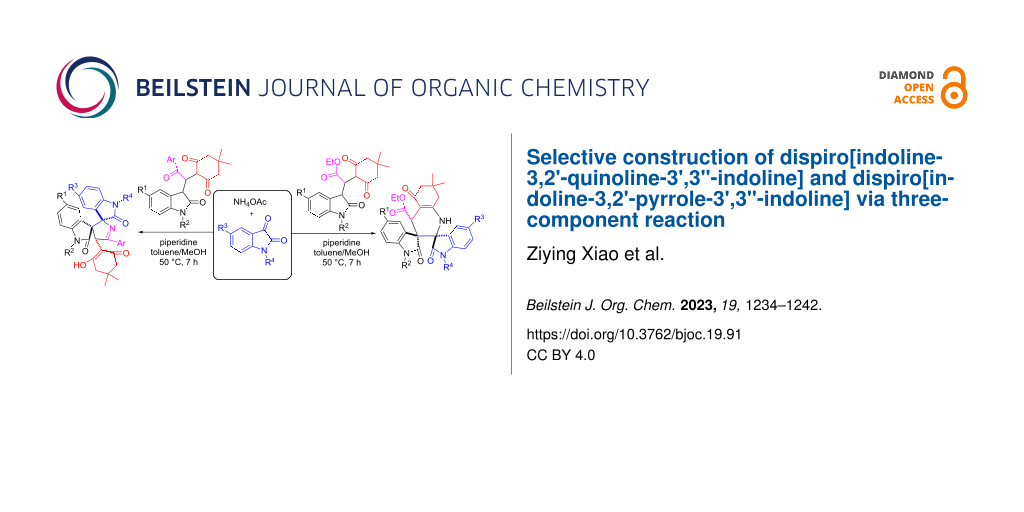
Abstract
A convenient synthetic procedure for the construction of novel dispirooxindole motifs was successfully developed by base-promoted three-component reaction of ammonium acetate, isatins and in situ-generated 3-isatyl-1,4-dicarbonyl compounds. The piperidine-promoted three-component reaction of ammonium acetate, isatins and the in situ-generated dimedone adducts of 3-ethoxycarbonylmethyleneoxindoles afforded mutlifunctionalized dispiro[indoline-3,2'-quinoline-3',3''-indoline] derivatives in good yields and with high diastereoselectivity. On the other hand, a similar reaction of the dimedone adducts of 3-phenacylideneoxindoles afforded unique dispiro[indoline-3,2'-pyrrole-3',3''-indoline] derivatives with a cyclohexanedione substituent. A plausible reaction mechanism is proposed to explain the formation of the different spirooxindoles.

Graphical Abstract
Introduction
Spirooxindole is one important privileged structural skeleton and is found in many bioactive natural and synthetic compounds [1-3]. It is known that many spirooxindole derivatives show important biological activities [4-6]. On the other hand, a wide range of differently substituted spirooxindoles exist [7-9]. Therefore, the development of efficient synthetic methodologies for diverse spirooxindoles have become one of the hottest research fields in organic and pharmaceutical chemistry [10,11]. In order to synthesize diverse spirooxindole derivatives, commercially available isatins and easily obtainable 3-methyleneoxindolines were the most employed building blocks [12-15]. On the other hand, Morita–Baylis–Hillman (MBH) carbonates of isatins, which could be easily prepared by the MBH reactions of isatins with acrylonitrile or alkyl acrylates also became valuable synthons [16-25]. The active 3-methyleneoxindoles could act as Michael acceptors, 1,3-dipolarophiles, 1,4-dienophiles and other multiple substrates. They have been widely employed as key component to finish many multicomponent and domino reactions [26-35]. Recently, the Michael adduct of 3-methyleneoxindoles with various nucleophiles, especially active methylene compounds, have attracted much attentions [36-41]. This kind of Michael adduct has more than one reactive site and could proceed domino reactions with various electrophiles [42-49]. In this respect, several elegant domino or multicomponent reactions have been successfully developed to construct multifunctionalized or polycyclic spirooxindoles. For example, Zhang successfully developed a recyclable bifunctional cinchona/thiourea-catalyzed four-component Michael/Mannich cyclization sequence for the asymmetric synthesis of spirooxindoles, in which the in situ-generated Michael adduct of 3-ethoxycarbonylmethyleneoxindole underwent a Mannich reaction and annulation reaction with in situ-generated aldimines (reaction 1 in Scheme 1) [50,51]. Tanaka reported chiral quinidine derivative-catalyzed Michael–Henry cascade reactions of nitrostyrenes with previously prepared oxindole-functionalized dihydrobenzofuranones (reaction 2 in Scheme 1) [52]. We reported a piperidine-promoted domino reaction of thiophenols and two molecules of 3-phenacylideneoxindoles to give multifunctionalized dispirocyclopentanebisoxindoles (reaction 3 in Scheme 1), in which the in situ-generated adduct of thiophenol and 3-phenacylideneoxindole was believed to be the key intermediate [53-55]. Inspired by these elegant synthetic protocols and in continuation of our aim to develop convenient reactions for the synthesis of diverse spiro compounds [56-62], we investigated the base-promoted annulation reaction of dimedone adducts of 3-methyleneoxindoles, with isatin and ammonium acetate. It was unexpectedly found that novel dispiro[indoline-3,2'-quinoline-3',3''-indoline] and dispiro[indoline-3,2'-pyrrole-3',3''-indoline] were selectively produced by using differently substituted 3-methyleneoxindoles (reaction 4 in Scheme 1). Herein, we wish to report these interesting results.
![[1860-5397-19-91-i1]](/bjoc/content/inline/1860-5397-19-91-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative cascade reactions of Michael adducts of 3-methyleneoxindoles.
Scheme 1: Representative cascade reactions of Michael adducts of 3-methyleneoxindoles.
