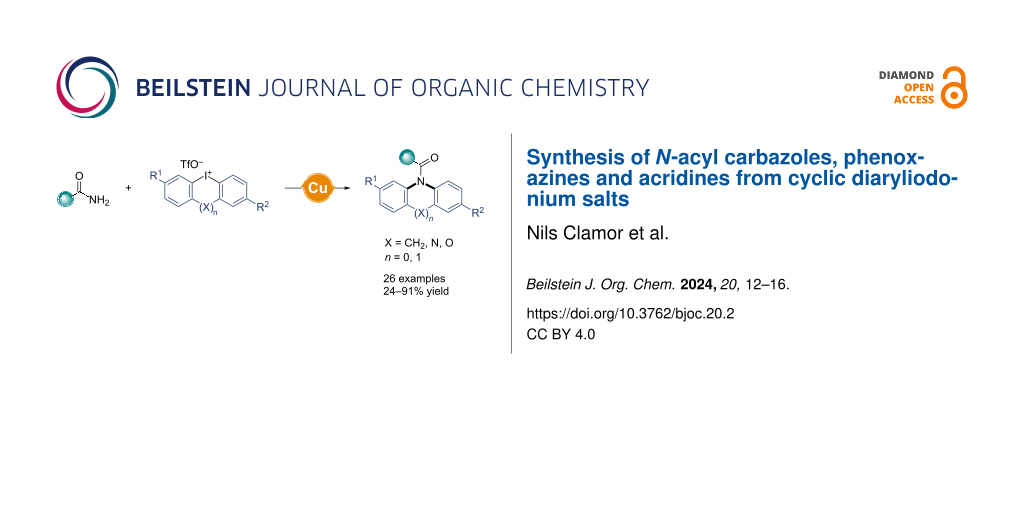
Abstract
N-Acyl carbazoles can be efficiently produced through a single-step process using amides and cyclic diaryliodonium triflates. This convenient reaction is facilitated by copper iodide in p-xylene, using the commonly found activating ligand diglyme. We have tested this method with a wide range of amides and iodonium triflates, proving its versatility with numerous substrates. Beyond carbazoles, we also produced a variety of other N-heterocycles, such as acridines, phenoxazines, or phenazines, showcasing the robustness of our technique. In a broader sense, this new method creates two C–N bonds simultaneously based on a mono-halogenated starting material, thus allowing heterocycle formation with diminished halogen waste.

Graphical Abstract
Introduction
N-Acyl carbazoles are effective fluorophors, previously shown to exhibit strong organic phosphorescence when mixed with specific additives [1-5]. Carbazole units are also found in drugs and natural products. They are also used in electrochemistry and as reagents in transamidation reactions [6-12]. The traditional method to produce this versatile N-acyl carbazole motif involves combining 9H-carbazoles with acyl chlorides or similar activated acyl derivatives in the presence of a base (Scheme 1a) [13,14]. As an alternative, acyl carbazoles can be synthesized through step-wise metal-catalysed C–X-amidations followed by a ring-closure starting from 2,2'-diiodo-1,1'-biphenyls [15-17]. Related one-pot procedures are also described (Scheme 1b) [18,19].
![[1860-5397-20-2-i1]](/bjoc/content/inline/1860-5397-20-2-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Examples for direct syntheses of N-acyl heteroaromatic compounds.
Scheme 1: Examples for direct syntheses of N-acyl heteroaromatic compounds.
Such 2,2´-dihalobiphenyls are established starting materials for synthesizing a variety of heterocycles. Usually, their utilization leads to the production of two equivalents of halogen salt waste. Their substitution with iodolium salts will be more sustainable since it reduces these unproductive halogenide salts by half. Due to our recent activity in the field of synthesis and applications of 5- and 6-membered cyclic iodonium salts, we searched for an efficient method to synthesize N-acyl carbazoles from readily available iodolium salt and amides via a ring-opening/intramolecular coupling cascade (Scheme 1c) [20-30]. Our group recently explored principle synthetic pathways of hetero- and carbocyclic 5- and 6-membered diaryliodonium salts [29,31], as well as Pd-catalysed methods for synthesizing N-aryl carbazoles [32]. Similar procedures were published for the Cu-catalysed synthesis of aryl carbazoles from amines as well as other heterocycles such as N-acyl acridanes with nitriles using cyclic iodonium salts by Wen and Chen [33,34].
