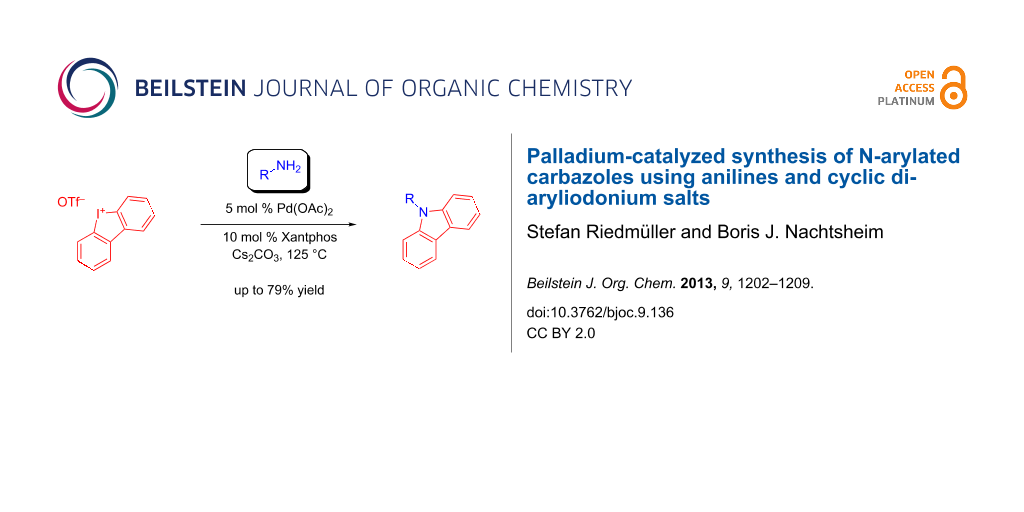
Abstract
The direct synthesis of N-arylated carbazoles through a palladium-catalyzed amination of cyclic iodonium salts with anilines is described. In particular, electron-poor aniline derivatives reacted smoothly with only 5 mol % of Pd(OAc)2 as catalyst to give the desired products in up to 71% yield. Furthermore, the reactivity of cyclic iodonium salts is compared with the reactivity of the corresponding cyclic bromonium analogues.

Graphical Abstract
Introduction
Carbazoles play an important role as core structural elements in natural products (e.g., alkaloids) and pharmaceuticals [1]. In addition, the carbazole motif constitutes an immense class of materials in the rapidly growing field of molecular electronics. In particular N-arylcarbazoles have promising electroluminescent properties and have subsequently found diverse applications as hole-transport, or as host or luminescent-materials in electronic devices (OLEDs) (Figure 1) [2-7]. Representative examples are the host molecules mCP, CBP and CBZ1-F2, the hole transporter BCz2 [8] or the recently described thermally activated delayed fluorescence (TADF) emitter 4CzIPN [9].
![[1860-5397-9-136-1]](/bjoc/content/figures/1860-5397-9-136-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative examples of carbazoles with hole-transport, host or luminescent properties.
Figure 1: Representative examples of carbazoles with hole-transport, host or luminescent properties.
Therefore, the efficient synthesis of N-arylated carbazoles is an attractive goal and numerous synthetic methods are known so far from the literature. The main synthetic routes are shown in Scheme 1. The majority are transition-metal mediated. Starting from functionalized 2,2'-biphenyls (path A) [10-13] or the direct arylation [14,15] of the free NH-functionality of carbazole (path B).
![[1860-5397-9-136-i1]](/bjoc/content/inline/1860-5397-9-136-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic access to N-arylated carbazoles.
Scheme 1: Synthetic access to N-arylated carbazoles.
In the past decade, hypervalent iodine chemistry has undergone a renaissance and has developed to become a powerful area in synthetic organic chemistry. Open-chained iodonium salts are well explored in transition-metal-mediated reactions to construct new C–N bonds [16-19], whereas examples dealing with cyclic iodonium salts are underrepresented [20]. Our group is interested in the development of new C–X coupling strategies via (hypo)iodite or hypervalent iodine catalysis [21-23]. Here, we wish to present an alternative Pd-catalyzed method for the construction of N-substituted carbazoles based on a stable, cyclic iodonium salt and electron-deficient anilines [24,25].
In the initial C–N bond-forming step of this cascade reaction, a ring opening of the cyclic iodonium salt through the amine is proposed to give 2'-iodobiphenyl-2-phenylamine (I). In a second, Pd-mediated intramolecular cross-coupling, 9-phenyl-9H-carbazole (3a) should be observed (Scheme 2).