Results and Discussion
At first, 3-isatyl-1,4-dicarbonyl compound 1 was prepared by DBU-catalyzed Michael addition reaction of dimedone and ethyl 2-(2-oxoindolin-3-ylidene)acetate in toluene according to the published method [52]. Then, the reaction conditions of the three-component reaction of isatyl adduct 1a (0.20 mmol), isatin 2a (0.20 mmol) and ammonium acetate (0.5 mmol) were examined according to Zhang and co-workers reported reaction (reaction 1 in Scheme 1) [12]. In the presence of piperidine, the reaction in methanol at room temperature did not yield the product (Table 1, entry 1). However, the reaction in methanol at elevated temperature gave the expected spiro compound 3a in 51% and 48% yields, respectively (Table 1, entries 2 and 3). The reaction in other solvents such as acetonitrile, toluene and ethyl acetate at 50 °C gave the desired product 3a in very low yields (Table 1, entries 4–6). When the reaction was carried out in a mixture of toluene and methanol (v/v = 2:1) in the presence of piperidine, the yield of 3a increased to 60% (Table 1, entry 7). When DABCO or DBU was employed as base, the yield of 3a decreased to 36% and 29% yield, respectively (Table 1, entries 8 and 9). When the loading of ammonium acetate was increased to 0.8 mmol and 1.0 mmol, the yield of 3a increased to 85% and 82% (Table 1, entries 10 and 11). Prolonging the reaction time did not increase the yield of product 3a (Table 1, entry 12). Therefore, the optimized reaction conditions found for this three-component reaction are the use of a mixture of methanol and toluene at 50 °C for seven hours in the presence of piperidine.
Table 1: Optimizing reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-91-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base | Solvent | T (°C) | Time (h) | Yield (%)b |
| 1 | piperidine | MeOH | 25 | 7 | nr |
| 2 | piperidine | MeOH | 50 | 7 | 51 |
| 3 | piperidine | MeOH | 60 | 7 | 48 |
| 4 | piperidine | CH3CN | 50 | 7 | 20 |
| 5 | piperidine | toluene | 50 | 7 | 32 |
| 6 | piperidine | EtOAc | 50 | 7 | 26 |
| 7 | piperidine | toluene/MeOH | 50 | 7 | 60 |
| 8 | DABCO | toluene/MeOH | 50 | 7 | 36 |
| 9 | DBU | toluene/MeOH | 50 | 7 | 29 |
| 10 | piperidinec | toluene/MeOH | 50 | 7 | 85 |
| 11 | piperidined | toluene/MeOH | 50 | 7 | 82 |
| 12 | piperidinec | toluene/MeOH | 50 | 12 | 84 |
aReaction conditions: 3-isatyl-1,4-dicarbonyl compound 1 (0.20 mmol), isatin 2 (0.20 mmol), NH4OAc (0.50 mmol), base (0.30 mmol), solvent (4.0 mL). bIsolated yields. cNH4OAc (0.8 mmol) was used; dNH4OAc (1.0 mmol) was used.
With the optimized reaction conditions in hand, the scope of the reaction was investigated by using various substrates. The results are summarized in Table 2. All reactions proceeded smoothly to give the desired dispiro compounds 3a–m in satisfactory yields. Various isatins with different substituents can be successfully used in the reaction. The substituents showed marginal effects on the yields. On the other hand, the dimedone adducts of alkyl 2-(2-oxoindolin-3-ylidene)acetate with various substituents were also successfully employed in the reaction to give the desired products. It can be seen that the dimedone adducts of alkyl 2-(2-oxoindolin-3-ylidene)acetate with 5-chloro and 5-fluoro substituent gave the spiro compounds 3a–k in satisfactory yields. However, the dimedone adducts of ethyl 2-(2-oxoindolin-3-ylidene)acetate itself and its derivatives with 5-methyl group gave the products 3l and 3m in moderate yields. The chemical structures of the obtained dispiro compounds 3a–m were fully characterized by IR, HRMS, 1H and 13C NMR spectroscopy. Because of the three chiral carbon atoms in the product, several diastereomers might be formed in the reaction. However, TLC monitoring and 1H NMR spectra of the crude products clearly indicated that only one diastereoisomer was predominately produced in the reaction, while the other possible diastereomers were not detected. This result shows that this reaction has a high diastereoselectivity due to the large steric effect of two oxindole scaffolds and the thermodynamically controlling effect. The single crystal structure of compound 3a was determined by X-ray crystallographic diffraction (Figure 1). From Figure 1 it can be seen that the two scaffolds of oxindole at neighboring positions are in trans-configuration. The ethoxycarbonyl group is also in trans-position to the carbonyl group in the neighboring oxindole scaffold. Therefore, it can be concluded that the obtained dispiro compounds 3a–m have this kind of relative configuration on the basis of 1H NMR spectra and crystal structure determination. It should be pointed out that ammonium acetate was employed as nitrogen source in this three-component reaction. For investigating the scope of this reaction, aniline was also used in the reaction, but in this case no expected dispirooxindoles could be obtained.
Table 2: Synthesis of the dispirooxindoles 3a–m.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-91-i4.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Compd | R1 | R3 | R2 | R4 | R5 | Yield (%)b |
| 1 | 3a | Cl | C2H5 | Bn | CH3 | Bn | 85 |
| 2 | 3b | Cl | C2H5 | Bn | Cl | Bn | 78 |
| 3 | 3c | Cl | C2H5 | Bn | Cl | n-Bu | 82 |
| 4 | 3d | Cl | C2H5 | Bn | CH3 | n-Bu | 81 |
| 5 | 3e | Cl | C2H5 | Bn | H | H | 69 |
| 6 | 3f | Cl | C2H5 | Bn | CH3 | H | 70 |
| 7 | 3g | Cl | C2H5 | Bn | Cl | H | 72 |
| 8 | 3h | Cl | C2H5 | Bn | F | H | 70 |
| 9 | 3i | Cl | C2H5 | H | CH3 | Bn | 68 |
| 10 | 3j | Cl | CH3 | Bn | CH3 | Bn | 72 |
| 11 | 3k | F | CH3 | Bn | CH3 | Bn | 69 |
| 12 | 3l | H | C2H5 | Bn | Cl | Bn | 42 |
| 13 | 3m | CH3 | C2H5 | Bn | Cl | Bn | 48 |
aReaction conditions: 3-isatyl-1,4-dicarbonyl compound 1 (0.20 mmol), isatin 2 (0.20 mmol), NH4OAc (0.80 mmol), piperidine (0.30 mmol), toluene (2.0 mL), MeOH (2.0 mL), 50 °C, 7 h. bIsolated yields.
![[1860-5397-19-91-1]](/bjoc/content/figures/1860-5397-19-91-1.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 1: Crystal structure of dispiro compound 3a.
Figure 1: Crystal structure of dispiro compound 3a.
