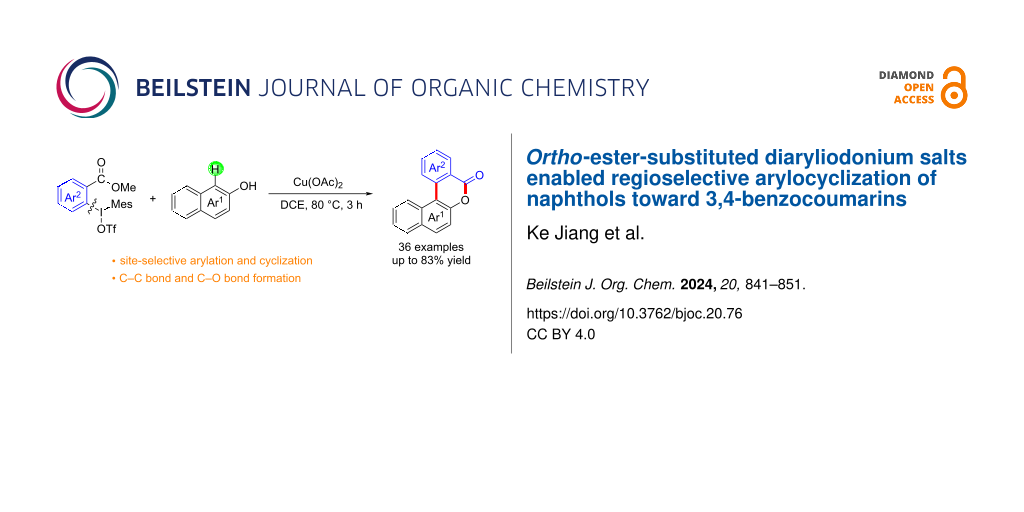
Abstract
Cyclic annulation involving diaryliodonium salts is an efficient tool for the construction of two or more chemical bonds in a one-pot process. Ortho-functionalized diaryliodonium salts have showcased distinct reactivity in the exploration of benzocyclization or arylocyclization. With this strategy of ortho-ester-substituted diaryliodonium salts, herein, we utilized a copper catalyst to activate the C–I bond of diaryliodonium salts in the generation of aryl radicals, thus resulting in an annulation reaction with naphthols and substituted phenols. This approach yielded a diverse array of 3,4-benzocoumarin derivatives bearing various substituents.

Graphical Abstract
Introduction
Diaryliodonium salts as electrophilic reagents have attracted significant attention in the field of organic synthesis owing to their efficiency and selectivity [1-7]. Particularly, they have been employed in benzocyclization and arylocyclization reactions, enabling intramolecular cyclization by forming aromatic or heterocyclic rings as a part of cyclic structures [8]. In these reactions, the dual activation of a C–I bond and vicinal C–H bonds/functional groups features a distinct advantage, facilitating the formation of two or more chemical bonds in a step-economic manner [9-13]. In a prior study, we reported a palladium-catalyzed efficient activation of both C–I bond and the adjacent C–H bond of diaryliodonium salts in the formation of 4,5-benzocoumarin derivatives, expanding the benzocoumarin family (Scheme 1b) [14]. Recently, ortho-functionalized diaryliodonium salts, due to their coordinating and electrophilic effects, have exhibited unique reactivity and chemoselectivity [15]. As such, a wide range of functional groups including the trimethylsilyl group, boronic acid, trifluoroborate moiety, trifluoromethanesulfonate, aryl sulfonamides, and heterocycles, have been incorporated into the ortho-position of diaryliodonium structures [16-21]. Ortho-trimethylsilyl or boronic acid-substituted diaryliodonium salts can serve as aryne precursors. Ortho-trifluoroborate-substituted diaryliodonium salts furnished iodonium zwitterions as bifunctional reagents [22-25]. Additionally, ortho-trifluoromethanesulfonate, N-sulfonyl, or tosylmethylene-substituted diaryliodonium salts can undergo intramolecular aryl migrations [26-28]. More recently, we explored the reactivity of ortho-functionalized diaryliodonium salts containing electron-withdrawing groups (EWGs) such as fluorine and nitro groups [29,30]. These ortho-substituted diaryliodonium salts undergo selective benzocyclizations and arylocyclizations with aromatic acids, leading to 3,4-benzocoumarin skeletons in the presence of palladium catalysts (Scheme 1b). Furthermore, Olofsson and colleagues described an unprecedented reaction pathway using ortho-fluoro-substituted diaryliodonium salts bearing strong electron-withdrawing groups, leading to novel diarylations of N-, O-, and S-nucleophiles [31-33]. Building on our great interest in ortho-functionalized diaryliodonium salts and their dual activation capabilities, we sought to incorporate carboxylic ester groups into the structures of ortho-substituted diaryliodonium salts to explore their properties and reactivity. Our previous investigations demonstrated the ability of diaryliodonium salts for selective mono-arylation of 2-naphthols [34]. In this context, we embark on a strategy to modify the neighbouring position of the diaryliodonium salt with an ester group, presenting a novel copper-catalysed regioselective arylocyclization of naphthols and substituted phenols. This method represents an efficient approach to access 3,4-benzocoumarin derivatives (Scheme 1c).
