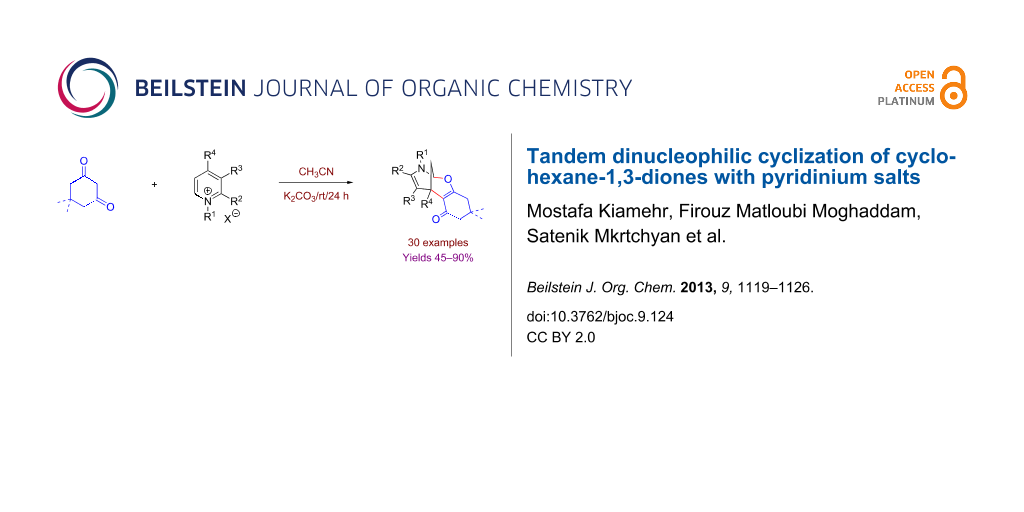
Abstract
The cyclization of cyclohexane-1,3-diones with various substituted pyridinium salts afforded functionalized 8-oxa-10-aza-tricyclo[7.3.1.02,7]trideca-2(7),11-dienes. The reaction proceeds by regioselective attack of the central carbon atom of the 1,3-dicarbonyl unit to 4-position of the pyridinium salt and subsequent cyclization by base-assisted proton migration and nucleophilic addition of the oxygen atom to the 2-position, as was elucidated by DFT computations. Fairly extensive screening of bases and additives revealed that the presence of potassium cations is essential for formation of the product.

Graphical Abstract
Introduction
The nucleophilic addition to pyridinium salts has recently been proven to be a powerful method for the construction of various dihydropyridines [1-4], which are not only valuable intermediates in organic synthesis [5-12], but also interesting compounds in medicinal [13] and bioorganic chemistry [14-16]. The tetrahydropyridine ring is an essential building block for numerous natural products, synthetic pharmaceuticals, and various biologically active compounds [17]. The tetrahydropyridine scaffold is inherent for pharmacologically relevant representatives [18-24]. At the same time, heterocyclic systems containing an annulated pyridinium core, such as quinolinium and isoquinolinium salts, are of considerable importance as building blocks for the synthesis of various alkaloid frameworks [10,25-35].
During the past decade the reaction of dinucleophiles with dielectrophiles has been a major research subject in both our laboratories. For example, we have studied reactions of quinolinium [36,37], isoquinolinium [38-40], quinazolinium [41,42] and quinoxalinium [43] salts with 1,3-bis(silyl enol ethers), i.e., masked 1,3-dicarbonyl compounds, which provided facile access to a number of bicyclic systems. Some of these reactions, such as the cyclization with quinazolinium salts, proceeded as one-pot cyclizations. Other cyclizations, such as the reaction with isoquinolinium salts, had to be carried out in two steps (formation of open-chained condensation products, which are subsequently cyclized by the addition of acid). In some cases the cyclization step failed, e.g., the reaction of 1,3-bis(silyl enol ethers) with pyridine in the presence of methyl chloroformate resulted in the formation of 1,4-dihydropyridines, which however, could not be cyclized by the addition of acid due to decomposition [36]. Additionally, we have shown a broad application of the quinolinium [44-48] and isoquinolinium [49] salts for the synthesis of a wide variety of alkaloid-like frameworks.
Results and Discussion
Reaction optimization
During the course of the above-mentioned study we discovered an interesting reaction that represents an external dinucleophilic addition of the dimedone molecule to the pyridinium salt 2a, taken as a random example, delivering 8-oxa-10-aza-tricyclo[7.3.1.02,7]trideca-2(7),11-diene (3a) in modest yield (Scheme 1). Previously, we have communicated a similar cycloaddition reaction using quinolinium salts [45], and recently Li and Yan [50] reported this reaction with phenanthrolinium salts.
![[1860-5397-9-124-i1]](/bjoc/content/inline/1860-5397-9-124-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (i) CH3CN, K2CO3, rt, 24 h.
Scheme 1: Reagents and conditions: (i) CH3CN, K2CO3, rt, 24 h.
This efficient cycloaddition encouraged us to investigate this process, to improve the reaction conditions, and to extend the reaction with other pyridinium salts and diketones. Herein we report the results of that study. First, we studied the reaction of pyridinium salt 2a with dimedone (1a) using various bases in different solvents (Table 1). At the same time, different ratios of 1a and 2a were examined. To reach the optimal yields, dimedone and pyridinium salt 2a should be used in a ratio of two to one. In addition, the optimization study showed the essential role of a non-nucleophilic base as well as that of the potassium cation.