![[1860-5397-9-136-i2]](/bjoc/content/inline/1860-5397-9-136-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanistic motivation towards the formation of 3a.
Scheme 2: Proposed mechanistic motivation towards the formation of 3a.
Results and Discussion
First, we decided to prepare cyclic iodonium salt 1 as the triflate salt, to avoid unwanted side-reactions in solution with a concurrent nucleophilic counterion [26]. However, 1 was synthesized from 2-iodobiphenyl according to the established one-pot procedure for the synthesis of diaryliodonium triflates [27,28] by Olofsson and co-workers (Supporting Information File 1).
After promising initial experiments, we systematically optimized the reaction conditions of a reaction between aniline and cyclic iodonium salt 1 (Table 1). Various reaction parameters, in particular the Pd catalyst, catalyst loading, the phosphine ligand, and the temperature had a significant influence on the outcome of this transformation. Starting with Pd2(dba)3, SPhos (2 mol % and 4 mol %, respectively), NaOt-Bu or Cs2CO3 in toluene resulted only in trace amounts of 3a (Table 1, entry 1 and 2). After increasing the catalyst/ligand ratio (palladium to phosphine 5 mol % and 10 mol %, respectively) and using Cs2CO3 as the base, 3a could be isolated in 35% yield (Table 1, entry 3). Next, we varied the phosphine ligands (Table 1, entries 4–9). Xantphos was the most efficient bidentate ligand yielding 3a in 46% yield (Table 1, entry 6). Xylenes as the solvent, for example p-xylene, were also suitable for the reaction; in contrast to DME, where the yield slightly decreases (Table 1, entry 11). We also tested t-Bu-Xantphos (Table 1, entry 8) as a common ligand with a higher bite angle. However, this Xantphos derivative is totally inefficient in our coupling reaction. Dppf and BINAP gave very similar results in isolated yield (natural bite angle of the phosphines 99° and 93°, respectively) (Table 1, entry 5 and 7). When using bidentate ligands with bite angles higher than 100° (DPEphos 104°, Xantphos 108°) the reaction is more efficient and the yield increases significantly. Next, we asked ourselves, whether other palladium salts could be equal or better in efficiency and yield. Changing Pd2(dba)3 to Pd(OAc)2 had no significant impact on product yields (Table 1, entry 12). Further increase of the catalyst ratio from 5 to 10 mol % had little effect (Table 1, entry 15).
Table 1: Optimizing the reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-136-i5.svg?max-width=637&scale=1.0)
|
|||||||
| entry |
catalyst
5 mol % |
phosphine
10 mol % |
base | solvent | time [h] | temp [°C] | yield [%]b |
|---|---|---|---|---|---|---|---|
| 1c | Pd2dba3 | SPhos | NaOt-Bu | toluene | 19 | 105 | traced |
| 2c | Pd2dba3 | SPhos | Cs2CO3 | toluene | 19 | 105 | traced |
| 3 | Pd2dba3 | SPhos | Cs2CO3 | toluene | 5 | 105 | 35 |
| 4 | Pd2dba3 | P(t-Bu)3 | Cs2CO3 | toluene | 4 | 105 | 34 |
| 5 | Pd2dba3 | dppf | Cs2CO3 | toluene | 13 | 105 | 14 |
| 6 | Pd2dba3 | Xantphos | Cs2CO3 | toluene | 14 | 105 | 46 |
| 7 | Pd2dba3 | BINAP | Cs2CO3 | p-xylene | 16 | 125 | 16 |
| 8 | Pd2dba3 | t-Bu-Xantphos | Cs2CO3 | p-xylene | 16 | 125 | traced |
| 9 | Pd2dba3 | DPE-Phos | Cs2CO3 | p-xylene | 14 | 125 | 34 |
| 10 | Pd2dba3 | Xantphos | Cs2CO3 | p-xylene | 12 | 125 | 42 |
| 11 | Pd2dba3 | Xantphos | Cs2CO3 | DME | 13 | 79 | 39 |
| 12 | Pd(OAc)2 | Xantphos | Cs2CO3 | p-xylene | 2.5 | 125 | 45e |
| 13 | Pd(OAc)2 | Xantphos | Cs2CO3 | DME | 3 | 79 | 12 |
| 14 | Pd(OAc)2 | Xantphos | NaOt-Bu | p-xylene | 3 | 125 | traced |
| 15f | Pd(OAc)2 | Xantphos | Cs2CO3 | p-xylene | 4 | 126 | 51 |
aAll reactions were run using iodonium salt 1 (0.35 mmol), 1.2 equiv of aniline 2a, 2.7 equiv of base, and 5 mL of solvent. bIsolated yield after column chromatography. c2 mol % Pd2dba3 and 4 mol % SPhos were used. dProduct not isolated. e1.0 equiv of aniline 2a was used. f10 mol % Pd(OAc)2 and 20 mol % Xantphos were used. Pd2dba3 = tris(dibenzylideneacetone)dipalladium(0), SPhos = 2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl, dppf = 1,1'-bis(diphenylphosphino)ferrocene, Xantphos = 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene, BINAP = 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl, t-Bu-Xantphos = 4,5-bis(di-tert-butylphosphino)-9,9-dimethylxanthene, DPEphos = bis[(2-diphenylphosphino)phenyl] ether.