![[1860-5397-20-76-i1]](/bjoc/content/inline/1860-5397-20-76-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Arylation reactions of aromatic compounds and reaction patterns of ortho-functionalized diaryliodonium salts.
Scheme 1: Arylation reactions of aromatic compounds and reaction patterns of ortho-functionalized diaryliodon...
Results and Discussion
To start the study, we used 2-naphthol (1a) and 1.1 equivalents of ortho-methyl formate-substituted diaryliodonium salt 2a as template substrates. The reaction was performed in the presence of 10 mol % Cu(OTf)2 and 1.0 equivalent of K2CO3 in DCE at a temperature of 80 °C. To our delight, the reaction afforded 3,4-benzocoumarin 3aa in a 27% yield (Table 1, entry 1). The structure of 3aa was confirmed through NMR spectroscopy and mass spectra analysis. Subsequently, we started to screen various bases such as Na2CO3, Cs2CO3, KOH, NaOt-Bu, LiHMDS, and DMAP (Table 1, entries 2–7). Fortunately, it was found that the reaction yield was increased to 50% in the absence of any base (Table 1, entry 8). Further investigations for assessing the influence of various solvents including dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), toluene, acetic acid (AcOH) and water (Table 1, entries 9–13) were carried out. However, polar solvents such as AcOH and H2O were proved to be unsuitable for this reaction. For catalysts, we found that Cu(OAc)2 gave the best results (Table 1, entries 15–18). Finally, the reaction temperature and time were optimized, 3aa was produced in 61% yield at a temperature of 80 °C after 3 hours (Table 1, entry 15).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-76-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Base | Catalyst | 3aa (%)b |
| 1 | DCE | K2CO3 | Cu(OTf)2 | 27 |
| 2 | DCE | Na2CO3 | Cu(OTf)2 | 25 |
| 3 | DCE | Cs2CO3 | Cu(OTf)2 | 16 |
| 4 | DCE | KOH | Cu(OTf)2 | 24 |
| 5 | DCE | DMAP | Cu(OTf)2 | 26 |
| 6 | DCE | NaOt-Bu | Cu(OTf)2 | 35 |
| 7 | DCE | LiHMDS | Cu(OTf)2 | 30 |
| 8 | DCE | – | Cu(OTf)2 | 50 |
| 9 | DMSO | – | Cu(OTf)2 | 45c (40)d |
| 10 | DMF | – | Cu(OTf)2 | 23 |
| 11 | toluene | – | Cu(OTf)2 | 10 |
| 12 | AcOH | – | Cu(OTf)2 | 0 |
| 13 | H2O | – | Cu(OTf)2 | 0 |
| 14e | DCE | – | Cu(OTf)2 | 48 |
| 15 | DCE | – | Cu(OAc)2 | 61 |
| 16 | DCE | – | Pd(OAc)2 | 22 |
| 17 | DCE | – | PdCl2 | 40 |
| 18 | DCE | – | AgOAc | 20 |
aReaction conditions: 1a (0.3 mmol, 1 equiv), 2a (0.33 mmol, 1.1 equiv), base (0.3 mmol; 1 equiv), catalyst (10 mol %), solvent (2 mL), 80 °C, 3 hours. bIsolated yields were obtained after purification by column chromatography. cThe reaction temperature was 110 °C. dThe reaction temperature was 130 °C. eThe reaction was quenched after 12 hours.