Table 1: Optimization of the reaction conditions.
| Entry | Conditionsa | Yield 3a (%) |
|---|---|---|
| 1 | 1a (1 equiv)/CH3CN/K2CO3/rt/24 h | 68 |
| 2 | 1a (2 equiv)/CH3CN/K2CO3/rt/24 h | 85 |
| 3 | 1a (2 equiv)/CH3CN/K2CO3/rt/12 h | 72 |
| 4 | 1a (2 equiv)/CH3CN/KHCO3/rt/4 h | 74 |
| 5 | 1a (2 equiv)/CH3CN/KF/rt/24 h | 0 |
| 6 | 1a (2 equiv)/CH3CN/K3PO4/rt/24 h | 0 |
| 7 | 1a (2 equiv)/AcOH/AcOK/rt/12 h | 0 |
| 8 | 1a (2 equiv)/AcOH/AcOK/100 °C/3 h | 0 |
| 9 | 1a (2 equiv)/CH3CN/NaHCO3/rt/4 d | 12 |
| 10 | 1a (2 equiv)/CH3CN/Na2CO3/rt/24 h | 17 |
| 11 | 1a (2 equiv)/CH3CN/Na2CO3/KI (0.5 equiv) | 32 |
| 12 | 1a (2 equiv)/CH3CN/Na2CO3/KI (1 equiv) | 42 |
| 13 | 1a (2 equiv)/CH3CN/Na2CO3/KI (2 equiv) | 68 |
| 14 | 1a (2 equiv)/CH3CN/Na2CO3/LiI (2 equiv) | 0 |
| 15 | 1a (2 equiv)/CH3CN/Cs2CO3/rt/12 h | 32 |
| 16 | 1a (2 equiv)/CH3CN/Cs2CO3/KI (0.5 equiv) | 34 |
| 17 | 1a (2 equiv)/CH3CN/Cs2CO3/KI (1 equiv) | 44 |
| 18 | 1a (2 equiv)/CH3CN/Cs2CO3/KI (2 equiv) | 68 |
| 19 | 1a (2 equiv)/CH3CN/Cs2CO3/NaI (0.5 equiv) | 8 |
| 20 | 1a (2 equiv)/CH3CN/Cs2CO3/NaI (1 equiv) | 10 |
| 21 | 1a (2 equiv)/CH3CN/Cs2CO3/NaI (2 equiv) | 12 |
| 22 | 1a (2 equiv)/CH3CN/Cs2CO3/LiI (2 equiv) | 0 |
| 23 | 1a (2 equiv)/CH3CN/Li2CO3/rt/24 h | 0 |
| 24 | 1a (2 equiv)/CH3CN/Li2CO3/KI (0.5 equiv) | 35 |
| 25 | 1a (2 equiv)/CH3CN/Li2CO3/KI (1 equiv) | 40 |
| 26 | 1a (2 equiv)/CH3CN/Li2CO3/KI (2 equiv) | 71 |
| 27 | 1a (2 equiv)/CH3CN/Li2CO3/NaI (0.5 equiv) | 7 |
| 28 | 1a (2 equiv)/CH3CN/Li2CO3/NaI (1 equiv) | 10 |
| 29 | 1a (2 equiv)/CH3CN/Li2CO3/NaI (2 equiv) | 10 |
| 30 | 1a (2 equiv)/CH3CN/organic baseb/rt/24 h | 0 |
| 31 | 1a (2 equiv)/CH3CN/organic baseb (except DIPEA)/KI, KBr, NaI or LiI/rt/24 h | 0 |
| 32 | 1a (2 equiv)/CH3CN/DIPEA/KI (0.5 equiv) | 45 |
| 33 | 1a (2 equiv)/CH3CN/DIPEA/KI (1 equiv) | 58 |
| 34 | 1a (2 equiv)/CH3CN/DIPEA/KI (2 equiv) | 75 |
| 35 | 1a (2 equiv)/CH3CN/DIPEA/KBr (2 equiv) | 68 |
| 36 | 1a (2 equiv)/CH3CN/DIPEA/NaI (0.5 equiv) | 22 |
| 37 | 1a (2 equiv)/CH3CN/DIPEA/NaI (1 equiv) | 28 |
| 38 | 1a (2 equiv)/CH3CN/DIPEA/NaI (2 equiv) | 34 |
| 39 | 1a (2 equiv)/CH3CN/DIPEA/LiI (0.5–2 equiv) | 0 |
aFor screening we used 2 equiv of base; bas organic bases we tested pyridine, 2,4,6-trimethylpyridine, NEt3, DABCO and DIPEA; in all cases 2 equiv of base were added.
This optimization study afforded a general procedure and optimal reaction conditions: 2.0 mmol of diketone, 1.0 mmol of the appropriate pyridinium salt and 1 mmol of K2CO3 in 7 mL of CH3CN are stirred at room temperature for 24 hours.
Reaction scope
Having found the optimal reaction conditions, we concentrated on studying the scope and limitations of the discovered cycloaddition. A variety of pyridinium salts with different substituents in positions 1, 2, 3 and 4 were prepared by alkylation of the parental commercially available pyridines (see Supporting Information File 1, Table S1). The results of this cyclization reaction (Scheme 2) are summarized in Figure 1. Generally the reaction was complete in 24 hours and afforded the desired products in good to excellent yields. It is worth mentioning that only cyclohexanediones were prone to reaction with the tested pyridinium salts with formation of the corresponding tetracyclic products 3, 4, 6 and 7. In the frame of the current project a variety of 1,3-diketones and CF3-containing 1,3-diketones were investigated, but these failed for all mentioned cases. For 3-cyanopyridinium salts a departure from the general trend was observed in terms of reaction conditions regarding the base used; here the highest efficiency was observed for NaHCO3, and the reaction took 4 days to reach completion. In general, the reaction proceeded with high regio- and diastereoselectivity.
![[1860-5397-9-124-i2]](/bjoc/content/inline/1860-5397-9-124-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of compounds 3–8. Reagents and conditions: (i): CH3CN, K2CO3, rt, 24 h. In the case of 3-cyanopyridinium salts, NaHCO3 (2 equiv) was used as a base and the reaction time was 4 days.
Scheme 2: Synthesis of compounds 3–8. Reagents and conditions: (i): CH3CN, K2CO3, rt, 24 h. In the case of 3-...
![[1860-5397-9-124-1]](/bjoc/content/figures/1860-5397-9-124-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthesized compounds 3, 4, 6, 7.
Figure 1: Synthesized compounds 3, 4, 6, 7.
Additionally, the reaction was monitored by HPLC, and for most pyridinium salts investigated no byproducts were detected. However, in the case of 3-cyanopyridinium salts the formation of additional products, namely the constitutive isomers of type 5 and 8, was observed. These compounds were isolated and identified by using standard 2D NMR methods and X-ray analysis [51].
At the same time, to our great disappointment 4-substituted pyridinium salts appeared to be unreactive, and testing different reaction conditions as well as bases with various combinations of time and temperature did not deliver any positive results.