Next, we decided to analyze the byproducts of this reaction by GC–MS analysis. For this experiment the reaction was performed according to conditions given in entry 12, Table 1. Besides the desired product 3a (56%), we could identify the masses of 2-iodobiphenyl (22%), 2,2'-diiodobiphenyl (4%), 2-(2,5-dimethylphenyl)-2'-iodobiphenyl (5%), and 2'-iodo-N-phenylbiphenyl-2-amine (<2%) in significant amounts (Figure 2).
![[1860-5397-9-136-2]](/bjoc/content/figures/1860-5397-9-136-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: FID chromatogram of the reaction mixture. Only the most intense peaks were structurally assigned. x-Axis = retention time.
Figure 2: FID chromatogram of the reaction mixture. Only the most intense peaks were structurally assigned. x...
After a deeper literature research we came to the conclusion, that those byproducts should probably not only arise from side reactions within the catalytic cycle (for instance, 2-iodobiphenyl from β-H elimination) but also from homolytic or heterolytic decomposition pathways of the diaryliodonium salt (2-iodobiphenyl, 2,2'-diiodobiphenyl, and 2-(2,5-dimethylphenyl)-2'-iodobiphenyl) [29-31]. To further verify these observations, we reacted 1 in the presence of aniline (2a) for three days at elevated temperature without adding a Pd catalyst. Again, after GC–MS analysis, we could detect 2-iodobiphenyl, 2,2'-diiodobiphenyl, 2-(2,5-dimethylphenyl)-2'-iodobiphenyl, and 2'-iodo-N-phenylbiphenyl-2-amine in the absence of any produced 3a. Contrary to that observation, when we conducted an analogous experiment without aniline, we observed no decomposition or byproduct formation. These results led us to conclude that byproduct formation is, at least partially, induced through the nucleophilic and/or basic nature of aniline [26,32-35]. We therefore had to accept that a significant amount of byproducts are formed during the formation of 3a, reducing our isolated yield.
After we had gained a better understanding about byproduct formation, we decided to explore various substituted anilines under our optimized reaction conditions (Figure 3). Electron-rich p-toluidine (2b) gave the corresponding carbazole 3b in only 45% yield. Comparable results were obtained when benzylamine (2c) was used (41% yield). Even more electron-donating aniline derivatives, such as p-anisidine (p-methoxyaniline), resulted in the formation of trace amounts of the carbazole product (not shown). The aliphatic primary amines tert-butylamine (2d) and propylamine (2e) were also investigated. Amine 2d was completely inefficient and yielded 3% of 3d compared to a 79% yield of 3e, when using propylamine. However, when electron-withdrawing anilines were utilized, isolated yields of the corresponding carbazole increased significantly. As examples, p-cyano-, p-chloro- or p-COOMe-substituted anilines yielded 3g, 3h, and 3k in 53%, 55% and 62% yield; p-fluoroaniline yielded 3f in 71%. Other fluorine-substituted anilines, such as 4-(trifluoromethyl)aniline (2i) or 3,5-difluoroaniline (2j), were also suitable with slightly decreased yields of 3i (61%) and 3j (54%), respectively.
![[1860-5397-9-136-3]](/bjoc/content/figures/1860-5397-9-136-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Substrate scope. All reactions were performed using iodonium salt 1 (0.35 mmol), 1.2 equiv of primary amine 2, 2.7 equiv of Cs2CO3, 5 mol % Pd(OAc)2, 10 mol % Xantphos, and 5–8 mL p-xylene at 125 °C. Reaction times 2–4 h. Isolated yields are given in parentheses.
Figure 3: Substrate scope. All reactions were performed using iodonium salt 1 (0.35 mmol), 1.2 equiv of prima...