In order to investigate the scope of this reaction, similar dimedone adducts of 3-phenacylideneoxindoles were used in the three-component reaction. To our surprise, instead of the above mentioned dispiro[indoline-3,2'-quinoline-3',3''-indolines] 3a–m, the novel dispiro[indoline-3,2'-pyrrole-3',3''-indoline] derivatives 4a–i were obtained in high yields. The results are summarized in Table 3. The structural analysis showed that the carbonyl group of the dimedone does not take part in the further cyclization reaction, while the carbonyl group of the benzoyl group participated in the annulation reaction to give the pyrrolidyl ring. This result clearly indicated that the adducts of 3-phenacylideneoxindoles showed different reactivity to that of the adducts of 3-ethoxycarbonylmethyleneoxindoles. For confirming the chemical structures of dispirooxindoles 4a–i, the single crystal structure of compound 4a was determined by X-ray diffraction (Figure 2). In Figure 2, the two oxindole scaffolds are in trans-position. The dimedone moiety is also in trans-position to the carbonyl group in the neighboring oxindole scaffold. To demonstrate the synthetic value of this three-component reaction, 3-phenacylideneoxindole adducts of 1,3-cyclohexanedione were also employed in the reaction. In the presence of piperidine, the three-component reaction proceeded smoothly at room temperature in twelve hours to give the expected dispiro[indoline-3,2'-pyrrole-3',3''-indoline] derivatives 4j–p in satisfactory yields. The 1,3-cyclohexanedione moiety does not take part in the further cyclization process. These results show that this reaction is largely general. The 1H NMR spectra of the obtained compounds 4j–p clearly show similar chemical shifts of the characteristic groups as the spiro compounds 4a–i. Therefore, it can be concluded that the spiro compounds 4j–p have the same relative configuration as the spiro compounds 4a–i.
Table 3: Synthesis of dispirooxindoles 4a–p.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-91-i5.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Compd | R1 | R2 | Ar | R3 | R4 | R5 | Yield (%)b |
| 1 | 4a | Cl | Bn | p-CH3C6H4 | CH3 | CH3 | Bn | 85 |
| 2 | 4b | Cl | Bn | p-CH3C6H4 | CH3 | Cl | Bn | 68 |
| 3 | 4c | Cl | Bn | p-CH3C6H4 | CH3 | H | Bn | 75 |
| 4 | 4d | Cl | Bn | p-CH3C6H4 | CH3 | CH3 | n-Bu | 72 |
| 5 | 4e | Cl | Bn | p-ClC6H4 | CH3 | CH3 | Bn | 63 |
| 6 | 4f | CH3 | Bn | p-ClC6H4 | CH3 | CH3 | Bn | 51 |
| 7 | 4g | Cl | Bn | p-CH3C6H4 | CH3 | H | H | 34 |
| 8 | 4h | Cl | n-Bu | p-CH3OC6H4 | CH3 | CH3 | Bn | 65 |
| 9 | 4i | Cl | H | p-CH3OC6H4 | CH3 | CH3 | Bn | 62 |
| 10 | 4j | Cl | Bn | p-CH3C6H4 | H | CH3 | Bn | 71 |
| 11 | 4k | Cl | Bn | p-CH3C6H4 | H | Cl | Bn | 68 |
| 12 | 4l | Cl | Bn | p-CH3C6H4 | H | H | Bn | 64 |
| 13 | 4m | Cl | Bn | p-CH3C6H4 | H | Cl | n-Bu | 65 |
| 14 | 4n | Cl | Bn | p-CH3C6H4 | H | CH3 | H | 52 |
| 15 | 4o | Cl | Bn | p-CH3C6H4 | H | CH3 | n-Bu | 63 |
| 16 | 4p | Cl | Bn | p-ClC6H4 | H | CH3 | Bn | 73 |
aReaction conditions: 3-isatyl-1,4-dicarbonyl compound 1 (0.20 mmol), isatin 2 (0.20 mmol), NH4OAc (0.80 mmol), piperidine (0.30 mmol), toluene (2.0 mL), MeOH (2.0 mL), 50 °C, 7 h. bIsolated yields.
![[1860-5397-19-91-2]](/bjoc/content/figures/1860-5397-19-91-2.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of compound 4a.
Figure 2: Crystal structure of compound 4a.
In order to explain the formation of different cyclic compounds, a plausible reaction mechanism was proposed in Scheme 2 on the basis of the present experiments and the previous works [51-53]. Firstly, 3-isatyl-1,4-dicarbonyl compound 1 was converted to a carbanion in the presence of base. In the meantime, the condensation of isatin 2 with ammonium acetate gave the 3-iminoisatin intermediate A. Secondly, Michael addition of the in situ-generated carbanion of the 3-isatyl-1,4-dicarbonyl compound 1 to 3- iminoisatin A gave intermediate B. In the case of intermediate B1 with an ethoxycarbonyl group, the nucleophilic addition of the amino anion to the carbonyl group in the of 1,3-cyclohexanedione scaffold resulted in cyclic intermediate C. Thirdly, the elimination of water from intermediate C gave the isolated product 3. In the case of the intermediate B2 with a benzoyl group, there are two kinds of reactive carbonyl groups in intermediate B2, one carbonyl group is in the benzoyl moiety and another carbonyl group is in the 1,3-cyclohexanedione part. In this case the amino group selectively attacked the benzoyl group to give cyclic intermediate D, while the two carbonyl groups in the 1,3-cyclohexanedione moiety remained unreacted. Then, the final product 4 was formed by elimination of water. Thus, the spiro compounds 3 and 4 were selectively produced due to the different reaction process. Thus, the 3-isatyl-1,4-dicarbonyl compounds derived from 3-ethoxycarbonylmethyloxindoles and 3-phenacylideneoxindoles resulted in the two different novel dispirooxindole skeletons.
![[1860-5397-19-91-i2]](/bjoc/content/inline/1860-5397-19-91-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have investigated the base-promoted multicomponent reaction of 3-methyleneoxindoles, dimedone, isatins and ammonium acetate. The reaction showed very interesting molecular diversity and diastereoselectivity. This reaction provided efficient synthetic protocols for the synthesis of dispiro[indoline-3,2'-quinoline-3',3''-indoline] and dispiro[indoline-3,2'-pyrrole-3',3''-indoline] derivatives. A plausible reaction mechanism was proposed to explain the selective formation of the different polycyclic compounds. This reaction has the advantages of using readily available materials, simple reaction conditions, satisfactory yields, high diastereoselectivity and atomic economy, which enable this reaction potential synthetic applications in heterocyclic chemistry and medicinal chemistry.