During the course of this work we encountered the low stability of 8-oxa-10-aza-tricyclo[7.3.1.02,7]trideca-2(7),11-dienes; decomposition was observed in polar solvents, i.e., in DMSO. In some cases this had an influence on the quality of the 1H NMR spectra. Heating also led to decomposition, which was observed after 2–3 hours at 60–70 °C.
The constitution of products 3, 4, 6, 7 was proven by 1D and 2D NMR experiments; that of compounds 6d, 7c and 8 was unambiguously corroborated by X-ray [51] diffraction analysis (Supporting Information File 1, Figures S2, S3, S4)
Reaction mechanism
In order to shed light on the reaction mechanism we used DFT computations at the B3LYP/6-31G(d,p) [52-54] level, as implemented in the Gaussian 09 program [55], and acetonitrile was included implicitly within the IEFPCM method (more details on inclusion of the solvent can be found in Supporting Information File 2). For simplicity 1,3-cyclohexanedione rather than dimedone was included in the computations.
Our ability to calculate the mechanism as well as to make any further predictions about reaction products is limited by the obscure structure of the real reaction product. In fact the reaction of pyridinium salt 2a with cyclic 1,3-diketones in the presence of either K2CO3 or DIPEA/KI yields an orange precipitate in acetonitrile. Redissolved in DMSO-d6, this precipitate gave rise to broad peaks in the 1H NMR spectra; thus its structure was not defined and is supposed to be of polymeric nature. Potassium with a counter-ion (HCO3− or I−) is also included in the precipitate since that cannot be burned completely and yields an unburned material. On the other hand the orange precipitate is quickly decomposed by silica gel liberating the desired product 3a or 6a, which was finally eluted from silica. The formation of a precipitate must be a driving force of the reaction promoted by DIPEA/KI (see Supporting Information File 2, thermodynamic analysis).
The reaction starts with 1,3-diketone deprotonation and nucleophilic attack of potassium 3-oxocyclohex-1-enolate on pyridinium cation, which yields an intermediate with a new C–C bond, and this is a key stage in the reaction. The C–O bond formation can be excluded in the initial stage (see Supporting Information File 2 for the explanation). Bearing this in mind, we computed the local softness of 3-acetyl-1-methylpyridinium (2a+), 2-cyano-1-methylpyridinium (2b+) and 3-cyano-1-methylpyridinium (2e+) cations towards nucleophilic attack. According to the local softness indexes (see Supporting Information File 2) with 2b+ cation we should observe a regioselective formation of product 4a or 7a, while for cations 2a+ and 2e+ a regioisomeric mixture should be expected (but is really only observed for 2e+). In reality, regioisomeric mixtures of compounds 3/5 or 6/8 are produced by cation 2e+, while for cations 2a+ and 2b+ regioselective reaction is inherent. Intrigued by this discrepancy, we computed the full reaction mechanism for cation 2a+ with potassium 3-oxocyclohex-1-enolate in the presence of potassium cation and hydrogencarbonate anion as base. We included HCO3− in our computation, both because it has only five atoms and because this base is presumed to operate; it is formed from neutral carbonate when it deprotonates the cyclic diketone. However, the mechanism involving any other base should not differ significantly.
A plausible mechanism is presented in Figure 2. After initial deprotonation of the diketone and formation of the hydrogencarbonate anion, the latter is exchanged with iodine in the salt of 2a, and an initial complex consisting of 2a+HCO3− with potassium 3-oxocyclohex-1-enolate is assembled (intermediate 9). Nucleophilic attack yields intermediate 11 through TS 10. The latter TS was first localized without HCO3− (TS K-S1a-1, see Supporting Information File 2); however, an inclusion of HCO3− resulted in a slight energy drop. Cyclohexanedione ring inversion (intermediates 13, 15 and TSs 12, 14) and HCO3− semi-decoordination (TS 16) lead to intermediate 17, which undergoes proton abstraction under action of hydrogencarbonate (TS 18 and intermediate 19). Structural rearrangement of 19 through TS 20 shows an intermediate 21. Finally 21 is protonated by coordinated H2CO3 (highest rate-determining TS 22a) to yield the final product 23 with a great release of free energy. It is worth mentioning that the last reaction is a protonation of enamine in essence; the imaginary frequency inherent for TS 22a shows only proton displacement, without formation of a C–O bond. The latter was formed during IRC computation, starting from TS 22a. Previously, Hui Li and Chao-Guo Yan [50] proposed the nucleophilic attack of oxygen onto the carbon atom with subsequent protonation. We have explored this process by decreasing the C–O distance in relaxed scans: the scan from intermediate 19 showed only a constant rise of energy, while the scan from intermediate 21 showed a maximum, which was not optimized to any TS. Hence, the attack by oxygen on the last stage must be excluded. Within the scope of this mechanism the activation energy is 21.3 kcal/mol (energy gap between 22a and 19).
![[1860-5397-9-124-2]](/bjoc/content/figures/1860-5397-9-124-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Other TSs (K-S1a-2, K-S1a-3, see Supporting Information File 2), inherent for the initial stage of new C–C bond formation turned out to be unimportant, because in both cases reactions run again through TS 22a (rate limiting), as was shown by additional computations. On the other hand, we have computed four conformers of TS 22: the conformation of the cyclohexane ring and that of the acetyl group were changed. While the cyclohexane-ring conformation has only a negligible effect on the TS energy (TS 22b, see Supporting Information File 2), the change of the acetyl group conformation prevents potassium from coordinating to it and makes the TS energy somewhat higher (TSs 22c and 22d from Supporting Information File 2).
In the proposed mechanism the regioselective formation of product 6a is not explained (see Supporting Information File 2 for discussion). We attribute this to a higher thermodynamic stabilization of compound 6a by a potassium cation (Scheme 3), as compared with that of regioisomeric 6aa (its formation was not observed); the energetic distance between 6a-K+ and 6aa-K+ is 2.5 kcal/mol at the B3LYP/6-31G(d,p) level and 2.0 kcal/mol at the B3LYP/6-311++G(d,p)//B3LYP/6-31G(d,p) level. As mentioned before, the assumption is hard to prove due to lack of information about the real product composition.
![[1860-5397-9-124-i3]](/bjoc/content/inline/1860-5397-9-124-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The influence of K+ on the product free energy.