Furthermore, meta-substituted anilines 2l and 2m were tested, giving the N-arylated carbazoles 3l and 3m in good yields of 64% and 61%, respectively. In general, the use of fluorine-substituted anilines showed the best results so far in this study. However, with the perfluorinated derivative 2p, the isolated yield of 3p was diminished to 37%. Furthermore, we used our protocol to synthesize the N-arylcarbazole based electronic materials 3n and 3o in 40% (3n) and 23% (3o) yield.
After an extensive exploration of the reaction conditions and the substrate scope with iodonium salt 1, we wanted to compare our results with the corresponding bromonium analogue. Cyclic diarylbromonium salts are considerably less explored than their iodonium congeners as can be seen by only a handful of synthetic methods described in the literature [32,36-41]. In general, bromonium salts are more reactive but have similar reaction behaviour [32,36]. Thus they could be helpful substrates for the synthesis of N-arylcarbazoles from anilines. With this in mind, we initially focussed on the synthesis of dibenzo[b,d]bromolium chloride (5) using a procedure published by Sandin and Hay in 1952 [41] (Scheme 3).
![[1860-5397-9-136-i3]](/bjoc/content/inline/1860-5397-9-136-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of dibenzo[b,d]bromolium trifluoromethanesulfonate (6).
Scheme 3: Synthesis of dibenzo[b,d]bromolium trifluoromethanesulfonate (6).
The biphenyl derivative 4*HCl was prepared by Suzuki coupling of 2-iodoaniline and 2-bromophenylboronic acid. Diazotation of 4*HCl and cyclization gave the cyclic diarylbromonium chloride 5 as an off-white powder in good isolated yield (52%) (Supporting Information File 1) [41]. Finally, 5 could be converted into the corresponding diarylbromonium triflate 6 with silver trifluoromethanesulfonate in quantitative yield (Supporting Information File 1). To verify whether dibenzo[b,d]bromolium trifluoromethanesulfonate (6) is indeed a more reactive surrogate for the construction of N-arylated carbazoles, we reacted 6 with p-fluoroaniline (2f) according to our previously described optimized conditions (Scheme 4).
![[1860-5397-9-136-i4]](/bjoc/content/inline/1860-5397-9-136-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Dibenzo[b,d]bromolium trifluoromethanesulfonate (6) and p-fluoroaniline (2f) to construct carbazole 3f.
Scheme 4: Dibenzo[b,d]bromolium trifluoromethanesulfonate (6) and p-fluoroaniline (2f) to construct carbazole ...
However, 3f was obtained in only 25% yield (Scheme 4), compared to 71% when using the corresponding diaryliodonium salt 1 (Figure 3). Apart from the desired product, we were able to isolate two byproducts from the crude reaction mixture. After a systematic structure determination by one- and two-dimensional NMR techniques as well as mass spectrometry, we elucidated the two byproducts as the two regioisomers 2'-bromo-N-(4-fluorophenyl)biphenyl-2-amine (7a, 12%) and 2'-bromo-N-(4-fluorophenyl)biphenyl-3-amine (7b, 18%) (Scheme 4). Compound 7a was either formed during the catalytic cycle as a reaction intermediate, which had not reacted further to the final product 3f, or is the result of a nucleophilic attack, caused by the nucleophilic nature of aniline 2f at the electrophilic ipso-position in 6. The formation of the other regioisomer 7b, is not evident at first glance. One plausible explanation could be the emergence of a benzyne intermediate during synthesis, generated by β-elimination using aniline as a base. Subsequent nucleophilic trapping of the benzyne with aniline, this time reacting as a nucleophile, results in the formation of 7b. A very similar reactivity was described recently for a nitro-substituted diarylbromonium salt [42]. However, the results of these experiments demonstrate, that the higher reactivity of diarylbromonium salts towards nucleophilic ring opening is accompanied, to a significant degree, by an undesired β-elimination pathway, leading to more complex reaction mixtures and subsequently lower yields of the desired N-arylcarbazole.
Conclusion
In summary, we have developed a novel synthesis of synthetically highly useful N-arylcarbazoles starting from cyclic diaryliodonium salts by a ring opening/Buchwald-amination cascade using anilines and aliphatic amines as nitrogen-containing substrates. With 5 mol % of Pd(OAc)2 the desired N-arylcarbazoles could be isolated in up to 71% yield. Finally, the corresponding cyclic diarylbromonium derivatives were tested in the same reaction. Significantly lower yields were observed due to undesired side reactions involving benzyne intermediates by β-elimination.