Experimental
General procedure for the preparation of compounds 3a–m
To a round flask was added 3-isatyl-1,4-dicarbonyl compound 1 (0.20 mmol), isatin 2 (0.20 mmol), ammonium acetate (0.80 mmol), piperidine (0.30 mmol), toluene (2.0 mL) and methanol (2.0 mL). The mixture was heated at 50 °C for seven hours. After removing the solvent by rotatory evaporation at reduced pressure, the residue was subjected to column chromatography (silicon gel, 300–400 mesh) with petroleum ether and ethyl acetate (v/v = 1:1) to give the pure product for analysis.
Ethyl rel-(3R,3'S,4'R)-1,1''-dibenzyl-5,5''-dichloro-7',7'-dimethyl-2,2'',5'-trioxo-1',4',5',6',7',8'-hexahydrodispiro[indoline-3,2'-quinoline-3',3''-indoline]-4'-carboxylate (3a): White solid, 78%, mp 280–282 °C; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 2.0 Hz, 1H, ArH), 7.34–7.28 (m, 6H, ArH), 7.51 (s, 1H, ArH), 7.24 (s, 2H, ArH), 7.23 (s, 1H, ArH), 7.20–7.17 (m, 3H, ArH), 7.11–7.09 (m, 1H, ArH), 7.0–6.98 (m, 1H, ArH), 6.40 (d, J = 8.4 Hz, 1H, ArH), 6.26 (d, J = 8.4 Hz, 1H, ArH), 5.01 (d, J = 16.4, 1H, CH2), 4.89 (d, J = 15.6 Hz, 1H, CH2), 4.78 (d, J = 15.6 Hz, 1H, CH2), 4.76 (s, 1H, NH), 4.74 (s, 1H, CH), 4.66 (d, J = 16.4 Hz, 1H, CH2), 3.94–3.87 (m, 1H, CH2), 3.84–3.77 (m, 1H, CH2), 2.46 (d, J = 16.0 Hz, 1H, CH2), 2.39 (d, J = 16.0 Hz, 1H, CH2), 2.37–2.34 (m, 2H, CH2), 1.30 (s, 3H, CH3), 1.15 (s, 3H, CH3), 0.76 (t, J = 7.2 Hz, 3H, CH3) ppm; 13C NMR (101 MHz, CDCl3) δ 193.0, 173.8, 171.9, 171.2, 155.6, 142.0, 141.8, 134.8, 134.1, 131.0, 129.3, 129.1, 128.8, 128.7, 128.6, 127.9, 127.5, 127.5, 127.1, 127.1, 127.0, 126.2, 125.9, 125.6, 124.9, 110.9, 110.3, 102.3, 62.3, 60.3, 50.0, 49.4, 44.4, 44.2, 42.5, 42.4, 32.9, 29.0, 27.6, 13.5 ppm; IR (KBr) ν: 3504, 3024, 3010, 2995, 2985, 1847, 1711, 1603, 1517, 1400, 1299, 1250, 1053, 953, 841 cm−1; HRMS (ESI-TOF): [M + Na]+ calcd. for C42H37Cl2N3O5, 756.2002; found, 756.1989.
General procedure for the preparation of compounds 4a–p
To a round flask was added 3-isatyl-1,4-dicarbonyl compound 1 (0.20 mmol), isatin 2 (0.20 mmol), ammonium acetate (0.80 mmol), piperidine (0.30 mmol), toluene (2.0 mL) and methanol (2.0 mL). The mixture was heated at 50 °C for seven hours. After removing the solvent by rotatory evaporation at reduced pressure, the residue was subjected to column chromatography (silicon gel, 300–400 mesh) with petroleum ether and ethyl acetate (v/v = 4:1) to give the pure product for analysis.
rel-(3R,3'S,4'R)-1,1''-Dibenzyl-5''-chloro-4'-(2-hydroxy-4,4-dimethyl-6-oxocyclohex-1-en-1-yl)-5-methyl-5'-(p-tolyl)-4'H-dispiro[indoline-3,2'-pyrrole-3',3''-indoline]-2,2''-dione (4a): White solid, 60%, mp 250–251 °C; 1H NMR (400 MHz, CDCl3) δ 10.86 (s, 1H, OH), 7.80 (d, J = 8.0 Hz, 2H, ArH), 7.51 (s, 1H, ArH), 7.21 (d, J = 8.4 Hz, 2H, ArH), 7.18–7.11 (m, 4H, ArH), 7.10–7.05 (m, 3H, ArH), 7.00–6.97 (m, 2H, ArH), 6.76 (d, J = 7.2 Hz, 2H, ArH), 6.65 (d, J = 7.6 Hz, 2H, ArH), 6.46 (d, J = 8.0 Hz, 1H, ArH), 6.28 (d, J = 8.4 Hz, 1H, ArH), 5.65 (s, 1H, CH), 5.18 (d, J = 16.4 Hz, 1H, CH2), 5.09 (d, J = 16.0, 1H, CH2), 4.47 (d, J = 4.8 Hz, 1H, CH2), 4.43 (d, J = 5.2 Hz, 1H, CH2), 2.41 (d, J = 26.8 Hz, 1H, CH2), 2.40 (s, 3H, CH3), 2.21 (d, J = 18.4 Hz, 1H, CH2), 2.10 (s, 1H, CH3), 2.05 (s, 1H, CH2), 1.87 (d, J = 16.0 Hz, 1H, CH2), 1.00 (s, 3H, CH3), 0.99 (s, 3H, CH3) ppm; 13C NMR (101 MHz, CDCl3) δ 197.5, 181.6, 177.4, 177.2, 173.0, 142.8, 142.2, 140.6, 134.7, 134.3, 133.9, 130.2, 130.0, 129.1, 128.8, 128.7, 128.5, 128.4, 127.5, 127.3, 127.1, 127.0, 126.4, 126.2, 126.1, 126.0, 112.1, 109.8, 109.7, 87.2, 61.8, 53.7, 50.0, 44.5, 44.4, 43.6, 30.7, 29.9, 26.3, 21.6, 20.9 ppm; IR (KBr) ν: 3756, 3056, 3023, 2984, 2988, 1832, 1792, 1526, 1545, 1368, 1285, 1145, 1025, 956, 882 cm−1; HRMS (ESI-TOF): [M + H]+ calcd. for C48H44ClN3O5, 760.2937; found, 760.2921.