Scheme 3: The influence of K+ on the product free energy.
Conclusion
In summary, we have studied the dicomponent binucleophilic cycloaddition of 1,3-diketones with diverse substituted pyridinium salts. The reaction took place under mild conditions and provided an efficient route to 8-oxa-10-aza-tricyclo[7.3.1.02,7]trideca-2(7),11-dienes with a wide range of substituents. Further studies to extend the scope of the synthetic utility of these cyclizations are in progress in both of our laboratories.
Experimental
General Information
Solvents were purchased from ACROS and directly used without further purification. Analytical thin-layer chromatography was performed on 0.20 mm 60 Å silica gel plates. Column chromatography was performed by using 60 Å silica gel (60–200 mesh). Pyridinium salts were synthesized according to the published procedure [56].
The density functional B3LYP [52-54] with 6-31G(d,p) basis set for all atoms except iodine and solvation by acetonitrile within the IEFPCM method as implemented in the Gaussian 09 package [55], was employed for the calculations. For cases when iodine was included, the LANL2DZ basis set and core potential were assigned. All compounds were computed in the closed-shell singlet state (confirmed by “stable” calculation). Frequency calculations were performed on all optimized geometries to ensure only positive eigenvalues for minima and one imaginary frequency for transition states. Free energies at 298.15 K and 1 bar pressure were calculated according to the recommendation of Gaussian, Inc. [57,58], and the frequency scaling factor (0.9608) was applied. For every located transition state an IRC calculation at the same level of theory confirmed that this TS connects the corresponding educts/intermediates/products. The methodology of local softness computation [59,60] uses Fukui functions; for their approximation we used Mulliken, ESP, NPA and Hirshfeld charges. However, only the Fukui functions approximated by NPA and Hirshfeld charges were unambiguous and taken into consideration.
General procedures for the synthesis of compounds 3–8
Procedure (A)
In a 25 mL Schlenk flask, under argon flow, 2.0 mmol of diketone, 1.0 mmol of the appropriate pyridinium salt, and 1.0 mmol (138 mg) of K2CO3 were loaded. The flask was covered with a septum stopper and 7 mL of absolute CH3CN was added by syringe. The reaction mixture was left under intensive stirring at room temperature for 24 hours. Then the solvent was removed under reduced pressure and the crude material was subjected to column chromatography.
Procedure (B)
In the case of the 3-cyanopyridinium salt, NaHCO3 (2.0 mmol, 168 mg) was used as a base, and the reaction mixture was left over 4 days.
Procedure (C)
In the case of 2-cyanopyridinium salts the reaction was completed within 1 hour.
Supporting Information
| Supporting Information File 1: Details on synthetic procedures, list of pyridinium salts, characterization of new compounds, copies of NMR spectra, X-ray structures of compounds 6d, 7c and 8. | ||
| Format: PDF | Size: 10.0 MB | Download |
| Supporting Information File 2: Computational results, optimized structures (atomic coordinates as reported by Gaussian 09), charges and local softness indexes (nucleophilic attack) for cations 2a+, 2b+ and 2e+, additional discussions related to computations. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Bennasar, M.-L.; Jiménez, J.-M.; Sufi, B. A.; Bosch, J. Tetrahedron Lett. 1996, 37, 7653–7656. doi:10.1016/0040-4039(96)01703-0
Return to citation in text: [1] -
Bennasar, M.-L.; Lavilla, R.; Alvarez, M.; Bosch, J. Heterocycles 1988, 27, 789–824. doi:10.3987/REV-87-374
Return to citation in text: [1] -
Spitzner, D.; Zaubitzer, T.; Shi, Y. J.; Wenkert, E. J. Org. Chem. 1988, 53, 2274–2278. doi:10.1021/jo00245a027
Return to citation in text: [1] -
Bieräugel, H.; Brands, K. M. J.; Pandit, U. K. Heterocycles 1988, 27, 1589–1593. doi:10.3987/COM-88-4564
Return to citation in text: [1] -
Comins, D. L.; Joseph, S. P. Alkaloid synthesis using 1-acylpyridinium salts as intermediates. In Advances in Nitrogen Heterocycles; Moody, C. J., Ed.; JAI Press: London, UK, 1996; Vol. 2, pp 251–294.
Return to citation in text: [1] -
Comins, D. L.; LaMunyon, D. H.; Chen, X. J. Org. Chem. 1997, 62, 8182–8187. doi:10.1021/jo971448u
Return to citation in text: [1] -
Comins, D. L.; Libby, A. H.; Al-awar, R. S.; Foti, C. J. J. Org. Chem. 1999, 64, 2184–2185. doi:10.1021/jo990192k
Return to citation in text: [1] -
Kuethe, J. T.; Comins, D. L. Org. Lett. 2000, 2, 855–857. doi:10.1021/ol0056271
Return to citation in text: [1] -
Pays, C.; Mangeney, P. Tetrahedron Lett. 2001, 42, 589–592. doi:10.1016/S0040-4039(00)02010-4
Return to citation in text: [1] -
Bennasar, M. L.; Zulaica, E.; Ramírez, A.; Bosch, J. J. Org. Chem. 1996, 61, 1239–1251. doi:10.1021/jo951456f
Return to citation in text: [1] [2] -
Bennasar, M. L.; Vidal, B.; Bosch, J. J. Org. Chem. 1996, 61, 1916–1917. doi:10.1021/jo9600058
Return to citation in text: [1] -
Amann, R.; Spitzner, D. Angew. Chem., Int. Ed. Engl. 1991, 30, 1320–1321. doi:10.1002/anie.199113201
Return to citation in text: [1] -
Lyle, R. E. Part B. Partially Reduced Pyridines. In Chemistry of Heterocyclic Compounds: Pyridine and its Derivatives; Abramovitch, R. A., Ed.; Supplement, Part One, Vol. 14; John Wiley & Sons, Inc.: Hoboken, New Jersey, 1974; pp 137–182.
