. Chem. Soc. 1995, 117, 12030–12049. doi:10.1021/ja00154a002
Return to citation in text: [1] -
Subbaraju, G. V.; Golakoti, T.; Patterson, G. M. L.; Moore, R. E. J. Nat. Prod. 1997, 60, 302–305. doi:10.1021/np960700a
Return to citation in text: [1] -
Chaganty, S.; Golakoti, T.; Heltzel, C.; Moore, R. E.; Yoshida, W. Y. J. Nat. Prod. 2004, 67, 1403–1406. doi:10.1021/np0499665
Return to citation in text: [1] -
Eggen, M.; Georg, G. I. Med. Res. Rev. 2002, 22, 85–101. doi:10.1002/med.10002
Return to citation in text: [1] [2] -
Hamel, E.; Covell, D. G. Curr. Med. Chem. 2002, 2, 19–53. doi:10.2174/1568011023354263
Return to citation in text: [1] -
Edelman, M. J.; Gandara, D. R.; Hausner, P.; Israel, V.; Thornton, D.; DeSanto, J.; Doyle, L. A. Lung Cancer 2003, 39, 197–199. doi:10.1016/S0169-5002(02)00511-1
Return to citation in text: [1] -
Sessa, C.; Weigang-Köhler, K.; Pagani, O.; Greim, G.; Mora, O.; De Pas, T.; Burgess, M.; Weimer, I.; Johnson, R. Eur. J. Cancer 2002, 38, 2388–2396. doi:10.1016/S0959-8049(02)00489-6
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Thayer, A. M. Chem. Eng. News 2006, 84 (33), 29–31.
Return to citation in text: [1] -
Rivkin, A.; Chou, T.-C.; Danishefsky, S. J. Angew. Chem. 2005, 117, 2898–2910. doi:10.1002/ange.200461751
Return to citation in text: [1] -
Chou, T.-C.; Dong, H.; Rivkin, A.; Yoshimura, F.; Gabarda, A. E.; Cho, Y. S.; Tong, W. P.; Danishefsky, S. J. Angew. Chem. 2003, 115, 4910–4915. doi:10.1002/ange.200352361
Return to citation in text: [1] -
Ojima, I.; Inoue, T.; Chakravarty, S. J. Fluorine Chem. 1999, 97, 3–10. doi:10.1016/S0022-1139(99)00058-5
Return to citation in text: [1] -
Eißler, S.; Stončius, A.; Nahrwold, M.; Sewald, N. Synthesis 2006, 3747–3789. doi:10.1055/s-2006-950332
Return to citation in text: [1] [2] [3] -
Nahrwold, M.; Eißler, S.; Sewald, N. Chim. Oggi – Chem. Today (Suppl. Focus on Peptides) 2008, 26, 13–16.
Return to citation in text: [1] [2] -
Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Chem.–Eur. J. 2009, 15, 11273–11287. doi:10.1002/chem.200901750
Return to citation in text: [1] [2] [3] [4] -
Al-Awar, R. S.; Ehlhardt, W. J.; Gottumukkala, S. V.; Martinelli, M. J.; Moher, E. D.; Moore, R. E.; Munroe, J. E.; Norman, B. H.; Patel, V. F.; Shih, C.; Toth, J. E.; Vasudevan, V.; Ray, J. E. (Eli Lilly and Company, University of Hawaii, Wayne State University) Pharmaceutical Compounds. WO9808505A1, March 3, 1998.
Return to citation in text: [1] [2] -
Moore, R. E.; Hemscheidt, T. K. (University of Hawaii, Wayne State University), Cryptophycins From Aberrant Biosynthesis. WO9708334A1, March 6, 1997.
Return to citation in text: [1] [2] -
Shih, C.; Williams, D. C. (Eli Lilly and Company). EP870510A2, 1998.