Supporting Information
The crystallographic data of compounds 3a (CCDC 2223538) and 4a (CCDC 2223539) have been deposited at the Cambridge Crystallographic Database Center (http://www.ccdc.cam.ac.uk).
| Supporting Information File 1: Characterization data, 1H NMR, 13C NMR, and HRMS spectra of the compounds. | ||
| Format: PDF | Size: 7.4 MB | Download |
References
-
Yang, Y.-T.; Zhu, J.-F.; Liao, G.; Xu, H.-J.; Yu, B. Curr. Med. Chem. 2018, 25, 2233–2244. doi:10.2174/0929867325666171129131311
Return to citation in text: [1] -
Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041
Return to citation in text: [1] -
Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056
Return to citation in text: [1] -
Arun, Y.; Saranraj, K.; Balachandran, C.; Perumal, P. T. Eur. J. Med. Chem. 2014, 74, 50–64. doi:10.1016/j.ejmech.2013.12.027
Return to citation in text: [1] -
Saranya, P. V.; Neetha, M.; Aneeja, T.; Anilkumar, G. RSC Adv. 2021, 11, 7146–7179. doi:10.1039/d1ra00139f
Return to citation in text: [1] -
Ma, Z.; Zhou, A.; Xia, C. Nat. Prod. Rep. 2022, 39, 1015–1044. doi:10.1039/d1np00060h
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808
Return to citation in text: [1] -
Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180
Return to citation in text: [1] -
Liu, Z.-M.; Li, N.-K.; Huang, X.-F.; Wu, B.; Li, N.; Kwok, C.-Y.; Wang, Y.; Wang, X.-W. Tetrahedron 2014, 70, 2406–2415. doi:10.1016/j.tet.2014.02.023
Return to citation in text: [1] -
Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r
Return to citation in text: [1] -
Wang, Y.; Cobo, A. A.; Franz, A. K. Org. Chem. Front. 2021, 8, 4315–4348. doi:10.1039/d1qo00220a
Return to citation in text: [1] [2] -
Santos, M. M. M. Tetrahedron 2014, 70, 9735–9757. doi:10.1016/j.tet.2014.08.005
Return to citation in text: [1] -
Saeed, R.; Sakla, A. P.; Shankaraiah, N. Org. Biomol. Chem. 2021, 19, 7768–7791. doi:10.1039/d1ob01176f
Return to citation in text: [1] -
Ganesh, M.; Suraj, S. Org. Biomol. Chem. 2022, 20, 5651–5693. doi:10.1039/d2ob00767c
Return to citation in text: [1] -
Chen, Z.-C.; Chen, Z.; Du, W.; Chen, Y.-C. Chem. Rec. 2020, 20, 541–555. doi:10.1002/tcr.201900058
Return to citation in text: [1] -
Yan, R.-J.; Liu, B.-X.; Xiao, B.-X.; Du, W.; Chen, Y.-C. Org. Lett. 2020, 22, 4240–4244. doi:10.1021/acs.orglett.0c01283
Return to citation in text: [1] -
Chen, Z.-C.; Chen, Z.; Yang, Z.-H.; Guo, L.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 15021–15025. doi:10.1002/anie.201907797
Return to citation in text: [1] -
Chen, P.; Chen, Z.-C.; Li, Y.; Ouyang, Q.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 4036–4040. doi:10.1002/anie.201814403
Return to citation in text: [1] -
Wang, Z.-H.; Lei, C.-W.; Zhang, X.-Y.; You, Y.; Zhao, J.-Q.; Yuan, W.-C. Org. Chem. Front. 2019, 6, 3342–3347. doi:10.1039/c9qo00890j
Return to citation in text: [1] -
Chen, Y.; Cui, B.-D.; Bai, M.; Han, W.-Y.; Wan, N.-W.; Chen, Y.-Z. Tetrahedron 2019, 75, 2971–2979. doi:10.1016/j.tet.2019.04.040
Return to citation in text: [1] -
Gu, J.; Xiao, B.-X.; Ouyang, Q.; Du, W.; Chen, Y.-C. Chin. J. Chem. 2019, 37, 155–160. doi:10.1002/cjoc.201800466
Return to citation in text: [1] -
Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h
Return to citation in text: [1] -
Zhou, T.; Xia, T.; Liu, Z.; Liu, L.; Zhang, J. Adv. Synth. Catal. 2018, 360, 4475–4479. doi:10.1002/adsc.201801152
Return to citation in text: [1] -
He, Z.-L.; Chen, P.; Chen, Z.-C.; Du, W.; Chen, Y.-C. Org. Lett. 2022, 24, 100–104. doi:10.1021/acs.orglett.1c03689
Return to citation in text: [1] -
Ramu, G.; Tangella, Y.; Ambala, S.; Nagendra Babu, B. J. Org. Chem. 2020, 85, 5370–5378. doi:10.1021/acs.joc.0c00078
Return to citation in text: [1] -
Dong, J.; Zhang, D.; Men, Y.; Zhang, X.; Hu, Z.; Xu, X. Org. Lett. 2019, 21, 166–169. doi:10.1021/acs.orglett.8b03646
Return to citation in text: [1] -
Chen, K.; Xu, T.; Liang, J.; Zhou, M.; Zhang, J.; Hao, W.-J.; Wang, J.; Tu, S.-J.; Jiang, B. Chem. Commun. 2019, 55, 14757–14760. doi:10.1039/c9cc08762a
Return to citation in text: [1] -
Pramanik, S.; Saha, P.; Ghosh, P.; Mukhopadhyay, C. J. Org. Chem. 2023, 88, 3386–3402. doi:10.1021/acs.joc.2c01523
Return to citation in text: [1] -