Return to citation in text: [1] -
Goldman, S.; Stoltefuss, J. Angew. Chem., Int. Ed. Engl. 1991, 30, 1559–1578. doi:10.1002/anie.199115591
Return to citation in text: [1] -
Sunkel, C. E.; Fau de Casa-Juana, M.; Santos, L.; Mar Gomes, M.; Villarroya, M.; Gonzalez-Morales, M. A.; Priego, J. G.; Ortega, M. P. J. Med. Chem. 1990, 33, 3205–3210. doi:10.1021/jm00174a017
Return to citation in text: [1] -
Jaing, J.-L.; Li, A.-H.; Jang, S.-Y.; Chang, L.; Melman, N.; Moro, S.; Ji, X.-d.; Lobkovsky, E.; Clardy, J.; Jacobson, K. A. J. Med. Chem. 1999, 42, 3055–3060. doi:10.1021/jm980688e
Return to citation in text: [1] -
Mateeva, N. N.; Wineld, L. L.; Redda, K. K. Curr. Med. Chem. 2005, 12, 551–571. doi:10.2174/0929867053362776
Return to citation in text: [1] -
Ito, S.; Satoh, A.; Nagatomi, Y.; Hirata, Y.; Suzuki, G.; Kimura, T.; Satow, A.; Maehara, S.; Hikichi, H.; Hata, M.; Kawamoto, H.; Ohta, H. Bioorg. Med. Chem. 2008, 16, 9817–9829. doi:10.1016/j.bmc.2008.09.060
Return to citation in text: [1] -
Jones, P.; Griffin, A. M.; Gawell, L.; Lavoie, R.; Delorme, D.; Roberts, E.; Brown, W.; Walpole, C.; Xiao, W.; Boulet, J.; Labarre, M.; Coupal, M.; Butterworth, J.; St-Onge, S.; Hodzic, L.; Salois, D. Bioorg. Med. Chem. Lett. 2009, 19, 5994–5998. doi:10.1016/j.bmcl.2009.09.072
Return to citation in text: [1] -
Liras, S.; McHardy, S. F.; Allen, M. P.; Segelstein, B. E.; Heck, S. D.; Bryce, D. K.; Schmidt, A. W.; Vanase-Frawley, M.; Callegari, E.; McLean, S. Bioorg. Med. Chem. Lett. 2010, 20, 503–507. doi:10.1016/j.bmcl.2009.11.113
Return to citation in text: [1] -
Chowdhury, M. A.; Abdellatif, K. R. A.; Dong, Y.; Knaus, E. E. Bioorg. Med. Chem. 2008, 16, 8882–8888. doi:10.1016/j.bmc.2008.08.059
Return to citation in text: [1] -
Misra, M.; Pandey, S. K.; Pandey, V. P.; Pandey, J.; Tripathi, R.; Tripathi, R. P. Bioorg. Med. Chem. 2009, 17, 625–633. doi:10.1016/j.bmc.2008.11.062
Return to citation in text: [1] -
Krishna, P. R.; Reddy, P. S. J. Comb. Chem. 2008, 10, 426–435. doi:10.1021/cc700171p
Return to citation in text: [1] -
Scott, J. D.; Li, S. W.; Wang, H. W.; Xia, Y.; Jayne, C. L.; Miller, M. W.; Duffy, R. A.; Boykow, G. C.; Kowalski, T. J.; Spar, B. D.; Stamford, A. W.; Chackalamannil, S.; Lachowicz, J. E.; Greenlee, W. J. Bioorg. Med. Chem. Lett. 2010, 20, 1278–1283. doi:10.1016/j.bmcl.2009.11.075
Return to citation in text: [1] -
Bosch, J.; Bennasar, M. L. Synlett 1995, 587–596. doi:10.1055/s-1995-5007
Return to citation in text: [1] -
Bennasar, M.-L.; Alvarez, M.; Lavilla, R.; Zulaica, E.; Bosch, J. J. Org. Chem. 1990, 55, 1156–1168. doi:10.1021/jo00291a013
Return to citation in text: [1] -
Bennasar, M.-L.; Zulaica, E.; Jiménez, J. M.; Bosch, J. J. Org. Chem. 1993, 58, 7756–7767. doi:10.1021/jo00079a021
Return to citation in text: [1] -
Alvarez, M.; Salas, M.; de Veciana, A.; Lavilla, R.; Bosch, J. Tetrahedron Lett. 1990, 31, 5089–5092. doi:10.1016/S0040-4039(00)97813-4
Return to citation in text: [1] -
Amat, M.; Linares, A.; Bosch, J. J. Org. Chem. 1990, 55, 6299–6312. doi:10.1021/jo00313a017
Return to citation in text: [1] -
Bennasar, M.-L.; Zulaica, E.; Ramírez, A.; Bosch, J. Tetrahedron 1999, 55, 3117–3128. doi:10.1016/S0040-4020(99)00070-8
Return to citation in text: [1] -
Bennasar, M.-L.; Vidal, B.; Bosch, J. Chem. Commun. 1996, 2755–2756. doi:10.1039/cc9960002755
Return to citation in text: [1] -
Bennasar, M.-L.; Vidal, B.; Bosch, J. J. Org. Chem. 1997, 62, 3597–3609. doi:10.1021/jo9623301
Return to citation in text: [1] -
Le Men, J.; Taylor, W. I. Experientia 1965, 21, 508–510. doi:10.1007/BF02138961
Return to citation in text: [1] -
Bennasar, M.-L.; Jiménez, J.-M.; Sufi, B. A.; Bosch, J. Tetrahedron Lett. 1996, 37, 9105–9109. doi:10.1016/S0040-4039(96)02097-7
Return to citation in text: [1] -
Bennasar, M.-L.; Vidal, B.; Sufi, B. A.; Bosch, J. Chem. Commun. 1998, 2639–2640. doi:10.1039/a807868h
Return to citation in text: [1] -
Albrecht, U.; Preuss, A.; Schmidt, A.; Fischer, C.; Langer, P. Heterocycles 2010, 80, 289–302. doi:10.3987/COM-09-S(S)12
Return to citation in text: [1] [2] -
Schmidt, A.; Michalik, D.; Rotzoll, S.; Ullah, E.; Fischer, C.; Reinke, H.; Görls, H.; Langer, P. Org. Biomol. Chem. 2008, 6, 2804–2814. doi:10.1039/b804139c
Return to citation in text: [1] -
Schmidt, A.; Gütlein, J.-P.; Preuss, A.; Albrecht, U.; Reinke, H.; Langer, P. Synlett 2005, 2489–2491. doi:10.1055/s-2005-872675