Supporting Information
| Supporting Information File 1: Experimental procedures and data of characterization of the described compounds. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Knölker, H.-J.; Reddy, K. R. Chem. Rev. 2002, 102, 4303–4428. doi:10.1021/cr020059j
Return to citation in text: [1] -
Tao, Y.; Yang, C.; Qin, J. Chem. Soc. Rev. 2011, 40, 2943–2970. doi:10.1039/C0CS00160K
Return to citation in text: [1] -
Duan, L.; Hou, L.; Lee, T.-W.; Qiao, J.; Zhang, D.; Dong, G.; Wang, L.; Qiu, Y. J. Mater. Chem. 2010, 20, 6392–6407. doi:10.1039/B926348A
Return to citation in text: [1] -
Meng, H.; Herron, N. Organic Small Molecule Materials for Organic Light-Emitting Diodes. In Organic Light-Emitting Materials and Devices; Li, Z.; Meng, H., Eds.; CRC Press: Boca Raton, 2007; pp 295–412.
Return to citation in text: [1] -
Shirota, Y. J. Mater. Chem. 2005, 15, 75–93. doi:10.1039/B413819H
Return to citation in text: [1] -
Shirota, Y. J. Mater. Chem. 2000, 10, 1–25. doi:10.1039/A908130E
Return to citation in text: [1] -
Shih, P.-I.; Chiang, C.-L.; Dixit, A. K.; Chen, C.-K.; Yuan, M.-C.; Lee, R.-Y.; Chen, C.-T.; Diau, E. W.-G.; Shu, C.-F. Org. Lett. 2006, 8, 2799–2802. doi:10.1021/ol060884c
Return to citation in text: [1] -
Tsai, M.-H.; Hong, Y.-H.; Chang, C.-H.; Su, H.-C.; Wu, C.-C.; Matoliukstyte, A.; Simokaitiene, J.; Grigalevicius, S.; Grazulevicius, J. V.; Hsu, C.-P. Adv. Mater. 2007, 19, 862–866. doi:10.1002/adma.200600822
Return to citation in text: [1] -
Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Nature 2012, 492, 234–238. doi:10.1038/nature11687
Return to citation in text: [1] -
Zhou, Y.; Verkade, J. G. Adv. Synth. Catal. 2010, 352, 616–620. doi:10.1002/adsc.200900846
Return to citation in text: [1] -
Kitawaki, T.; Hayashi, Y.; Ueno, A.; Chida, N. Tetrahedron 2006, 62, 6792–6801. doi:10.1016/j.tet.2006.04.087
Return to citation in text: [1] -
Kuwahara, A.; Nakano, K.; Nozaki, K. J. Org. Chem. 2005, 70, 413–419. doi:10.1021/jo048472+
Return to citation in text: [1] -
Nozaki, K.; Takahashi, K.; Nakano, K.; Hiyama, T.; Tang, H.-Z.; Fujiki, M.; Yamaguchi, S.; Tamao, K. Angew. Chem. 2003, 115, 2097–2099. doi:10.1002/ange.200250648
Angew. Chem., Int. Ed. 2003, 42, 2051–2053. doi:10.1002/anie.200250648
Return to citation in text: [1] -
Suzuki, K.; Hori, Y.; Kobayashi, T. Adv. Synth. Catal. 2008, 350, 652–656. doi:10.1002/adsc.200700543
Return to citation in text: [1] -
Xi, Z.; Liu, F.; Zhou, Y.; Chen, W. Tetrahedron 2008, 64, 4254–4259. doi:10.1016/j.tet.2008.02.082
Return to citation in text: [1] -
Kang, S.-K.; Lee, S.-H.; Lee, D. Synlett 2000, 1022–1024. doi:10.1055/s-2000-6673
Return to citation in text: [1] -
Beletskaya, I. P.; Davydov, D. V.; Moreno-Mañas, M. Tetrahedron Lett. 1998, 39, 5621–5622. doi:10.1016/S0040-4039(98)01063-6
Return to citation in text: [1] -
Davydov, D. V.; Beletskaya, I. P.; Semenov, B. B.; Smushkevich, Y. I. Tetrahedron Lett. 2002, 43, 6217–6219. doi:10.1016/S0040-4039(02)01326-6
Return to citation in text: [1] -
Beletskaya, I. P.; Davydov, D. V.; Gorovoy, M. S. Tetrahedron Lett. 2002, 43, 6221–6223. doi:10.1016/S0040-4039(02)01325-4
Return to citation in text: [1] -