Deng, Y.; Li, Y.; Wang, Y.; Sun, S.; Ma, S.; Jia, P.; Li, W.; Wang, K.; Yan, W. Org. Chem. Front. 2022, 9, 210–215. doi:10.1039/d1qo01392k
Return to citation in text: [1] -
Wang, B.; Yan, X.; Zhong, H.; Ouyang, Q.; Tian, X. Org. Chem. Front. 2022, 9, 1621–1627. doi:10.1039/d1qo01708j
Return to citation in text: [1] -
Cruché, C.; Neiderer, W.; Collins, S. K. ACS Catal. 2021, 11, 8829–8836. doi:10.1021/acscatal.1c01983
Return to citation in text: [1] -
Wang, J.; Zheng, X.-Z.; Xiao, J.-A.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2021, 23, 963–968. doi:10.1021/acs.orglett.0c04164
Return to citation in text: [1] -
Yuan, W.-C.; Zuo, J.; Yuan, S.-P.; Zhao, J.-Q.; Wang, Z.-H.; You, Y. Org. Chem. Front. 2021, 8, 784–791. doi:10.1039/d0qo01335h
Return to citation in text: [1] -
Ren, J.-W.; Xie, Z.-Z.; Zheng, L.; Ye, Z.-P.; Deng, Z.-X.; Zhao, Q.-L.; Xiao, J.-A.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Chem. Front. 2021, 8, 778–783. doi:10.1039/d0qo01364a
Return to citation in text: [1] -
Shanthi, G.; Perumal, P. T. Tetrahedron Lett. 2008, 49, 7139–7142. doi:10.1016/j.tetlet.2008.09.152
Return to citation in text: [1] -
Tan, B.; Candeias, N. R.; Barbas, C. F., III. Nat. Chem. 2011, 3, 473–477. doi:10.1038/nchem.1039
Return to citation in text: [1] -
Tan, F.; Cheng, H.-G.; Feng, B.; Zou, Y.-Q.; Duan, S.-W.; Chen, J.-R.; Xiao, W.-J. Eur. J. Org. Chem. 2013, 2071–2075. doi:10.1002/ejoc.201300081
Return to citation in text: [1] -
Sun, W.; Hong, L.; Zhu, G.; Wang, Z.; Wei, X.; Ni, J.; Wang, R. Org. Lett. 2014, 16, 544–547. doi:10.1021/ol4034226
Return to citation in text: [1] -
Suman, K.; Thennarasu, S. RSC Adv. 2015, 5, 23291–23302. doi:10.1039/c5ra00283d
Return to citation in text: [1] -
You, Z.-H.; Zou, S.; Song, Y.-L.; Song, X.-Q. Org. Lett. 2022, 24, 7183–7187. doi:10.1021/acs.orglett.2c02920
Return to citation in text: [1] -
Sun, J.; Sun, Y.; Gong, H.; Xie, Y.-J.; Yan, C.-G. Org. Lett. 2012, 14, 5172–5175. doi:10.1021/ol302530m
Return to citation in text: [1] -
Han, Y.; Sheng, Y.-J.; Yan, C.-G. Org. Lett. 2014, 16, 2654–2657. doi:10.1021/ol5008394
Return to citation in text: [1] -
Lu, L.-J.; Fu, Q.; Sun, J.; Yan, C.-G. Tetrahedron 2014, 70, 2537–2545. doi:10.1016/j.tet.2014.02.050
Return to citation in text: [1] -
Shen, G.-L.; Sun, J.; Yan, C.-G. RSC Adv. 2015, 5, 4475–4483. doi:10.1039/c4ra13760d
Return to citation in text: [1] -
Huang, Y.; Fang, H.-L.; Huang, Y.-X.; Sun, J.; Yan, C.-G. J. Org. Chem. 2019, 84, 12437–12451. doi:10.1021/acs.joc.9b01920
Return to citation in text: [1] -
Zhan, S.-C.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Biomol. Chem. 2020, 18, 163–168. doi:10.1039/c9ob02013f
Return to citation in text: [1] -
Yan, C.; Sun, J.; Yan, C.-G. Chin. Chem. Lett. 2021, 32, 1253–1256. doi:10.1016/j.cclet.2020.08.052
Return to citation in text: [1] -
Wang, D.; Sun, J.; Liu, R.-Z.; Wang, Y.; Yan, C.-G. J. Org. Chem. 2021, 86, 5616–5629. doi:10.1021/acs.joc.1c00103
Return to citation in text: [1] -
Huang, X.; Pham, K.; Yi, W.; Zhang, X.; Clamens, C.; Hyatt, J. H.; Jasinsk, J. P.; Tayvah, U.; Zhang, W. Adv. Synth. Catal. 2015, 357, 3820–3824. doi:10.1002/adsc.201500748
Return to citation in text: [1] -
Huang, X.; Liu, M.; Pham, K.; Zhang, X.; Yi, W.-B.; Jasinski, J. P.; Zhang, W. J. Org. Chem. 2016, 81, 5362–5369. doi:10.1021/acs.joc.6b00653
Return to citation in text: [1] [2] -
Sohail, M.; Tanaka, F. Angew. Chem., Int. Ed. 2021, 60, 21256–21260. doi:10.1002/anie.202108734
Return to citation in text: [1] [2] [3] -
Sun, J.; Xie, Y.-J.; Yan, C.-G. J. Org. Chem. 2013, 78, 8354–8365. doi:10.1021/jo4010603
Return to citation in text: [1] [2] -
Shen, G.-L.; Sun, J.; Xie, Y.-J.; Yan, C.-G. ChemistrySelect 2016, 1, 1447–1451. doi:10.1002/slct.201600209
Return to citation in text: [1] -
Yang, R.-Y.; Sun, J.; Jin, G.; Yan, C.-G. Mol. Diversity 2018, 22, 21–36. doi:10.1007/s11030-017-9786-z
Return to citation in text: [1] -
Liu, D.; Cao, J.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Lett. 2020, 22, 8931–8936. doi:10.1021/acs.orglett.0c03331
Return to citation in text: [1] -
Liu, D.; Liu, X.; Sun, J.; Yan, C.-G. J. Org. Chem. 2021, 86, 14705–14719. doi:10.1021/acs.joc.1c01513
Return to citation in text: [1] -
Zheng, H.; Han, Y.; Sun, J.; Yan, C.-G. Beilstein J. Org. Chem. 2022, 18, 669–679. doi:10.3762/bjoc.18.68
Return to citation in text: [1] -