Return to citation in text: [1] -
Ullah, E.; Rotzoll, S.; Schmidt, A.; Michalik, D.; Langer, P. Tetrahedron Lett. 2005, 46, 8997–8999. doi:10.1016/j.tetlet.2005.10.108
Return to citation in text: [1] -
Yawer, M. A.; Hussain, I.; Gütlein, J.-P.; Schmidt, A.; Jiao, H.; Reinke, H.; Spannenberg, A.; Fischer, C.; Langer, P. Eur. J. Org. Chem. 2008, 4193–4199. doi:10.1002/ejoc.200800478
Return to citation in text: [1] -
Karapetyan, V.; Mkrtchyan, S.; Schmidt, A.; Gütlein, J. P.; Villinger, A.; Reinke, H.; Jiao, H.; Fischer, C.; Langer, P. Org. Biomol. Chem. 2008, 6, 2961–2968. doi:10.1039/b803141j
Return to citation in text: [1] -
Schmidt, A.; Gütlein, J.-P.; Mkrtchyan, S.; Görls, H.; Langer, P. Synlett 2007, 1305–1307. doi:10.1055/s-2007-977443
Return to citation in text: [1] -
Schmidt, A.; Gütlein, J.-P.; Langer, P. Tetrahedron Lett. 2007, 48, 2067–2069. doi:10.1016/j.tetlet.2007.01.147
Return to citation in text: [1] -
Moghaddam, F. M.; Mirjafary, Z.; Saeidian, H.; Taheri, S.; Khodabakhshi, M. R. Tetrahedron Lett. 2010, 51, 2704–2707. doi:10.1016/j.tetlet.2010.03.038
Return to citation in text: [1] -
Moghaddam, F. M.; Mirjafary, Z.; Saeidian, H.; Taheri, S.; Doulabi, M.; Kiamehr, M. Tetrahedron 2010, 66, 134–138. doi:10.1016/j.tet.2009.11.032
Return to citation in text: [1] [2] -
Moghaddam, F. M.; Mirjafary, Z.; Saeidian, H.; Taheri, S.; Soltanzadeh, B. Tetrahedron 2010, 66, 3678–3681. doi:10.1016/j.tet.2010.03.078
Return to citation in text: [1] -
Moghaddam, F. M.; Taheri, S.; Mirjafary, Z.; Saeidian, H.; Kiamehr, M.; Tafazzoli, M. Helv. Chim. Acta 2011, 94, 142–147. doi:10.1002/hlca.201000144
Return to citation in text: [1] -
Moghaddam, F. M.; Saeidian, H.; Kiamehr, M.; Mirjafary, Z.; Taheri, S. ARKIVOC 2010, xi, 91–100. doi:10.3998/ark.5550190.0011.b09
Return to citation in text: [1] -
Moghaddam, F. M.; Taheri, S.; Mirjafary, Z.; Saeidian, H. Synlett 2010, 123–127. doi:10.1055/s-0029-1218534
Return to citation in text: [1] -
Li, H.; Yan, C.-G. Tetrahedron 2011, 67, 2863–2869. doi:10.1016/j.tet.2011.02.063
Return to citation in text: [1] [2] -
X-ray crystallographic data (excluding structure factors) for the structures 6d, 7c, 8, reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. 844028-844030 and can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44(1223)336033; E-mail: deposit@ccdc.cam.ac.uk or via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] [2] -
Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304
Return to citation in text: [1] [2] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] [2] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text: [1] [2] -
Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] [2] -
Kiamehr, M.; Moghaddam, F. M.; Gormay, P. V.; Semeniuchenko, V.; Villinger, A.; Langer, P.; Iaroshenko, V. O. Tetrahedron 2012, 68, 9685–9693. doi:10.1016/j.tet.2012.09.059
Return to citation in text: [1] -
Foresman, J. B.; Frish, A. Exploring chemistry with electronic structure methods, 2nd ed.; Gaussian, Inc.: Pittsburgh, 1993; pp 1995–1996.
Return to citation in text: [1] -
Ochterski, J. W. Thermochemistry in Gaussian. http://www.gaussian.com/g_whitepap/thermo.htm (accessed May 7, 2013).
Return to citation in text: [1] -
Semeniuchenko, V.; Exner, T. E.; Khilya, V.; Groth, U. Appl. Organomet. Chem. 2011, 25, 804–809. doi:10.1002/aoc.1841
Return to citation in text: [1] -
Meza, R.; Gordillo, B.; Galván, M. Int. J. Quantum Chem. 2005, 104, 29–37. doi:10.1002/qua.20577
Return to citation in text: [1]
| 1. | Bennasar, M.-L.; Jiménez, J.-M.; Sufi, B. A.; Bosch, J. Tetrahedron Lett. 1996, 37, 7653–7656. doi:10.1016/0040-4039(96)01703-0 |
| 2. | Bennasar, M.-L.; Lavilla, R.; Alvarez, M.; Bosch, J. Heterocycles 1988, 27, 789–824. doi:10.3987/REV-87-374 |
| 3. | Spitzner, D.; Zaubitzer, T.; Shi, Y. J.; Wenkert, E. J. Org. Chem. 1988, 53, 2274–2278. doi:10.1021/jo00245a027 |
| 4. | Bieräugel, H.; Brands, K. M. J.; Pandit, U. K. Heterocycles 1988, 27, 1589–1593. doi:10.3987/COM-88-4564 |
| 17. | Mateeva, N. N.; Wineld, L. L.; Redda, K. K. Curr. Med. Chem. 2005, 12, 551–571. doi:10.2174/0929867053362776 |
| 45. | Moghaddam, F. M.; Mirjafary, Z.; Saeidian, H.; Taheri, S.; Doulabi, M.; Kiamehr, M. Tetrahedron 2010, 66, 134–138. doi:10.1016/j.tet.2009.11.032 |