With the optimized reaction conditions in hand, we started to explore the substrate scope of the cyclization to construct a variety of 3,4-benzocoumarin derivatives. Our investigations commenced with 2-naphthol (1), and the results are presented in Table 2. Various substituted naphthols with a broad range of substituents on the naphthalene unit were well tolerated in the reaction, affording the corresponding products 3aa–aq in generally moderate to good yields of 22–83% (Table 2, entries 1–17). These substituents included halogen (Br), methyl, phenyl, aldehyde, ester, and methoxy groups, all of which were compatible with the reaction conditions. Notably, compounds 3ab, 3ah, 3aj, 3am and 3ap bearing bromine are very useful modules for the synthesis of functional materials via cross-coupling reactions. Next, we extended our investigation to 1-naphthol in this reaction, and found that the arylation of 1-naphthol was achieved selectively at the C-2 position. The cascade cyclization resulted in the corresponding products 3an and 3ao in yields of 49% and 40%, respectively (Table 2, entries 14 and 15). When 5,6,7,8-tetrahydro-2-naphthol was subjected to the reaction, we obtained products 3ar and 3as as a mixture (40% and 10% yield, respectively, Table 2, entry 18). However, when naphthol bearing a strong electron-withdrawing group (such as a nitro group) in the para position was reacted, the corresponding product could not be obtained, but instead the O-arylated product 3at was obtained (Table 2, entry 19). Apart from naphthol, we also tested substituted phenols under the standard conditions. The corresponding products of 3au and 3av were produced in 34% and 39% yields, respectively, in which methoxy and tert-butyl groups were located in the para position to the hydroxy group (Table 2, entries 20 and 21). In the case of 3al, the mono-arylation of naphthol generated 3al’ in 20% isolated yield, which is the reason for the low yield of 3al.
Table 2: Scope of naphthols and phenols for the synthesis of 3,4-benzocoumarins.a,b.
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-76-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | 1 | Product | Yield (%)b | |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-76-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-76-i6.svg?max-width=637&scale=1.0)
3aa |
61 | |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-76-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-76-i8.svg?max-width=637&scale=1.0)
3ab |
63 | |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-20-76-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-20-76-i10.svg?max-width=637&scale=1.0)
3ac |
80 | |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-20-76-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-20-76-i12.svg?max-width=637&scale=1.0)
3ad |
77 | |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-20-76-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-20-76-i14.svg?max-width=637&scale=1.0)
3ae |
26 | |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-20-76-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-20-76-i16.svg?max-width=637&scale=1.0)
3af |
31 | |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-20-76-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-20-76-i18.svg?max-width=637&scale=1.0)
3ag |
28 | |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-20-76-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-20-76-i20.svg?max-width=637&scale=1.0)
3ah |
54 | |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-20-76-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-20-76-i22.svg?max-width=637&scale=1.0)
3ai |
25 | |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-20-76-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-20-76-i24.svg?max-width=637&scale=1.0)
3aj |
22 | |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-20-76-i25.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-20-76-i26.svg?max-width=637&scale=1.0)
3ak |
49 | |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-20-76-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-20-76-i28.svg?max-width=637&scale=1.0)
3al |
![[Graphic 27]](/bjoc/content/inline/1860-5397-20-76-i29.svg?max-width=637&scale=1.0)
3al’ |
48 (20) |
| 13 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-20-76-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 29]](/bjoc/content/inline/1860-5397-20-76-i31.svg?max-width=637&scale=1.0)
3am |
3 | |
| 14 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-20-76-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 31]](/bjoc/content/inline/1860-5397-20-76-i33.svg?max-width=637&scale=1.0)
3an |
49 | |
| 15 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-20-76-i34.svg?max-width=637&scale=1.0)
|
![[Graphic 33]](/bjoc/content/inline/1860-5397-20-76-i35.svg?max-width=637&scale=1.0)
3ao |
40 | |
| 16 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-20-76-i36.svg?max-width=637&scale=1.0)
|
![[Graphic 35]](/bjoc/content/inline/1860-5397-20-76-i37.svg?max-width=637&scale=1.0)
3ap |
25 | |
| 17 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-20-76-i38.svg?max-width=637&scale=1.0)
|
![[Graphic 37]](/bjoc/content/inline/1860-5397-20-76-i39.svg?max-width=637&scale=1.0)
3aq |
43 | |
| 18 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-20-76-i40.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-20-76-i41.svg?max-width=637&scale=1.0)
3ar |
![[Graphic 40]](/bjoc/content/inline/1860-5397-20-76-i42.svg?max-width=637&scale=1.0)
3as |
45 (10) |
| 19 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-20-76-i43.svg?max-width=637&scale=1.0)
|
![[Graphic 42]](/bjoc/content/inline/1860-5397-20-76-i44.svg?max-width=637&scale=1.0)
3at |
51 | |
| 20 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-20-76-i45.svg?max-width=637&scale=1.0)
|
![[Graphic 44]](/bjoc/content/inline/1860-5397-20-76-i46.svg?max-width=637&scale=1.0)
3au |
34 | |
| 21 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-20-76-i47.svg?max-width=637&scale=1.0)
|
![[Graphic 46]](/bjoc/content/inline/1860-5397-20-76-i48.svg?max-width=637&scale=1.0)
3av |
39 | |
aReaction conditions: 1 (0.3 mmol, 1 equiv), 2a (0.33 mmol, 1.1 equiv), Cu(OAc)2 (10 mol %), DCE (2 mL), 80 °C, 3 hours. bIsolated yields were obtained after purification with column chromatography. Mes = 2,4,6-trimethylphenyl, OTf = trifluoromethansulfonate.