Liang, Y.; Luo, S.; Liu, C.; Wu, X.; Ma, Y. Tetrahedron 2000, 56, 2961–2965. doi:10.1016/S0040-4020(00)00166-6
Return to citation in text: [1] -
Kloeckner, U.; Weckenmann, N. M.; Nachtsheim, B. J. Synlett 2012, 97–100. doi:10.1055/s-0031-1289902
Return to citation in text: [1] -
Hempel, C.; Weckenmann, N. M.; Maichle-Moessmer, C.; Nachtsheim, B. J. Org. Biomol. Chem. 2012, 10, 9325–9329. doi:10.1039/C2OB26815A
Return to citation in text: [1] -
Froehr, T.; Sindlinger, C. P.; Kloeckner, U.; Finkbeiner, P.; Nachtsheim, B. J. Org. Lett. 2011, 13, 3754–3757. doi:10.1021/ol201439t
Return to citation in text: [1] -
Letessier, J.; Detert, H. Synthesis 2012, 290–296. doi:10.1055/s-0031-1289652
Return to citation in text: [1] -
Sandin, R. B. J. Org. Chem. 1969, 34, 456–457. doi:10.1021/jo01254a043
Return to citation in text: [1] -
Beringer, F. M.; Falk, R. A. J. Chem. Soc. 1964, 4442–4451. doi:10.1039/JR9640004442
Return to citation in text: [1] [2] -
Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/B701864A
Return to citation in text: [1] -
Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373
Return to citation in text: [1] -
Beringer, F. M.; Chang, L. L. J. Org. Chem. 1971, 36, 4055–4060. doi:10.1021/jo00825a011
Return to citation in text: [1] -
Sato, T.; Shimizu, K.; Moriya, H. J. Chem. Soc., Perkin Trans. 1 1974, 1537–1539. doi:10.1039/P19740001537
Return to citation in text: [1] -
Sato, T.; Shimada, S.; Shimizu, K.; Hata, K. Bull. Chem. Soc. Jpn. 1970, 43, 1918. doi:10.1246/bcsj.43.1918
Return to citation in text: [1] -
Olah, G. A.; Sakakibara, T.; Asensio, G. J. Org. Chem. 1978, 43, 463–468. doi:10.1021/jo00397a018
Return to citation in text: [1] [2] [3] -
Grushin, V. V.; Demkina, I. I.; Tolstaya, T. P. J. Chem. Soc., Perkin Trans. 2 1992, 505–511. doi:10.1039/P29920000505
Return to citation in text: [1] -
Vanchikov, A. N.; Bobyleva, M. S.; Komissarova, E. E.; Kulikov, N. S.; Tolstaya, T. P. Chem. Heterocycl. Compd. 1998, 34, 371–377. doi:10.1007/BF02290735
Return to citation in text: [1] -
Yamada, Y.; Kashima, K.; Okawara, M. Bull. Chem. Soc. Jpn. 1974, 47, 3179–3180. doi:10.1246/bcsj.47.3179
Return to citation in text: [1] -
Farooq, U.; Shah, A.-u.-H. A.; Wirth, T. Angew. Chem. 2009, 121, 1036–1038. doi:10.1002/ange.200805027
Angew. Chem. Int. Ed. 2009, 48, 1018-1020. doi:10.1002/anie.200805027
Return to citation in text: [1] [2] -
Nesmeyanov, A. N.; Lisichkina, I. N.; Vanchikov, A. N.; Tolstaya, T. P. Russ. Chem. Bull. 1977, 26, 1110. doi:10.1007/BF01152740
Return to citation in text: [1] -
Nesmeyanov, A. N.; Lisichkina, I. N.; Vanchikov, A. N.; Tolstaya, T. P. Russ. Chem. Bull. 1976, 25, 224. doi:10.1007/BF00925666
Return to citation in text: [1] -
Nesmeyanov, A. N.; Lisichkina, I. N.; Tolstaya, T. P. Russ. Chem. Bull. 1973, 22, 2123. doi:10.1007/BF00929431
Return to citation in text: [1] -
Nesmeyanov, A. N.; Tolstaya, T. P.; Lisichkina, I. N. Russ. Chem. Bull. 1968, 17, 189–190. doi:10.1007/BF00914669
Return to citation in text: [1] -
Sandin, R. B.; Hay, A. S. J. Am. Chem. Soc. 1952, 74, 274–275. doi:10.1021/ja01121a524
Return to citation in text: [1] [2] [3] -
Hou, Z. J. P.; He, L. X. H. Chin. Chem. Lett. 2002, 13, 189–192.