| 14. | Goldman, S.; Stoltefuss, J. Angew. Chem., Int. Ed. Engl. 1991, 30, 1559–1578. doi:10.1002/anie.199115591 |
| 15. | Sunkel, C. E.; Fau de Casa-Juana, M.; Santos, L.; Mar Gomes, M.; Villarroya, M.; Gonzalez-Morales, M. A.; Priego, J. G.; Ortega, M. P. J. Med. Chem. 1990, 33, 3205–3210. doi:10.1021/jm00174a017 |
| 16. | Jaing, J.-L.; Li, A.-H.; Jang, S.-Y.; Chang, L.; Melman, N.; Moro, S.; Ji, X.-d.; Lobkovsky, E.; Clardy, J.; Jacobson, K. A. J. Med. Chem. 1999, 42, 3055–3060. doi:10.1021/jm980688e |
| 50. | Li, H.; Yan, C.-G. Tetrahedron 2011, 67, 2863–2869. doi:10.1016/j.tet.2011.02.063 |
| 13. | Lyle, R. E. Part B. Partially Reduced Pyridines. In Chemistry of Heterocyclic Compounds: Pyridine and its Derivatives; Abramovitch, R. A., Ed.; Supplement, Part One, Vol. 14; John Wiley & Sons, Inc.: Hoboken, New Jersey, 1974; pp 137–182. |
| 44. | Moghaddam, F. M.; Mirjafary, Z.; Saeidian, H.; Taheri, S.; Khodabakhshi, M. R. Tetrahedron Lett. 2010, 51, 2704–2707. doi:10.1016/j.tetlet.2010.03.038 |
| 45. | Moghaddam, F. M.; Mirjafary, Z.; Saeidian, H.; Taheri, S.; Doulabi, M.; Kiamehr, M. Tetrahedron 2010, 66, 134–138. doi:10.1016/j.tet.2009.11.032 |
| 46. | Moghaddam, F. M.; Mirjafary, Z.; Saeidian, H.; Taheri, S.; Soltanzadeh, B. Tetrahedron 2010, 66, 3678–3681. doi:10.1016/j.tet.2010.03.078 |
| 47. | Moghaddam, F. M.; Taheri, S.; Mirjafary, Z.; Saeidian, H.; Kiamehr, M.; Tafazzoli, M. Helv. Chim. Acta 2011, 94, 142–147. doi:10.1002/hlca.201000144 |
| 48. | Moghaddam, F. M.; Saeidian, H.; Kiamehr, M.; Mirjafary, Z.; Taheri, S. ARKIVOC 2010, xi, 91–100. doi:10.3998/ark.5550190.0011.b09 |
| 5. | Comins, D. L.; Joseph, S. P. Alkaloid synthesis using 1-acylpyridinium salts as intermediates. In Advances in Nitrogen Heterocycles; Moody, C. J., Ed.; JAI Press: London, UK, 1996; Vol. 2, pp 251–294. |
| 6. | Comins, D. L.; LaMunyon, D. H.; Chen, X. J. Org. Chem. 1997, 62, 8182–8187. doi:10.1021/jo971448u |
| 7. | Comins, D. L.; Libby, A. H.; Al-awar, R. S.; Foti, C. J. J. Org. Chem. 1999, 64, 2184–2185. doi:10.1021/jo990192k |
| 8. | Kuethe, J. T.; Comins, D. L. Org. Lett. 2000, 2, 855–857. doi:10.1021/ol0056271 |
| 9. | Pays, C.; Mangeney, P. Tetrahedron Lett. 2001, 42, 589–592. doi:10.1016/S0040-4039(00)02010-4 |
| 10. | Bennasar, M. L.; Zulaica, E.; Ramírez, A.; Bosch, J. J. Org. Chem. 1996, 61, 1239–1251. doi:10.1021/jo951456f |
| 11. | Bennasar, M. L.; Vidal, B.; Bosch, J. J. Org. Chem. 1996, 61, 1916–1917. doi:10.1021/jo9600058 |
| 12. | Amann, R.; Spitzner, D. Angew. Chem., Int. Ed. Engl. 1991, 30, 1320–1321. doi:10.1002/anie.199113201 |
| 49. | Moghaddam, F. M.; Taheri, S.; Mirjafary, Z.; Saeidian, H. Synlett 2010, 123–127. doi:10.1055/s-0029-1218534 |
| 38. | Schmidt, A.; Gütlein, J.-P.; Preuss, A.; Albrecht, U.; Reinke, H.; Langer, P. Synlett 2005, 2489–2491. doi:10.1055/s-2005-872675 |
| 39. | Ullah, E.; Rotzoll, S.; Schmidt, A.; Michalik, D.; Langer, P. Tetrahedron Lett. 2005, 46, 8997–8999. doi:10.1016/j.tetlet.2005.10.108 |
| 40. | Yawer, M. A.; Hussain, I.; Gütlein, J.-P.; Schmidt, A.; Jiao, H.; Reinke, H.; Spannenberg, A.; Fischer, C.; Langer, P. Eur. J. Org. Chem. 2008, 4193–4199. doi:10.1002/ejoc.200800478 |
| 43. | Schmidt, A.; Gütlein, J.-P.; Langer, P. Tetrahedron Lett. 2007, 48, 2067–2069. doi:10.1016/j.tetlet.2007.01.147 |
| 36. | Albrecht, U.; Preuss, A.; Schmidt, A.; Fischer, C.; Langer, P. Heterocycles 2010, 80, 289–302. doi:10.3987/COM-09-S(S)12 |
| 37. | Schmidt, A.; Michalik, D.; Rotzoll, S.; Ullah, E.; Fischer, C.; Reinke, H.; Görls, H.; Langer, P. Org. Biomol. Chem. 2008, 6, 2804–2814. doi:10.1039/b804139c |
| 36. | Albrecht, U.; Preuss, A.; Schmidt, A.; Fischer, C.; Langer, P. Heterocycles 2010, 80, 289–302. doi:10.3987/COM-09-S(S)12 |
| 10. | Bennasar, M. L.; Zulaica, E.; Ramírez, A.; Bosch, J. J. Org. Chem. 1996, 61, 1239–1251. doi:10.1021/jo951456f |
| 25. | Bosch, J.; Bennasar, M. L. Synlett 1995, 587–596. doi:10.1055/s-1995-5007 |
| 26. | Bennasar, M.-L.; Alvarez, M.; Lavilla, R.; Zulaica, E.; Bosch, J. J. Org. Chem. 1990, 55, 1156–1168. doi:10.1021/jo00291a013 |
| 27. | Bennasar, M.-L.; Zulaica, E.; Jiménez, J. M.; Bosch, J. J. Org. Chem. 1993, 58, 7756–7767. doi:10.1021/jo00079a021 |
| 28. | Alvarez, M.; Salas, M.; de Veciana, A.; Lavilla, R.; Bosch, J. Tetrahedron Lett. 1990, 31, 5089–5092. doi:10.1016/S0040-4039(00)97813-4 |
| 29. | Amat, M.; Linares, A.; Bosch, J. J. Org. Chem. 1990, 55, 6299–6312. doi:10.1021/jo00313a017 |