We subsequently turned our attention to explore the effect of structural diversity of the ortho-ester-substituted diaryliodonium salts. Firstly, a family of substituted diaryliodonium salts were synthesized in a one-pot procedure. These ortho-substituted diaryliodonium salts were isolated as stable solids, whose structures were fully characterized by NMR spectroscopy. As shown in Table 3, we utilized 2-naphthol and 1-naphthol as template substrates to react with various unsymmetrical 2-ester-substituted diaryliodonium salts. Remarkably, iodonium salts 2 proved to be versatile in this reaction, regardless of the electronic nature and position of the substituents. The desired 3,4-benzocoumarin products 3ba–ma were obtained in yields of 21–59%. Notably, substituents such as halogens (F, Cl, and Br), methyl, methoxy, and trifluoromethyl groups at the ortho-, meta-, or para-positions to the ester group were all well-tolerated (Table 3).
Table 3: Scope of ortho-ester-substituted diaryliodonium salts.a
![[Graphic 47]](/bjoc/content/inline/1860-5397-20-76-i49.svg?max-width=637&scale=1.0)
|
|||
| Entry | 2 | Product | Yield (%)b |
| 1 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-20-76-i50.svg?max-width=637&scale=1.0)
2b |
![[Graphic 49]](/bjoc/content/inline/1860-5397-20-76-i51.svg?max-width=637&scale=1.0)
3aa |
55 |
| 2 |
![[Graphic 50]](/bjoc/content/inline/1860-5397-20-76-i52.svg?max-width=637&scale=1.0)
2c |
![[Graphic 51]](/bjoc/content/inline/1860-5397-20-76-i53.svg?max-width=637&scale=1.0)
3ca |
32 |
| 3 |
![[Graphic 52]](/bjoc/content/inline/1860-5397-20-76-i54.svg?max-width=637&scale=1.0)
2d |
![[Graphic 53]](/bjoc/content/inline/1860-5397-20-76-i55.svg?max-width=637&scale=1.0)
3da |
50 |
| 4 |
![[Graphic 54]](/bjoc/content/inline/1860-5397-20-76-i56.svg?max-width=637&scale=1.0)
2e |
![[Graphic 55]](/bjoc/content/inline/1860-5397-20-76-i57.svg?max-width=637&scale=1.0)
3ea |
46 |
| 5 |
![[Graphic 56]](/bjoc/content/inline/1860-5397-20-76-i58.svg?max-width=637&scale=1.0)
2f |
![[Graphic 57]](/bjoc/content/inline/1860-5397-20-76-i59.svg?max-width=637&scale=1.0)
3fa |
49 |
| 6 |
![[Graphic 58]](/bjoc/content/inline/1860-5397-20-76-i60.svg?max-width=637&scale=1.0)
2g |
![[Graphic 59]](/bjoc/content/inline/1860-5397-20-76-i61.svg?max-width=637&scale=1.0)
3ga |
55 |
| 7 |
![[Graphic 60]](/bjoc/content/inline/1860-5397-20-76-i62.svg?max-width=637&scale=1.0)
2h |
![[Graphic 61]](/bjoc/content/inline/1860-5397-20-76-i63.svg?max-width=637&scale=1.0)
3ha |
43 |
| 8 |
![[Graphic 62]](/bjoc/content/inline/1860-5397-20-76-i64.svg?max-width=637&scale=1.0)
2i |
![[Graphic 63]](/bjoc/content/inline/1860-5397-20-76-i65.svg?max-width=637&scale=1.0)
3ia |
21 |
| 9 |
![[Graphic 64]](/bjoc/content/inline/1860-5397-20-76-i66.svg?max-width=637&scale=1.0)
2j |
![[Graphic 65]](/bjoc/content/inline/1860-5397-20-76-i67.svg?max-width=637&scale=1.0)
3ja |
59 |
| 10c |
![[Graphic 66]](/bjoc/content/inline/1860-5397-20-76-i68.svg?max-width=637&scale=1.0)
2j |
![[Graphic 67]](/bjoc/content/inline/1860-5397-20-76-i69.svg?max-width=637&scale=1.0)
3jb |
28 |
| 11 |
![[Graphic 68]](/bjoc/content/inline/1860-5397-20-76-i70.svg?max-width=637&scale=1.0)
2k |
![[Graphic 69]](/bjoc/content/inline/1860-5397-20-76-i71.svg?max-width=637&scale=1.0)
3ka |
37 |
| 12c |
![[Graphic 70]](/bjoc/content/inline/1860-5397-20-76-i72.svg?max-width=637&scale=1.0)
2k |
![[Graphic 71]](/bjoc/content/inline/1860-5397-20-76-i73.svg?max-width=637&scale=1.0)
3kb |
45 |
| 13 |
![[Graphic 72]](/bjoc/content/inline/1860-5397-20-76-i74.svg?max-width=637&scale=1.0)
2l |
![[Graphic 73]](/bjoc/content/inline/1860-5397-20-76-i75.svg?max-width=637&scale=1.0)
3la |
35 |
| 14 |
![[Graphic 74]](/bjoc/content/inline/1860-5397-20-76-i76.svg?max-width=637&scale=1.0)
2m |
![[Graphic 75]](/bjoc/content/inline/1860-5397-20-76-i77.svg?max-width=637&scale=1.0)
3ma |
50 |
aReaction conditions: 1 (0.3 mmol, 1 equiv), 2 (0.33 mmol, 1.1 equiv), Cu(OAc)2 (10 mol %), DCE (2 mL), 80 °C, 3 hours. bIsolated yields were obtained after purification with column chromatography. c1-Naphthol was used instead of 2-naphthol. Mes = 2,4,6-trimethylphenyl, OTf = trifluoromethansulfonate.