Return to citation in text: [1] [2] -
Eißler, S.; Nahrwold, M.; Neumann, B.; Stammler, H.-G.; Sewald, N. Org. Lett. 2007, 9, 817–819. doi:10.1021/ol063032l
Return to citation in text: [1] [2] [3] [4] [5] -
Mast, C. A.; Eißler, S.; Stončius, A.; Stammler, H.-G.; Neumann, B.; Sewald, N. Chem.–Eur. J. 2005, 11, 4667–4677. doi:10.1002/chem.200500282
Return to citation in text: [1] -
Eißler, S.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2007, 273–277. doi:10.1055/s-2007-1000868
Return to citation in text: [1] -
Sammet, B.; Radzey, H.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2009, 417–420. doi:10.1055/s-0028-1087541
Return to citation in text: [1] -
Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. Org. Lett. 2010, 12, 1064–1067. doi:10.1021/ol1000473
Return to citation in text: [1] -
Sammet, B.; Bogner, T.; Nahrwold, M.; Weiß, C.; Sewald, N. J. Org. Chem. 2010, 75, 6953–6960. doi:10.1021/jo101563s
Return to citation in text: [1] -
Sammet, B.; Brax, M.; Sewald, N. Beilstein J. Org. Chem. 2011, 7, 243–245. doi:10.3762/bjoc.7.32
Return to citation in text: [1] -
Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preuße, T.; Sewald, N. J. Med. Chem. 2012.
in revision.
Return to citation in text: [1] -
Ragoussis, N.; Ragoussis, V. J. Chem. Soc., Perkin Trans. 1 1998, 3529–3534. doi:10.1039/A806173D
Return to citation in text: [1] -
Eißler, S. Synthese von Cryptophycinen für SAR-Studien. Ph.D. Thesis, Bielefeld University, Bielefeld, Germany, 2008.
Return to citation in text: [1] -
Weiß, C. Synthese von Cryptophycin-Analoga für SAR- und subzelluläre Lokalisationsstudien. Ph.D. Thesis, Bielefeld University, Bielefeld, Germany, 2012.
Return to citation in text: [1] -
Hoard, D. W.; Moher, E. D.; Martinelli, M. J.; Norman, B. H. Org. Lett. 2002, 4, 1813–1815. doi:10.1021/ol025933+
Return to citation in text: [1] [2]
| 17. | Eißler, S.; Stončius, A.; Nahrwold, M.; Sewald, N. Synthesis 2006, 3747–3789. doi:10.1055/s-2006-950332 |
| 1. | Schwartz, R. E.; Hirsch, C. F.; Sesin, D. F.; Flor, J. E.; Chartrain, M.; Fromtling, R. E.; Harris, G. H.; Salvatore, M. J.; Liesch, J. M.; Yudin, K. J. Ind. Microbiol. Biotechnol. 1990, 5, 113–123. doi:10.1007/BF01573860 |
| 8. | Eggen, M.; Georg, G. I. Med. Res. Rev. 2002, 22, 85–101. doi:10.1002/med.10002 |
| 9. | Hamel, E.; Covell, D. G. Curr. Med. Chem. 2002, 2, 19–53. doi:10.2174/1568011023354263 |
| 21. | Moore, R. E.; Hemscheidt, T. K. (University of Hawaii, Wayne State University), Cryptophycins From Aberrant Biosynthesis. WO9708334A1, March 6, 1997. |
| 5. | Golakoti, T.; Ogino, J.; Heltzel, C. E.; Le Husebo, T.; Jensen, C. M.; Larsen, L. K.; Patterson, G. M. L.; Moore, R. E.; Mooberry, S. L. J. Am. Chem. Soc. 1995, 117, 12030–12049. doi:10.1021/ja00154a002 |
| 6. | Subbaraju, G. V.; Golakoti, T.; Patterson, G. M. L.; Moore, R. E. J. Nat. Prod. 1997, 60, 302–305. doi:10.1021/np960700a |