Pan, L.-N.; Sun, J.; Liu, X.-Y.; Yan, C.-G. Org. Biomol. Chem. 2022, 20, 7099–7104. doi:10.1039/d2ob01257j
Return to citation in text: [1] -
Wang, D.; Sun, J.; Han, Y.; Sun, Q.; Yan, C.-G. Org. Lett. 2022, 24, 7790–7795. doi:10.1021/acs.orglett.2c03123
Return to citation in text: [1] -
Wang, D.; Sun, J.; Yan, C.-G. Green Synth. Catal. 2022, 3, 53–58. doi:10.1016/j.gresc.2021.10.004
Return to citation in text: [1] -
Liu, D.; Sun, J.; Sun, Q.; Yan, C.-G. Org. Chem. Front. 2023, 10, 540–547. doi:10.1039/d2qo01771g
Return to citation in text: [1]
| 1. | Yang, Y.-T.; Zhu, J.-F.; Liao, G.; Xu, H.-J.; Yu, B. Curr. Med. Chem. 2018, 25, 2233–2244. doi:10.2174/0929867325666171129131311 |
| 2. | Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041 |
| 3. | Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056 |
| 12. | Wang, Y.; Cobo, A. A.; Franz, A. K. Org. Chem. Front. 2021, 8, 4315–4348. doi:10.1039/d1qo00220a |
| 13. | Santos, M. M. M. Tetrahedron 2014, 70, 9735–9757. doi:10.1016/j.tet.2014.08.005 |
| 14. | Saeed, R.; Sakla, A. P.; Shankaraiah, N. Org. Biomol. Chem. 2021, 19, 7768–7791. doi:10.1039/d1ob01176f |
| 15. | Ganesh, M.; Suraj, S. Org. Biomol. Chem. 2022, 20, 5651–5693. doi:10.1039/d2ob00767c |
| 12. | Wang, Y.; Cobo, A. A.; Franz, A. K. Org. Chem. Front. 2021, 8, 4315–4348. doi:10.1039/d1qo00220a |
| 10. | Liu, Z.-M.; Li, N.-K.; Huang, X.-F.; Wu, B.; Li, N.; Kwok, C.-Y.; Wang, Y.; Wang, X.-W. Tetrahedron 2014, 70, 2406–2415. doi:10.1016/j.tet.2014.02.023 |
| 11. | Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r |
| 51. | Huang, X.; Liu, M.; Pham, K.; Zhang, X.; Yi, W.-B.; Jasinski, J. P.; Zhang, W. J. Org. Chem. 2016, 81, 5362–5369. doi:10.1021/acs.joc.6b00653 |
| 52. | Sohail, M.; Tanaka, F. Angew. Chem., Int. Ed. 2021, 60, 21256–21260. doi:10.1002/anie.202108734 |
| 53. | Sun, J.; Xie, Y.-J.; Yan, C.-G. J. Org. Chem. 2013, 78, 8354–8365. doi:10.1021/jo4010603 |
| 7. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 8. | Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808 |
| 9. | Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180 |
| 56. | Liu, D.; Cao, J.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Lett. 2020, 22, 8931–8936. doi:10.1021/acs.orglett.0c03331 |
| 57. | Liu, D.; Liu, X.; Sun, J.; Yan, C.-G. J. Org. Chem. 2021, 86, 14705–14719. doi:10.1021/acs.joc.1c01513 |
| 58. | Zheng, H.; Han, Y.; Sun, J.; Yan, C.-G. Beilstein J. Org. Chem. 2022, 18, 669–679. doi:10.3762/bjoc.18.68 |
| 59. | Pan, L.-N.; Sun, J.; Liu, X.-Y.; Yan, C.-G. Org. Biomol. Chem. 2022, 20, 7099–7104. doi:10.1039/d2ob01257j |
| 60. | Wang, D.; Sun, J.; Han, Y.; Sun, Q.; Yan, C.-G. Org. Lett. 2022, 24, 7790–7795. doi:10.1021/acs.orglett.2c03123 |
| 61. | Wang, D.; Sun, J.; Yan, C.-G. Green Synth. Catal. 2022, 3, 53–58. doi:10.1016/j.gresc.2021.10.004 |
| 62. | Liu, D.; Sun, J.; Sun, Q.; Yan, C.-G. Org. Chem. Front. 2023, 10, 540–547. doi:10.1039/d2qo01771g |
| 4. | Arun, Y.; Saranraj, K.; Balachandran, C.; Perumal, P. T. Eur. J. Med. Chem. 2014, 74, 50–64. doi:10.1016/j.ejmech.2013.12.027 |
| 5. | Saranya, P. V.; Neetha, M.; Aneeja, T.; Anilkumar, G. RSC Adv. 2021, 11, 7146–7179. doi:10.1039/d1ra00139f |
| 6. | Ma, Z.; Zhou, A.; Xia, C. Nat. Prod. Rep. 2022, 39, 1015–1044. doi:10.1039/d1np00060h |
| 52. | Sohail, M.; Tanaka, F. Angew. Chem., Int. Ed. 2021, 60, 21256–21260. doi:10.1002/anie.202108734 |
| 42. | Sun, J.; Sun, Y.; Gong, H.; Xie, Y.-J.; Yan, C.-G. Org. Lett. 2012, 14, 5172–5175. doi:10.1021/ol302530m |
| 43. | Han, Y.; Sheng, Y.-J.; Yan, C.-G. Org. Lett. 2014, 16, 2654–2657. doi:10.1021/ol5008394 |
| 44. | Lu, L.-J.; Fu, Q.; Sun, J.; Yan, C.-G. Tetrahedron 2014, 70, 2537–2545. doi:10.1016/j.tet.2014.02.050 |
| 45. | Shen, G.-L.; Sun, J.; Yan, C.-G. RSC Adv. 2015, 5, 4475–4483. doi:10.1039/c4ra13760d |
| 46. | Huang, Y.; Fang, H.-L.; Huang, Y.-X.; Sun, J.; Yan, C.-G. J. Org. Chem. 2019, 84, 12437–12451. doi:10.1021/acs.joc.9b01920 |
| 47. | Zhan, S.-C.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Biomol. Chem. 2020, 18, 163–168. doi:10.1039/c9ob02013f |
| 48. | Yan, C.; Sun, J.; Yan, C.-G. Chin. Chem. Lett. 2021, 32, 1253–1256. doi:10.1016/j.cclet.2020.08.052 |
| 49. | Wang, D.; Sun, J.; Liu, R.-Z.; Wang, Y.; Yan, C.-G. J. Org. Chem. 2021, 86, 5616–5629. doi:10.1021/acs.joc.1c00103 |