To gain further insights into the reaction mechanism, we conducted control experiments. Given the utility of diaryliodonium salts in radical chemistry, we introduced 2 equivalents of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) or 2 equivalents of butylated hydroxytoluene (BHT) into the template reaction. Remarkably, we observed that the desired product was not formed, suggesting a radical pathway. Subsequently, we investigated the bond-formation sequence in the benzocyclization reaction. A possible intermediate of 3al’ was prepared and tested in the reaction under the standard conditions, however, product 3aa was not obtained.
Based on the literature known results and the experimental evidences [35,36], we proposed a plausible reaction mechanism (Scheme 2b). The reaction started with the formation of radical intermediate A from diaryliodonium salt 2a. Naphthol 1a forms intermediate B with A after participation with the Cu(II) catalyst. Intermediate B generates C by radical substitution. A final intramolecular transesterification yields the benzocoumarin product 3aa.
![[1860-5397-20-76-i2]](/bjoc/content/inline/1860-5397-20-76-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanism study. Standard conditions: 1 (0.3 mmol, 1 equiv), 2 (0.33 mmol, 1.1 equiv), Cu(OAc)2 (10 mol %), DCE (2 mL), 80 °C, 3 hours. TEMPO = 2,2,6,6-tetramethylpiperidine-1-oxyl; BHT = butylated hydroxytoluene.
Scheme 2: Mechanism study. Standard conditions: 1 (0.3 mmol, 1 equiv), 2 (0.33 mmol, 1.1 equiv), Cu(OAc)2 (10...
Conclusion
In summary, we have employed ortho-ester-substituted diaryliodonium salts in a cascade cyclization, the cyclization features a copper-catalyzed activation strategy involving the cleavage of the C–I bond and esterification. The resulting cascade of selective arylation/intramolecular cyclization facilitated the synthesis of 3,4-benzocoumarin derivatives. The protocol enables the efficient formation of two chemical bonds in one pot, representing a valuable tool for the synthesis of polycyclic benzocoumarins. Our ongoing research endeavours are dedicated to explore the detailed reaction mechanism with the ultimate aim of broadening the scope and applicability of this approach.
Supporting Information
| Supporting Information File 1: Experimental procedures, LC–MS spectra and characterization data of all products, copies of 1H, 13C, 19F NMR spectra of all compounds. | ||
| Format: PDF | Size: 12.2 MB | Download |
Acknowledgements
The authors thank the Research Center of Analysis and Test of the East China University of Science and Technology for the help on the characterization and Professor Zhen-Jiang Xu from SIOC, CAS for helpful discussion and instrumental analysis.
Funding
The work was supported by the Industry-University-Research Collaborative Innovation Fund for Chinese Universities-DeZhou Project (2021DZ030) and the Natural Science Foundation of Shanghai (20ZR1413500); Shanghai Municipal Science and Technology Major Project (grant no. 2018SHZDZX03) and Program of Introducing Talents of Discipline to Universities (B16017).
Data Availability Statement
The data that supports the findings of this study is available from the corresponding author upon reasonable request.