Return to citation in text: [1]
| 41. | Sandin, R. B.; Hay, A. S. J. Am. Chem. Soc. 1952, 74, 274–275. doi:10.1021/ja01121a524 |
| 41. | Sandin, R. B.; Hay, A. S. J. Am. Chem. Soc. 1952, 74, 274–275. doi:10.1021/ja01121a524 |
| 1. | Knölker, H.-J.; Reddy, K. R. Chem. Rev. 2002, 102, 4303–4428. doi:10.1021/cr020059j |
| 10. | Zhou, Y.; Verkade, J. G. Adv. Synth. Catal. 2010, 352, 616–620. doi:10.1002/adsc.200900846 |
| 11. | Kitawaki, T.; Hayashi, Y.; Ueno, A.; Chida, N. Tetrahedron 2006, 62, 6792–6801. doi:10.1016/j.tet.2006.04.087 |
| 12. | Kuwahara, A.; Nakano, K.; Nozaki, K. J. Org. Chem. 2005, 70, 413–419. doi:10.1021/jo048472+ |
| 13. |
Nozaki, K.; Takahashi, K.; Nakano, K.; Hiyama, T.; Tang, H.-Z.; Fujiki, M.; Yamaguchi, S.; Tamao, K. Angew. Chem. 2003, 115, 2097–2099. doi:10.1002/ange.200250648
Angew. Chem., Int. Ed. 2003, 42, 2051–2053. doi:10.1002/anie.200250648 |
| 32. | Olah, G. A.; Sakakibara, T.; Asensio, G. J. Org. Chem. 1978, 43, 463–468. doi:10.1021/jo00397a018 |
| 36. |
Farooq, U.; Shah, A.-u.-H. A.; Wirth, T. Angew. Chem. 2009, 121, 1036–1038. doi:10.1002/ange.200805027
Angew. Chem. Int. Ed. 2009, 48, 1018-1020. doi:10.1002/anie.200805027 |
| 37. | Nesmeyanov, A. N.; Lisichkina, I. N.; Vanchikov, A. N.; Tolstaya, T. P. Russ. Chem. Bull. 1977, 26, 1110. doi:10.1007/BF01152740 |
| 38. | Nesmeyanov, A. N.; Lisichkina, I. N.; Vanchikov, A. N.; Tolstaya, T. P. Russ. Chem. Bull. 1976, 25, 224. doi:10.1007/BF00925666 |
| 39. | Nesmeyanov, A. N.; Lisichkina, I. N.; Tolstaya, T. P. Russ. Chem. Bull. 1973, 22, 2123. doi:10.1007/BF00929431 |
| 40. | Nesmeyanov, A. N.; Tolstaya, T. P.; Lisichkina, I. N. Russ. Chem. Bull. 1968, 17, 189–190. doi:10.1007/BF00914669 |
| 41. | Sandin, R. B.; Hay, A. S. J. Am. Chem. Soc. 1952, 74, 274–275. doi:10.1021/ja01121a524 |
| 9. | Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Nature 2012, 492, 234–238. doi:10.1038/nature11687 |
| 32. | Olah, G. A.; Sakakibara, T.; Asensio, G. J. Org. Chem. 1978, 43, 463–468. doi:10.1021/jo00397a018 |
| 36. |
Farooq, U.; Shah, A.-u.-H. A.; Wirth, T. Angew. Chem. 2009, 121, 1036–1038. doi:10.1002/ange.200805027
Angew. Chem. Int. Ed. 2009, 48, 1018-1020. doi:10.1002/anie.200805027 |
| 8. | Tsai, M.-H.; Hong, Y.-H.; Chang, C.-H.; Su, H.-C.; Wu, C.-C.; Matoliukstyte, A.; Simokaitiene, J.; Grigalevicius, S.; Grazulevicius, J. V.; Hsu, C.-P. Adv. Mater. 2007, 19, 862–866. doi:10.1002/adma.200600822 |
| 29. | Beringer, F. M.; Chang, L. L. J. Org. Chem. 1971, 36, 4055–4060. doi:10.1021/jo00825a011 |
| 30. | Sato, T.; Shimizu, K.; Moriya, H. J. Chem. Soc., Perkin Trans. 1 1974, 1537–1539. doi:10.1039/P19740001537 |
| 31. | Sato, T.; Shimada, S.; Shimizu, K.; Hata, K. Bull. Chem. Soc. Jpn. 1970, 43, 1918. doi:10.1246/bcsj.43.1918 |
| 2. | Tao, Y.; Yang, C.; Qin, J. Chem. Soc. Rev. 2011, 40, 2943–2970. doi:10.1039/C0CS00160K |
| 3. | Duan, L.; Hou, L.; Lee, T.-W.; Qiao, J.; Zhang, D.; Dong, G.; Wang, L.; Qiu, Y. J. Mater. Chem. 2010, 20, 6392–6407. doi:10.1039/B926348A |