| 7. | Chaganty, S.; Golakoti, T.; Heltzel, C.; Moore, R. E.; Yoshida, W. Y. J. Nat. Prod. 2004, 67, 1403–1406. doi:10.1021/np0499665 |
| 3. | Kobayashi, M.; Kurosu, M.; Ohyabu, N.; Wang, W.; Fujii, S.; Kitagawa, I. Chem. Pharm. Bull. 1994, 42, 2196–2198. doi:10.1248/cpb.42.2196 |
| 4. | Kobayashi, M.; Aoki, S.; Ohyabu, N.; Kurosu, M.; Wang, W.; Kitagawa, I. Tetrahedron Lett. 1994, 35, 7969–7972. doi:10.1016/S0040-4039(00)78398-5 |
| 17. | Eißler, S.; Stončius, A.; Nahrwold, M.; Sewald, N. Synthesis 2006, 3747–3789. doi:10.1055/s-2006-950332 |
| 18. | Nahrwold, M.; Eißler, S.; Sewald, N. Chim. Oggi – Chem. Today (Suppl. Focus on Peptides) 2008, 26, 13–16. |
| 2. | Trimurtulu, G.; Ohtani, I.; Patterson, G. M. L.; Moore, R. E.; Corbett, T. H.; Valeriote, F. A.; Demchik, L. J. Am. Chem. Soc. 1994, 116, 4729–4737. doi:10.1021/ja00090a020 |
| 20. | Al-Awar, R. S.; Ehlhardt, W. J.; Gottumukkala, S. V.; Martinelli, M. J.; Moher, E. D.; Moore, R. E.; Munroe, J. E.; Norman, B. H.; Patel, V. F.; Shih, C.; Toth, J. E.; Vasudevan, V.; Ray, J. E. (Eli Lilly and Company, University of Hawaii, Wayne State University) Pharmaceutical Compounds. WO9808505A1, March 3, 1998. |
| 16. | Ojima, I.; Inoue, T.; Chakravarty, S. J. Fluorine Chem. 1999, 97, 3–10. doi:10.1016/S0022-1139(99)00058-5 |
| 19. | Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Chem.–Eur. J. 2009, 15, 11273–11287. doi:10.1002/chem.200901750 |
| 14. | Rivkin, A.; Chou, T.-C.; Danishefsky, S. J. Angew. Chem. 2005, 117, 2898–2910. doi:10.1002/ange.200461751 |
| 15. | Chou, T.-C.; Dong, H.; Rivkin, A.; Yoshimura, F.; Gabarda, A. E.; Cho, Y. S.; Tong, W. P.; Danishefsky, S. J. Angew. Chem. 2003, 115, 4910–4915. doi:10.1002/ange.200352361 |
| 12. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 13. | Thayer, A. M. Chem. Eng. News 2006, 84 (33), 29–31. |
| 10. | Edelman, M. J.; Gandara, D. R.; Hausner, P.; Israel, V.; Thornton, D.; DeSanto, J.; Doyle, L. A. Lung Cancer 2003, 39, 197–199. doi:10.1016/S0169-5002(02)00511-1 |
| 11. | Sessa, C.; Weigang-Köhler, K.; Pagani, O.; Greim, G.; Mora, O.; De Pas, T.; Burgess, M.; Weimer, I.; Johnson, R. Eur. J. Cancer 2002, 38, 2388–2396. doi:10.1016/S0959-8049(02)00489-6 |
| 17. | Eißler, S.; Stončius, A.; Nahrwold, M.; Sewald, N. Synthesis 2006, 3747–3789. doi:10.1055/s-2006-950332 |
| 18. | Nahrwold, M.; Eißler, S.; Sewald, N. Chim. Oggi – Chem. Today (Suppl. Focus on Peptides) 2008, 26, 13–16. |
| 23. | Eißler, S.; Nahrwold, M.; Neumann, B.; Stammler, H.-G.; Sewald, N. Org. Lett. 2007, 9, 817–819. doi:10.1021/ol063032l |
| 31. | Ragoussis, N.; Ragoussis, V. J. Chem. Soc., Perkin Trans. 1 1998, 3529–3534. doi:10.1039/A806173D |