Results and Discussion
Initially, we investigated the synthesis of N-acyl carbazole by treatment of diaryliodonium salt 1a with valeramide using Cu(I) catalysts [18]. The results are shown in Table 1. In the first experiments in p-xylene at 120 °C with DMEDA as N,N-ligand, only modest amounts of 2a were observed (Table 1, entry 1). The predominant side products were 2,2'-diiodobiphenyl and 9H-carbazole. The formation of free carbazole indicated the formation and subsequent hydrolysis of 2a. The presence of 2,2'-diiodobiphenyl suggests the reaction of 1a with iodide released by each turnover of the desired reaction [35]. To mitigate this, using silver salts as iodide scavengers in the reaction was attempted but yielded none of the desired product (Table 1, entry 2). DMF as a solvent lowered the yield to 16% (Table 1, entry 3). Switching the catalyst system to Cu(OTf)2/glyme gave a significantly higher yield of 33% (Table 1, entry 4). Increasing the amount of iodolium salt to 1.5 equivalents yielded 2a in 42% (Table 1, entry 5). Further increasing the amount of 1a to 2 equivalents raised the yield only slightly (Table 1, entry 6), while finally exchanging the catalyst to CuI/diglyme at 15 mol % raised yields to synthetically useful 74% (Table 1, entry 7). The excess amount of 1a was still necessary as a significant amount of iodobiphenyl is formed under the reaction conditions as a result of an undesired heterolytic iodine–carbon bond cleavage. Other carbonate bases and changing the Cu(I) source resulted in a complete collapse of reactivity. In a further experiment, we investigated the influence of iodide on the reaction to confirm whether or not diiodobiphenyl plays a role as an intermediate. The addition of potassium iodide leads to only diiodobiphenyl as the product. To confirm the mechanism of opening of the iodane, we used 2,2'-diiodobiphenyl as the starting material, leading to no formation of 2a. Thus, we confirmed that our system does not activate 2,2'-diiodobiphenyl. Therefore, we applied the conditions described in Table 1, entry 7 for further investigation.
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-2-i4.svg?max-width=637&scale=1.0)
|
|||||
| Catalyst (mol %) | Ligand (mol %) | Base (equiv) | Equiv of 1a | Yield (%) | |
| 1 | CuI | DMEDA (20) | K3PO4 (1.5) | 1.0 | 22b |
| 2 | CuI (10) + 1 equiv AgNO3 | DMEDA (20) | K3PO4 (1.5) | 1.0 | n.d. |
| 3 | CuI (10) | DMEDA (20) | K2CO3 (1.5) | 1.0 | 16b,c |
| 4 | Cu(OTf)2 (10) | glyme (20) | K2CO3 (2.5) | 1.0 | 33b |
| 5 | Cu(OTf)2 (10) | diglyme (20) | K2CO3 (2.5) | 1.5 | 42b |
| 6 | Cu(OTf)2 (15) | diglyme (30) | K2CO3 (2.5) | 2.0 | 44b |
| 7 | CuI (15) | diglyme (30) | K2CO3 (3.0) | 2.0 |
76b
74d |
| 8 | CuI (15) | diglyme (30) | Cs2CO3 (3.0) | 2.0 | – |
| 9 | CuCl | diglyme (30) | K2CO3 (3.0) | 2.0 | – |
| 10 | CuBr | diglyme (30) | K2CO3 (3.0) | 2.0 | – |
| 11 | CuI (15) | diglyme (30) | K2CO3 (3.0) | 2.0 | –e |
| 12 | CuI (15) | diglyme (30) | K2CO3 (3.0) | – | –f |
aCommon reaction conditions: 18 h at 120 °C, in degassed p-xylene under Ar atmosphere. bYields determined via GC–MS at a 100 µmol scale. cReaction carried out in DMF. dIsolated yields on a 200 µmol scale. e2.0 equiv of KI added. f2,2-Diiodobiphenyl as starting material.
With the optimized conditions in hand, the substrate scope was explored. The variations of amides are outlined in Scheme 2. Switching from valeramide to benzamide as a substrate gave a more advantageous yield of 85% of 2b.