References
-
Silva, J. L. F.; Olofsson, B. Nat. Prod. Rep. 2011, 28, 1722–1754. doi:10.1039/c1np00028d
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Aradi, K.; Tóth, B.; Tolnai, G.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369
Return to citation in text: [1] -
Cao, C.; Sheng, J.; Chen, C. Synthesis 2017, 49, 5081–5092. doi:10.1055/s-0036-1589515
Return to citation in text: [1] -
Wang, Y.; An, G.; Wang, L.; Han, J. Curr. Org. Chem. 2020, 24, 2070–2105. doi:10.2174/1385272824999200507124328
Return to citation in text: [1] -
Yang, X.-G.; Hu, Z.-N.; Jia, M.-C.; Du, F.-H.; Zhang, C. Synlett 2021, 32, 1289–1296. doi:10.1055/a-1492-4943
Return to citation in text: [1] -
Peng, X.; Rahim, A.; Peng, W.; Jiang, F.; Gu, Z.; Wen, S. Chem. Rev. 2023, 123, 1364–1416. doi:10.1021/acs.chemrev.2c00591
Return to citation in text: [1] -
Pan, C.; Wang, L.; Han, J. Chem. Rec. 2023, 23, e202300138. doi:10.1002/tcr.202300138
Return to citation in text: [1] -
Wu, Y.; Peng, X.; Luo, B.; Wu, F.; Liu, B.; Song, F.; Huang, P.; Wen, S. Org. Biomol. Chem. 2014, 12, 9777–9780. doi:10.1039/c4ob02170c
Return to citation in text: [1] -
Mehra, M. K.; Sharma, S.; Rangan, K.; Kumar, D. Eur. J. Org. Chem. 2020, 2409–2413. doi:10.1002/ejoc.202000013
Return to citation in text: [1] -
An, G.; Wang, L.; Han, J. Org. Lett. 2021, 23, 8688–8693. doi:10.1021/acs.orglett.1c03016
Return to citation in text: [1] -
Xue, C.; Wang, L.; Han, J. J. Org. Chem. 2020, 85, 15406–15414. doi:10.1021/acs.joc.0c02192
Return to citation in text: [1] -
Kitamura, T.; Yamane, M. J. Chem. Soc., Chem. Commun. 1995, 983–984. doi:10.1039/c39950000983
Return to citation in text: [1] -
Wu, X.; Yang, Y.; Han, J.; Wang, L. Org. Lett. 2015, 17, 5654–5657. doi:10.1021/acs.orglett.5b02938
Return to citation in text: [1] -
Kikushima, K.; Elboray, E. E.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R.; Dohi, T. Org. Biomol. Chem. 2022, 20, 3231–3248. doi:10.1039/d1ob02501e
Return to citation in text: [1] -
Kitamura, T.; Gondo, K.; Oyamada, J. J. Am. Chem. Soc. 2017, 139, 8416–8419. doi:10.1021/jacs.7b04483
Return to citation in text: [1] -
Yoshimura, A.; Fuchs, J. M.; Middleton, K. R.; Maskaev, A. V.; Rohde, G. T.; Saito, A.; Postnikov, P. S.; Yusubov, M. S.; Nemykin, V. N.; Zhdankin, V. V. Chem. – Eur. J. 2017, 23, 16738–16742. doi:10.1002/chem.201704393
Return to citation in text: [1] -
Robidas, R.; Guérin, V.; Provençal, L.; Echeverria, M.; Legault, C. Y. Org. Lett. 2017, 19, 6420–6423. doi:10.1021/acs.orglett.7b03307
Return to citation in text: [1] -
Wu, Y.; Izquierdo, S.; Vidossich, P.; Lledós, A.; Shafir, A. Angew. Chem., Int. Ed. 2016, 55, 7152–7156. doi:10.1002/anie.201602569
Return to citation in text: [1] -
Boelke, A.; Vlasenko, Y. A.; Yusubov, M. S.; Nachtsheim, B. J.; Postnikov, P. S. Beilstein J. Org. Chem. 2019, 15, 2311–2318. doi:10.3762/bjoc.15.223
Return to citation in text: [1] -
Vlasenko, Y. A.; Kuczmera, T. J.; Antonkin, N. S.; Valiev, R. R.; Postnikov, P. S.; Nachtsheim, B. J. Adv. Synth. Catal. 2023, 365, 535–543. doi:10.1002/adsc.202201001
Return to citation in text: [1] -
Chen, H.; Wang, L.; Han, J. Chem. Commun. 2020, 56, 5697–5700. doi:10.1039/d0cc01766c
Return to citation in text: [1] -