| 4. | Meng, H.; Herron, N. Organic Small Molecule Materials for Organic Light-Emitting Diodes. In Organic Light-Emitting Materials and Devices; Li, Z.; Meng, H., Eds.; CRC Press: Boca Raton, 2007; pp 295–412. |
| 5. | Shirota, Y. J. Mater. Chem. 2005, 15, 75–93. doi:10.1039/B413819H |
| 6. | Shirota, Y. J. Mater. Chem. 2000, 10, 1–25. doi:10.1039/A908130E |
| 7. | Shih, P.-I.; Chiang, C.-L.; Dixit, A. K.; Chen, C.-K.; Yuan, M.-C.; Lee, R.-Y.; Chen, C.-T.; Diau, E. W.-G.; Shu, C.-F. Org. Lett. 2006, 8, 2799–2802. doi:10.1021/ol060884c |
| 26. | Beringer, F. M.; Falk, R. A. J. Chem. Soc. 1964, 4442–4451. doi:10.1039/JR9640004442 |
| 32. | Olah, G. A.; Sakakibara, T.; Asensio, G. J. Org. Chem. 1978, 43, 463–468. doi:10.1021/jo00397a018 |
| 33. | Grushin, V. V.; Demkina, I. I.; Tolstaya, T. P. J. Chem. Soc., Perkin Trans. 2 1992, 505–511. doi:10.1039/P29920000505 |
| 34. | Vanchikov, A. N.; Bobyleva, M. S.; Komissarova, E. E.; Kulikov, N. S.; Tolstaya, T. P. Chem. Heterocycl. Compd. 1998, 34, 371–377. doi:10.1007/BF02290735 |
| 35. | Yamada, Y.; Kashima, K.; Okawara, M. Bull. Chem. Soc. Jpn. 1974, 47, 3179–3180. doi:10.1246/bcsj.47.3179 |
| 21. | Kloeckner, U.; Weckenmann, N. M.; Nachtsheim, B. J. Synlett 2012, 97–100. doi:10.1055/s-0031-1289902 |
| 22. | Hempel, C.; Weckenmann, N. M.; Maichle-Moessmer, C.; Nachtsheim, B. J. Org. Biomol. Chem. 2012, 10, 9325–9329. doi:10.1039/C2OB26815A |
| 23. | Froehr, T.; Sindlinger, C. P.; Kloeckner, U.; Finkbeiner, P.; Nachtsheim, B. J. Org. Lett. 2011, 13, 3754–3757. doi:10.1021/ol201439t |
| 26. | Beringer, F. M.; Falk, R. A. J. Chem. Soc. 1964, 4442–4451. doi:10.1039/JR9640004442 |
| 20. | Liang, Y.; Luo, S.; Liu, C.; Wu, X.; Ma, Y. Tetrahedron 2000, 56, 2961–2965. doi:10.1016/S0040-4020(00)00166-6 |
| 27. | Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/B701864A |
| 28. | Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610–2618. doi:10.1002/adsc.200700373 |
| 16. | Kang, S.-K.; Lee, S.-H.; Lee, D. Synlett 2000, 1022–1024. doi:10.1055/s-2000-6673 |
| 17. | Beletskaya, I. P.; Davydov, D. V.; Moreno-Mañas, M. Tetrahedron Lett. 1998, 39, 5621–5622. doi:10.1016/S0040-4039(98)01063-6 |
| 18. | Davydov, D. V.; Beletskaya, I. P.; Semenov, B. B.; Smushkevich, Y. I. Tetrahedron Lett. 2002, 43, 6217–6219. doi:10.1016/S0040-4039(02)01326-6 |
| 19. | Beletskaya, I. P.; Davydov, D. V.; Gorovoy, M. S. Tetrahedron Lett. 2002, 43, 6221–6223. doi:10.1016/S0040-4039(02)01325-4 |
| 14. | Suzuki, K.; Hori, Y.; Kobayashi, T. Adv. Synth. Catal. 2008, 350, 652–656. doi:10.1002/adsc.200700543 |
| 15. | Xi, Z.; Liu, F.; Zhou, Y.; Chen, W. Tetrahedron 2008, 64, 4254–4259. doi:10.1016/j.tet.2008.02.082 |
| 24. | Letessier, J.; Detert, H. Synthesis 2012, 290–296. doi:10.1055/s-0031-1289652 |
| 25. | Sandin, R. B. J. Org. Chem. 1969, 34, 456–457. doi:10.1021/jo01254a043 |
© 2013 Riedmüller and Nachtsheim; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)