![[1860-5397-20-2-i2]](/bjoc/content/inline/1860-5397-20-2-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of amides. aIsolated yields, 200 µmol scale, all reactions carried out in p-xylene, with 1.00 equiv amide, 15 mol % CuI, 30 mol % diglyme, 3.00 equiv K2CO3 and 2.00 equiv dibenzo[b,d]iodol-5-ium trifluoromethanesulfonate.
Scheme 2: Scope of amides. aIsolated yields, 200 µmol scale, all reactions carried out in p-xylene, with 1.00...
We tested para-substituted benzamides in the reaction to further assess the diversity of possible products. We obtained p-halogenide- and p-pseudohalogenide-substituted compounds 2c–g in good yields of 76–87%. It is noteworthy that the para-chloro-substituted compound 2f is a known fluorophore [5]. The reaction tolerated methoxy- and methyl ester-substitution to give 2h and 2i in 84% and 90% yields. para-Amino- and boronic acid-substituted benzamides did not react. While meta-bromo-substituted benzamide gave 2j in 84% yield, ortho-bromination resulted in a diminished yield of 2k (55%). We obtained 3,5-disubstituted N-acyl carbazoles 2l and 2m in 91% and 88% yields. The same disubstitution with electron-donating methoxy groups gave product 2n in a diminished output of 56% yield. Other electron-rich 2,4,6-trimethyl- and 3,4,5-trimethoxy-substituted benzamides gave 2o in 91% and 2p in 69% yield. In an experiment at a larger scale (1 mmol), 2o was still generated in 81%, which underscores the robustness of this method. Substrates with multiple electron-withdrawing substituents such as trifluoromethyl and bromide gave good yields. The ethyl carbamate 2q proved to be a valid substrate for the reaction with a 74% yield. Phenylacetamide as the substrate resulted in the formation of 2r in 82% yield.
Next, we investigated structural variations in the cyclic iodonium salt. Due to the high reactivity of 2,4,6-trimethylbenzamide, we utilized this substrate as the nucleophile during these investigations. Scheme 3 displays the results. We started this investigation with the synthesis of nitrile-substituted carbazoles 2s and 2t, which are potent fluorophores. While 2s was isolated in 61% yield, the additional 6-bromo-substituent diminished the yield of 2t to 47%. When we subjected 6-membered 10H-dibenzo[b,e]iodinin-5-iums to our conditions, we synthesized the N-acyl dihydroacridane 3a with a 47% yield. A method for the synthesis of similar annulated N-heterocycles from iodanes with nitriles is described by Chen et al. [34].
![[1860-5397-20-2-i3]](/bjoc/content/inline/1860-5397-20-2-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of iodanes. aIsolated yields, 200 µmol scale, all reactions carried out in p-xylene, with 1.00 equiv amide 15 mol % CuI, 30 mol % diglyme, 3.00 equiv K2CO3 and 2.00 equiv iodane. bphenoxazines 3b–e required a higher reaction temperature of 135 °C.
Scheme 3: Scope of iodanes. aIsolated yields, 200 µmol scale, all reactions carried out in p-xylene, with 1.0...
Next, we investigated O-bridged dibenzo[b,e][1,4]iodaoxin-5-ium salts as substrates, as was shown in a recent work [29]. Following the trend, we already observed for the 5-membered iodoliums, electron-donating groups are beneficial, while electron-withdrawing groups adversely affect their reactivity. Hence, we isolated unsubstituted and methyl-substituted N-acyl phenoxazines 3b and 3c in 84% [29] and 74% yields and their bromo and cyano variants 3d and 3e in lower 25% and 26% yields. Lastly, we synthesized 3f using the standard procedure for iodoliums with more sophisticated benzimidazole-containing diaryliodonium salt, but we only observed a reproducible yield of 8%. The analysis of its solid-state structure showed the desired connectivity [36].