Han, J.; Chen, H.; An, G.; Sun, X.; Li, X.; Liu, Y.; Zhao, S.; Wang, L. J. Chem. Educ. 2021, 98, 3992–3998. doi:10.1021/acs.jchemed.1c00546
Return to citation in text: [1] -
Wang, Y.; Zhang, Y.; Wang, L.; Han, J. Asian J. Org. Chem. 2022, 11, e202100669. doi:10.1002/ajoc.202100669
Return to citation in text: [1] -
Liu, X.; Wang, L.; Wang, H.-Y.; Han, J. J. Org. Chem. 2023, 88, 13089–13101. doi:10.1021/acs.joc.3c01293
Return to citation in text: [1] -
Chen, H.; Han, J.; Wang, L. Angew. Chem., Int. Ed. 2018, 57, 12313–12317. doi:10.1002/anie.201806405
Return to citation in text: [1] -
Chen, H.; Wang, L.; Han, J. Org. Lett. 2020, 22, 3581–3585. doi:10.1021/acs.orglett.0c01024
Return to citation in text: [1] -
Wang, Y.; Pan, W.; Zhang, Y.; Wang, L.; Han, J. Angew. Chem., Int. Ed. 2023, 62, e202304897. doi:10.1002/anie.202304897
Return to citation in text: [1] -
Pan, C.; Wang, L.; Han, J. Org. Lett. 2020, 22, 4776–4780. doi:10.1021/acs.orglett.0c01577
Return to citation in text: [1] -
Liu, X.; Wang, L.; Han, J. Org. Biomol. Chem. 2022, 20, 8628–8632. doi:10.1039/d2ob01783k
Return to citation in text: [1] -
Linde, E.; Bulfield, D.; Kervefors, G.; Purkait, N.; Olofsson, B. Chem 2022, 8, 850–865. doi:10.1016/j.chempr.2022.01.009
Return to citation in text: [1] -
Mondal, S.; Di Tommaso, E. M.; Olofsson, B. Angew. Chem., Int. Ed. 2023, 62, e202216296. doi:10.1002/anie.202216296
Return to citation in text: [1] -
Linde, E.; Olofsson, B. Angew. Chem., Int. Ed. 2023, 62, e202310921. doi:10.1002/anie.202310921
Return to citation in text: [1] -
Qian, X.; Han, J.; Wang, L. Tetrahedron Lett. 2016, 57, 607–610. doi:10.1016/j.tetlet.2015.12.100
Return to citation in text: [1] -
Phipps, R. J.; McMurray, L.; Ritter, S.; Duong, H. A.; Gaunt, M. J. J. Am. Chem. Soc. 2012, 134, 10773–10776. doi:10.1021/ja3039807
Return to citation in text: [1] -
Wang, W.; Zhou, J.; Wang, C.; Zhang, C.; Zhang, X.-Q.; Wang, Y. Commun. Chem. 2022, 5, 145. doi:10.1038/s42004-022-00768-3
Return to citation in text: [1]
| 1. | Silva, J. L. F.; Olofsson, B. Nat. Prod. Rep. 2011, 28, 1722–1754. doi:10.1039/c1np00028d |
| 2. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 3. | Aradi, K.; Tóth, B.; Tolnai, G.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369 |
| 4. | Cao, C.; Sheng, J.; Chen, C. Synthesis 2017, 49, 5081–5092. doi:10.1055/s-0036-1589515 |
| 5. | Wang, Y.; An, G.; Wang, L.; Han, J. Curr. Org. Chem. 2020, 24, 2070–2105. doi:10.2174/1385272824999200507124328 |
| 6. | Yang, X.-G.; Hu, Z.-N.; Jia, M.-C.; Du, F.-H.; Zhang, C. Synlett 2021, 32, 1289–1296. doi:10.1055/a-1492-4943 |
| 7. | Peng, X.; Rahim, A.; Peng, W.; Jiang, F.; Gu, Z.; Wen, S. Chem. Rev. 2023, 123, 1364–1416. doi:10.1021/acs.chemrev.2c00591 |
| 15. | Kikushima, K.; Elboray, E. E.; Jiménez-Halla, J. O. C.; Solorio-Alvarado, C. R.; Dohi, T. Org. Biomol. Chem. 2022, 20, 3231–3248. doi:10.1039/d1ob02501e |
| 14. | Wu, X.; Yang, Y.; Han, J.; Wang, L. Org. Lett. 2015, 17, 5654–5657. doi:10.1021/acs.orglett.5b02938 |
| 9. | Wu, Y.; Peng, X.; Luo, B.; Wu, F.; Liu, B.; Song, F.; Huang, P.; Wen, S. Org. Biomol. Chem. 2014, 12, 9777–9780. doi:10.1039/c4ob02170c |
| 10. | Mehra, M. K.; Sharma, S.; Rangan, K.; Kumar, D. Eur. J. Org. Chem. 2020, 2409–2413. doi:10.1002/ejoc.202000013 |