| 30. | Bennasar, M.-L.; Zulaica, E.; Ramírez, A.; Bosch, J. Tetrahedron 1999, 55, 3117–3128. doi:10.1016/S0040-4020(99)00070-8 |
| 31. | Bennasar, M.-L.; Vidal, B.; Bosch, J. Chem. Commun. 1996, 2755–2756. doi:10.1039/cc9960002755 |
| 32. | Bennasar, M.-L.; Vidal, B.; Bosch, J. J. Org. Chem. 1997, 62, 3597–3609. doi:10.1021/jo9623301 |
| 33. | Le Men, J.; Taylor, W. I. Experientia 1965, 21, 508–510. doi:10.1007/BF02138961 |
| 34. | Bennasar, M.-L.; Jiménez, J.-M.; Sufi, B. A.; Bosch, J. Tetrahedron Lett. 1996, 37, 9105–9109. doi:10.1016/S0040-4039(96)02097-7 |
| 35. | Bennasar, M.-L.; Vidal, B.; Sufi, B. A.; Bosch, J. Chem. Commun. 1998, 2639–2640. doi:10.1039/a807868h |
| 18. | Ito, S.; Satoh, A.; Nagatomi, Y.; Hirata, Y.; Suzuki, G.; Kimura, T.; Satow, A.; Maehara, S.; Hikichi, H.; Hata, M.; Kawamoto, H.; Ohta, H. Bioorg. Med. Chem. 2008, 16, 9817–9829. doi:10.1016/j.bmc.2008.09.060 |
| 19. | Jones, P.; Griffin, A. M.; Gawell, L.; Lavoie, R.; Delorme, D.; Roberts, E.; Brown, W.; Walpole, C.; Xiao, W.; Boulet, J.; Labarre, M.; Coupal, M.; Butterworth, J.; St-Onge, S.; Hodzic, L.; Salois, D. Bioorg. Med. Chem. Lett. 2009, 19, 5994–5998. doi:10.1016/j.bmcl.2009.09.072 |
| 20. | Liras, S.; McHardy, S. F.; Allen, M. P.; Segelstein, B. E.; Heck, S. D.; Bryce, D. K.; Schmidt, A. W.; Vanase-Frawley, M.; Callegari, E.; McLean, S. Bioorg. Med. Chem. Lett. 2010, 20, 503–507. doi:10.1016/j.bmcl.2009.11.113 |
| 21. | Chowdhury, M. A.; Abdellatif, K. R. A.; Dong, Y.; Knaus, E. E. Bioorg. Med. Chem. 2008, 16, 8882–8888. doi:10.1016/j.bmc.2008.08.059 |
| 22. | Misra, M.; Pandey, S. K.; Pandey, V. P.; Pandey, J.; Tripathi, R.; Tripathi, R. P. Bioorg. Med. Chem. 2009, 17, 625–633. doi:10.1016/j.bmc.2008.11.062 |
| 23. | Krishna, P. R.; Reddy, P. S. J. Comb. Chem. 2008, 10, 426–435. doi:10.1021/cc700171p |
| 24. | Scott, J. D.; Li, S. W.; Wang, H. W.; Xia, Y.; Jayne, C. L.; Miller, M. W.; Duffy, R. A.; Boykow, G. C.; Kowalski, T. J.; Spar, B. D.; Stamford, A. W.; Chackalamannil, S.; Lachowicz, J. E.; Greenlee, W. J. Bioorg. Med. Chem. Lett. 2010, 20, 1278–1283. doi:10.1016/j.bmcl.2009.11.075 |
| 41. | Karapetyan, V.; Mkrtchyan, S.; Schmidt, A.; Gütlein, J. P.; Villinger, A.; Reinke, H.; Jiao, H.; Fischer, C.; Langer, P. Org. Biomol. Chem. 2008, 6, 2961–2968. doi:10.1039/b803141j |
| 42. | Schmidt, A.; Gütlein, J.-P.; Mkrtchyan, S.; Görls, H.; Langer, P. Synlett 2007, 1305–1307. doi:10.1055/s-2007-977443 |
| 52. | Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304 |
| 53. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 54. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 51. | X-ray crystallographic data (excluding structure factors) for the structures 6d, 7c, 8, reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. 844028-844030 and can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44(1223)336033; E-mail: deposit@ccdc.cam.ac.uk or via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 51. | X-ray crystallographic data (excluding structure factors) for the structures 6d, 7c, 8, reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. 844028-844030 and can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44(1223)336033; E-mail: deposit@ccdc.cam.ac.uk or via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 59. | Semeniuchenko, V.; Exner, T. E.; Khilya, V.; Groth, U. Appl. Organomet. Chem. 2011, 25, 804–809. doi:10.1002/aoc.1841 |
| 60. | Meza, R.; Gordillo, B.; Galván, M. Int. J. Quantum Chem. 2005, 104, 29–37. doi:10.1002/qua.20577 |
| 57. | Foresman, J. B.; Frish, A. Exploring chemistry with electronic structure methods, 2nd ed.; Gaussian, Inc.: Pittsburgh, 1993; pp 1995–1996. |
| 58. | Ochterski, J. W. Thermochemistry in Gaussian. http://www.gaussian.com/g_whitepap/thermo.htm (accessed May 7, 2013). |
| 56. | Kiamehr, M.; Moghaddam, F. M.; Gormay, P. V.; Semeniuchenko, V.; Villinger, A.; Langer, P.; Iaroshenko, V. O. Tetrahedron 2012, 68, 9685–9693. doi:10.1016/j.tet.2012.09.059 |
| 52. | Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304 |
| 53. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 54. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 50. | Li, H.; Yan, C.-G. Tetrahedron 2011, 67, 2863–2869. doi:10.1016/j.tet.2011.02.063 |
© 2013 Kiamehr et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)