| 52. | Sohail, M.; Tanaka, F. Angew. Chem., Int. Ed. 2021, 60, 21256–21260. doi:10.1002/anie.202108734 |
| 36. | Shanthi, G.; Perumal, P. T. Tetrahedron Lett. 2008, 49, 7139–7142. doi:10.1016/j.tetlet.2008.09.152 |
| 37. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. Nat. Chem. 2011, 3, 473–477. doi:10.1038/nchem.1039 |
| 38. | Tan, F.; Cheng, H.-G.; Feng, B.; Zou, Y.-Q.; Duan, S.-W.; Chen, J.-R.; Xiao, W.-J. Eur. J. Org. Chem. 2013, 2071–2075. doi:10.1002/ejoc.201300081 |
| 39. | Sun, W.; Hong, L.; Zhu, G.; Wang, Z.; Wei, X.; Ni, J.; Wang, R. Org. Lett. 2014, 16, 544–547. doi:10.1021/ol4034226 |
| 40. | Suman, K.; Thennarasu, S. RSC Adv. 2015, 5, 23291–23302. doi:10.1039/c5ra00283d |
| 41. | You, Z.-H.; Zou, S.; Song, Y.-L.; Song, X.-Q. Org. Lett. 2022, 24, 7183–7187. doi:10.1021/acs.orglett.2c02920 |
| 53. | Sun, J.; Xie, Y.-J.; Yan, C.-G. J. Org. Chem. 2013, 78, 8354–8365. doi:10.1021/jo4010603 |
| 54. | Shen, G.-L.; Sun, J.; Xie, Y.-J.; Yan, C.-G. ChemistrySelect 2016, 1, 1447–1451. doi:10.1002/slct.201600209 |
| 55. | Yang, R.-Y.; Sun, J.; Jin, G.; Yan, C.-G. Mol. Diversity 2018, 22, 21–36. doi:10.1007/s11030-017-9786-z |
| 26. | Ramu, G.; Tangella, Y.; Ambala, S.; Nagendra Babu, B. J. Org. Chem. 2020, 85, 5370–5378. doi:10.1021/acs.joc.0c00078 |
| 27. | Dong, J.; Zhang, D.; Men, Y.; Zhang, X.; Hu, Z.; Xu, X. Org. Lett. 2019, 21, 166–169. doi:10.1021/acs.orglett.8b03646 |
| 28. | Chen, K.; Xu, T.; Liang, J.; Zhou, M.; Zhang, J.; Hao, W.-J.; Wang, J.; Tu, S.-J.; Jiang, B. Chem. Commun. 2019, 55, 14757–14760. doi:10.1039/c9cc08762a |
| 29. | Pramanik, S.; Saha, P.; Ghosh, P.; Mukhopadhyay, C. J. Org. Chem. 2023, 88, 3386–3402. doi:10.1021/acs.joc.2c01523 |
| 30. | Deng, Y.; Li, Y.; Wang, Y.; Sun, S.; Ma, S.; Jia, P.; Li, W.; Wang, K.; Yan, W. Org. Chem. Front. 2022, 9, 210–215. doi:10.1039/d1qo01392k |
| 31. | Wang, B.; Yan, X.; Zhong, H.; Ouyang, Q.; Tian, X. Org. Chem. Front. 2022, 9, 1621–1627. doi:10.1039/d1qo01708j |
| 32. | Cruché, C.; Neiderer, W.; Collins, S. K. ACS Catal. 2021, 11, 8829–8836. doi:10.1021/acscatal.1c01983 |
| 33. | Wang, J.; Zheng, X.-Z.; Xiao, J.-A.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2021, 23, 963–968. doi:10.1021/acs.orglett.0c04164 |
| 34. | Yuan, W.-C.; Zuo, J.; Yuan, S.-P.; Zhao, J.-Q.; Wang, Z.-H.; You, Y. Org. Chem. Front. 2021, 8, 784–791. doi:10.1039/d0qo01335h |
| 35. | Ren, J.-W.; Xie, Z.-Z.; Zheng, L.; Ye, Z.-P.; Deng, Z.-X.; Zhao, Q.-L.; Xiao, J.-A.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Chem. Front. 2021, 8, 778–783. doi:10.1039/d0qo01364a |
| 16. | Chen, Z.-C.; Chen, Z.; Du, W.; Chen, Y.-C. Chem. Rec. 2020, 20, 541–555. doi:10.1002/tcr.201900058 |
| 17. | Yan, R.-J.; Liu, B.-X.; Xiao, B.-X.; Du, W.; Chen, Y.-C. Org. Lett. 2020, 22, 4240–4244. doi:10.1021/acs.orglett.0c01283 |
| 18. | Chen, Z.-C.; Chen, Z.; Yang, Z.-H.; Guo, L.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 15021–15025. doi:10.1002/anie.201907797 |
| 19. | Chen, P.; Chen, Z.-C.; Li, Y.; Ouyang, Q.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 4036–4040. doi:10.1002/anie.201814403 |
| 20. | Wang, Z.-H.; Lei, C.-W.; Zhang, X.-Y.; You, Y.; Zhao, J.-Q.; Yuan, W.-C. Org. Chem. Front. 2019, 6, 3342–3347. doi:10.1039/c9qo00890j |
| 21. | Chen, Y.; Cui, B.-D.; Bai, M.; Han, W.-Y.; Wan, N.-W.; Chen, Y.-Z. Tetrahedron 2019, 75, 2971–2979. doi:10.1016/j.tet.2019.04.040 |
| 22. | Gu, J.; Xiao, B.-X.; Ouyang, Q.; Du, W.; Chen, Y.-C. Chin. J. Chem. 2019, 37, 155–160. doi:10.1002/cjoc.201800466 |
| 23. | Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h |
| 24. | Zhou, T.; Xia, T.; Liu, Z.; Liu, L.; Zhang, J. Adv. Synth. Catal. 2018, 360, 4475–4479. doi:10.1002/adsc.201801152 |
| 25. | He, Z.-L.; Chen, P.; Chen, Z.-C.; Du, W.; Chen, Y.-C. Org. Lett. 2022, 24, 100–104. doi:10.1021/acs.orglett.1c03689 |
| 50. | Huang, X.; Pham, K.; Yi, W.; Zhang, X.; Clamens, C.; Hyatt, J. H.; Jasinsk, J. P.; Tayvah, U.; Zhang, W. Adv. Synth. Catal. 2015, 357, 3820–3824. doi:10.1002/adsc.201500748 |
| 51. | Huang, X.; Liu, M.; Pham, K.; Zhang, X.; Yi, W.-B.; Jasinski, J. P.; Zhang, W. J. Org. Chem. 2016, 81, 5362–5369. doi:10.1021/acs.joc.6b00653 |
© 2023 Xiao et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.