| 20. | Al-Awar, R. S.; Ehlhardt, W. J.; Gottumukkala, S. V.; Martinelli, M. J.; Moher, E. D.; Moore, R. E.; Munroe, J. E.; Norman, B. H.; Patel, V. F.; Shih, C.; Toth, J. E.; Vasudevan, V.; Ray, J. E. (Eli Lilly and Company, University of Hawaii, Wayne State University) Pharmaceutical Compounds. WO9808505A1, March 3, 1998. |
| 21. | Moore, R. E.; Hemscheidt, T. K. (University of Hawaii, Wayne State University), Cryptophycins From Aberrant Biosynthesis. WO9708334A1, March 6, 1997. |
| 22. | Shih, C.; Williams, D. C. (Eli Lilly and Company). EP870510A2, 1998. |
| 19. | Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Chem.–Eur. J. 2009, 15, 11273–11287. doi:10.1002/chem.200901750 |
| 23. | Eißler, S.; Nahrwold, M.; Neumann, B.; Stammler, H.-G.; Sewald, N. Org. Lett. 2007, 9, 817–819. doi:10.1021/ol063032l |
| 24. | Mast, C. A.; Eißler, S.; Stončius, A.; Stammler, H.-G.; Neumann, B.; Sewald, N. Chem.–Eur. J. 2005, 11, 4667–4677. doi:10.1002/chem.200500282 |
| 25. | Eißler, S.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2007, 273–277. doi:10.1055/s-2007-1000868 |
| 26. | Sammet, B.; Radzey, H.; Neumann, B.; Stammler, H.-G.; Sewald, N. Synlett 2009, 417–420. doi:10.1055/s-0028-1087541 |
| 27. | Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. Org. Lett. 2010, 12, 1064–1067. doi:10.1021/ol1000473 |
| 28. | Sammet, B.; Bogner, T.; Nahrwold, M.; Weiß, C.; Sewald, N. J. Org. Chem. 2010, 75, 6953–6960. doi:10.1021/jo101563s |
| 29. | Sammet, B.; Brax, M.; Sewald, N. Beilstein J. Org. Chem. 2011, 7, 243–245. doi:10.3762/bjoc.7.32 |
| 30. |
Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preuße, T.; Sewald, N. J. Med. Chem. 2012.
in revision. |
| 23. | Eißler, S.; Nahrwold, M.; Neumann, B.; Stammler, H.-G.; Sewald, N. Org. Lett. 2007, 9, 817–819. doi:10.1021/ol063032l |
| 34. | Hoard, D. W.; Moher, E. D.; Martinelli, M. J.; Norman, B. H. Org. Lett. 2002, 4, 1813–1815. doi:10.1021/ol025933+ |
| 19. | Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Chem.–Eur. J. 2009, 15, 11273–11287. doi:10.1002/chem.200901750 |
| 19. | Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Chem.–Eur. J. 2009, 15, 11273–11287. doi:10.1002/chem.200901750 |
| 33. | Weiß, C. Synthese von Cryptophycin-Analoga für SAR- und subzelluläre Lokalisationsstudien. Ph.D. Thesis, Bielefeld University, Bielefeld, Germany, 2012. |
| 34. | Hoard, D. W.; Moher, E. D.; Martinelli, M. J.; Norman, B. H. Org. Lett. 2002, 4, 1813–1815. doi:10.1021/ol025933+ |
| 23. | Eißler, S.; Nahrwold, M.; Neumann, B.; Stammler, H.-G.; Sewald, N. Org. Lett. 2007, 9, 817–819. doi:10.1021/ol063032l |
| 23. | Eißler, S.; Nahrwold, M.; Neumann, B.; Stammler, H.-G.; Sewald, N. Org. Lett. 2007, 9, 817–819. doi:10.1021/ol063032l |
| 32. | Eißler, S. Synthese von Cryptophycinen für SAR-Studien. Ph.D. Thesis, Bielefeld University, Bielefeld, Germany, 2008. |
© 2012 Weiß et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)