| 11. | An, G.; Wang, L.; Han, J. Org. Lett. 2021, 23, 8688–8693. doi:10.1021/acs.orglett.1c03016 |
| 12. | Xue, C.; Wang, L.; Han, J. J. Org. Chem. 2020, 85, 15406–15414. doi:10.1021/acs.joc.0c02192 |
| 13. | Kitamura, T.; Yamane, M. J. Chem. Soc., Chem. Commun. 1995, 983–984. doi:10.1039/c39950000983 |
| 8. | Pan, C.; Wang, L.; Han, J. Chem. Rec. 2023, 23, e202300138. doi:10.1002/tcr.202300138 |
| 29. | Pan, C.; Wang, L.; Han, J. Org. Lett. 2020, 22, 4776–4780. doi:10.1021/acs.orglett.0c01577 |
| 30. | Liu, X.; Wang, L.; Han, J. Org. Biomol. Chem. 2022, 20, 8628–8632. doi:10.1039/d2ob01783k |
| 34. | Qian, X.; Han, J.; Wang, L. Tetrahedron Lett. 2016, 57, 607–610. doi:10.1016/j.tetlet.2015.12.100 |
| 26. | Chen, H.; Han, J.; Wang, L. Angew. Chem., Int. Ed. 2018, 57, 12313–12317. doi:10.1002/anie.201806405 |
| 27. | Chen, H.; Wang, L.; Han, J. Org. Lett. 2020, 22, 3581–3585. doi:10.1021/acs.orglett.0c01024 |
| 28. | Wang, Y.; Pan, W.; Zhang, Y.; Wang, L.; Han, J. Angew. Chem., Int. Ed. 2023, 62, e202304897. doi:10.1002/anie.202304897 |
| 35. | Phipps, R. J.; McMurray, L.; Ritter, S.; Duong, H. A.; Gaunt, M. J. J. Am. Chem. Soc. 2012, 134, 10773–10776. doi:10.1021/ja3039807 |
| 36. | Wang, W.; Zhou, J.; Wang, C.; Zhang, C.; Zhang, X.-Q.; Wang, Y. Commun. Chem. 2022, 5, 145. doi:10.1038/s42004-022-00768-3 |
| 22. | Chen, H.; Wang, L.; Han, J. Chem. Commun. 2020, 56, 5697–5700. doi:10.1039/d0cc01766c |
| 23. | Han, J.; Chen, H.; An, G.; Sun, X.; Li, X.; Liu, Y.; Zhao, S.; Wang, L. J. Chem. Educ. 2021, 98, 3992–3998. doi:10.1021/acs.jchemed.1c00546 |
| 24. | Wang, Y.; Zhang, Y.; Wang, L.; Han, J. Asian J. Org. Chem. 2022, 11, e202100669. doi:10.1002/ajoc.202100669 |
| 25. | Liu, X.; Wang, L.; Wang, H.-Y.; Han, J. J. Org. Chem. 2023, 88, 13089–13101. doi:10.1021/acs.joc.3c01293 |
| 16. | Kitamura, T.; Gondo, K.; Oyamada, J. J. Am. Chem. Soc. 2017, 139, 8416–8419. doi:10.1021/jacs.7b04483 |
| 17. | Yoshimura, A.; Fuchs, J. M.; Middleton, K. R.; Maskaev, A. V.; Rohde, G. T.; Saito, A.; Postnikov, P. S.; Yusubov, M. S.; Nemykin, V. N.; Zhdankin, V. V. Chem. – Eur. J. 2017, 23, 16738–16742. doi:10.1002/chem.201704393 |
| 18. | Robidas, R.; Guérin, V.; Provençal, L.; Echeverria, M.; Legault, C. Y. Org. Lett. 2017, 19, 6420–6423. doi:10.1021/acs.orglett.7b03307 |
| 19. | Wu, Y.; Izquierdo, S.; Vidossich, P.; Lledós, A.; Shafir, A. Angew. Chem., Int. Ed. 2016, 55, 7152–7156. doi:10.1002/anie.201602569 |
| 20. | Boelke, A.; Vlasenko, Y. A.; Yusubov, M. S.; Nachtsheim, B. J.; Postnikov, P. S. Beilstein J. Org. Chem. 2019, 15, 2311–2318. doi:10.3762/bjoc.15.223 |
| 21. | Vlasenko, Y. A.; Kuczmera, T. J.; Antonkin, N. S.; Valiev, R. R.; Postnikov, P. S.; Nachtsheim, B. J. Adv. Synth. Catal. 2023, 365, 535–543. doi:10.1002/adsc.202201001 |
| 31. | Linde, E.; Bulfield, D.; Kervefors, G.; Purkait, N.; Olofsson, B. Chem 2022, 8, 850–865. doi:10.1016/j.chempr.2022.01.009 |
| 32. | Mondal, S.; Di Tommaso, E. M.; Olofsson, B. Angew. Chem., Int. Ed. 2023, 62, e202216296. doi:10.1002/anie.202216296 |
| 33. | Linde, E.; Olofsson, B. Angew. Chem., Int. Ed. 2023, 62, e202310921. doi:10.1002/anie.202310921 |
© 2024 Jiang et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.