Conclusion
In conclusion, we developed an effective method for synthesizing N-acyl carbazoles, phenoxazines, and acridines in a single-step reaction from 5- and 6-membered cyclic biaryliodonium salts. Based on the excellent synthetic availability of the underlying cyclic iodonium salts, this reaction provides fast and reliable access to these substrates, which are profound structural motifs with application in medicinal and materials chemistry.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.0 MB | Download |
References
-
Chen, C.; Chi, Z.; Chong, K. C.; Batsanov, A. S.; Yang, Z.; Mao, Z.; Yang, Z.; Liu, B. Nat. Mater. 2021, 20, 175–180. doi:10.1038/s41563-020-0797-2
Return to citation in text: [1] -
Song, X.; Lu, G.; Man, Y.; Zhang, J.; Chen, S.; Han, C.; Xu, H. Angew. Chem., Int. Ed. 2023, 62, e202300980. doi:10.1002/anie.202300980
Return to citation in text: [1] -
Hu, Y.; Cai, W.; Ying, L.; Chen, D.; Yang, X.; Jiang, X.-F.; Su, S.; Huang, F.; Cao, Y. J. Mater. Chem. C 2018, 6, 2690–2695. doi:10.1039/c7tc04064d
Return to citation in text: [1] -
Cai, S.; Shi, H.; Zhang, Z.; Wang, X.; Ma, H.; Gan, N.; Wu, Q.; Cheng, Z.; Ling, K.; Gu, M.; Ma, C.; Gu, L.; An, Z.; Huang, W. Angew. Chem., Int. Ed. 2018, 57, 4005–4009. doi:10.1002/anie.201800697
Return to citation in text: [1] -
Cai, S.; Shi, H.; Li, J.; Gu, L.; Ni, Y.; Cheng, Z.; Wang, S.; Xiong, W.-W.; Li, L.; An, Z.; Huang, W. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1701244. doi:10.1002/adma.201701244
Return to citation in text: [1] [2] -
Liu, Y.; Guo, Y.; Ji, F.; Gao, D.; Song, C.; Chang, J. J. Org. Chem. 2016, 81, 4310–4315. doi:10.1021/acs.joc.6b00729
Return to citation in text: [1] -
Utaipan, T.; Athipornchai, A.; Suksamrarn, A.; Jirachotikoon, C.; Yuan, X.; Lertcanawanichakul, M.; Chunglok, W. J. Nat. Med. 2017, 71, 158–169. doi:10.1007/s11418-016-1045-6
Return to citation in text: [1] -
Nishiyama, T.; Matsuoka, A.; Honda, R.; Kitamura, T.; Hatae, N.; Choshi, T. Tetrahedron 2020, 76, 131110. doi:10.1016/j.tet.2020.131110
Return to citation in text: [1] -
Crabtree, R. H. ACS Sustainable Chem. Eng. 2017, 5, 4491–4498. doi:10.1021/acssuschemeng.7b00983
Return to citation in text: [1] -
You, X.; Zhu, D.; Lu, W.; Sun, Y.; Qiao, S.; Luo, B.; Du, Y.; Pi, R.; Hu, Y.; Huang, P.; Wen, S. RSC Adv. 2018, 8, 17183–17190. doi:10.1039/c8ra02939c
Return to citation in text: [1] -
Kim, Y.; Yeom, M.; Lee, S.; Tae, J.; Kim, H. J.; Rhim, H.; Seong, J.; Choi, K. I.; Min, S.-J.; Choo, H. Bull. Korean Chem. Soc. 2018, 39, 1083–1089. doi:10.1002/bkcs.11555
Return to citation in text: [1] -
Kang, B.; Yasuno, Y.; Okamura, H.; Sakai, A.; Satoh, T.; Kuse, M.; Shinada, T. Bull. Chem. Soc. Jpn. 2020, 93, 993–999. doi:10.1246/bcsj.20200116
Return to citation in text: [1] -
Umehara, A.; Shimizu, S.; Sasaki, M. ChemCatChem 2023, 15, e202201596. doi:10.1002/cctc.202201596
Return to citation in text: [1] -
Umehara, A.; Shimizu, S.; Sasaki, M. Adv. Synth. Catal. 2023, 365, 2367–2376. doi:10.1002/adsc.202300487
Return to citation in text: [1] -
Kajiyama, D.; Inoue, K.; Ishikawa, Y.; Nishiyama, S. Tetrahedron 2010, 66, 9779–9784. doi:10.1016/j.tet.2010.11.015
Return to citation in text: [1] -
Wang, Q.; Zhang, X.; Wang, P.; Gao, X.; Zhang, H.; Lei, A. Chin. J. Chem. 2021, 39, 143–148. doi:10.1002/cjoc.202000407
Return to citation in text: [1] -
Antonchick, A. P.; Samanta, R.; Kulikov, K.; Lategahn, J. Angew. Chem., Int. Ed. 2011, 50, 8605–8608. doi:10.1002/anie.201102984
Return to citation in text: [1] -
Li, E.; Xu, X.; Li, H.; Zhang, H.; Xu, X.; Yuan, X.; Li, Y. Tetrahedron 2009, 65, 8961–8968. doi:10.1016/j.tet.2009.08.075
Return to citation in text: [1] [2] -
Liao, Q.; Zhang, L.; Wang, F.; Li, S.; Xi, C. Eur. J. Org. Chem. 2010, 5426–5431. doi:10.1002/ejoc.201000665
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689
Return to citation in text: [1] -
Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/b701864a
Return to citation in text: [1] -
Grushin, V. V. Chem. Soc. Rev. 2000, 29, 315–324. doi:10.1039/a909041j
Return to citation in text: [1] -
Liu, Z.; Luo, B.; Liu, X.; Hu, Y.; Wu, B.; Huang, P.; Wen, S. Eur. J. Org. Chem. 2016, 1110–1118. doi:10.1002/ejoc.201501544
Return to citation in text: [1] -
Zhu, D.; Wu, Z.; Liang, L.; Sun, Y.; Luo, B.; Huang, P.; Wen, S. RSC Adv. 2019, 9, 33170–33179. doi:10.1039/c9ra07288h
Return to citation in text: [1] -
Zhu, D.; Wu, Z.; Luo, B.; Du, Y.; Liu, P.; Chen, Y.; Hu, Y.; Huang, P.; Wen, S. Org. Lett. 2018, 20, 4815–4818. doi:10.1021/acs.orglett.8b01969
Return to citation in text: [1] -
Vlasenko, Y. A.; Postnikov, P. S.; Trusova, M. E.; Shafir, A.; Zhdankin, V. V.; Yoshimura, A.; Yusubov, M. S. J. Org. Chem. 2018, 83, 12056–12070. doi:10.1021/acs.joc.8b01995
Return to citation in text: [1] -
Wang, M.; Fan, Q.; Jiang, X. Org. Lett. 2018, 20, 216–219. doi:10.1021/acs.orglett.7b03564
Return to citation in text: [1] -
Chatterjee, N.; Goswami, A. Eur. J. Org. Chem. 2017, 3023–3032. doi:10.1002/ejoc.201601651
Return to citation in text: [1] -
Damrath, M.; Caspers, L. D.; Duvinage, D.; Nachtsheim, B. J. Org. Lett. 2022, 24, 2562–2566. doi:10.1021/acs.orglett.2c00691
Return to citation in text: [1] [2] [3] [4] -
Peng, X.; Rahim, A.; Peng, W.; Jiang, F.; Gu, Z.; Wen, S. Chem. Rev. 2023, 123, 1364–1416. doi:10.1021/acs.chemrev.2c00591
Return to citation in text: [1] -
Boelke, A.; Kuczmera, T. J.; Caspers, L. D.; Lork, E.; Nachtsheim, B. J. Org. Lett. 2020, 22, 7261–7266. doi:10.1021/acs.orglett.0c02593
Return to citation in text: [1] -
Riedmüller, S.; Nachtsheim, B. J. Beilstein J. Org. Chem. 2013, 9, 1202–1209. doi:10.3762/bjoc.9.136
Return to citation in text: [1] -
Zhu, D.; Liu, Q.; Luo, B.; Chen, M.; Pi, R.; Huang, P.; Wen, S. Adv. Synth. Catal. 2013, 355, 2172–2178. doi:10.1002/adsc.201300271
Return to citation in text: [1] -
Peng, X.; Li, L.; Ren, Y.; Xue, H.; Liu, J.; Wen, S.; Chen, J. Adv. Synth. Catal. 2020, 362, 2030–2038. doi:10.1002/adsc.201901460
Return to citation in text: [1] [2] -
Wu, B.; Yoshikai, N. Angew. Chem., Int. Ed. 2015, 54, 8736–8739. doi:10.1002/anie.201503134
Return to citation in text: [1] -
Kuczmera, T. J.; Dietz, A.; Boelke, A.; Nachtsheim, B. J. Beilstein J. Org. Chem. 2023, 19, 317–324. doi:10.3762/bjoc.19.27
Return to citation in text: [1]
| 1. | Chen, C.; Chi, Z.; Chong, K. C.; Batsanov, A. S.; Yang, Z.; Mao, Z.; Yang, Z.; Liu, B. Nat. Mater. 2021, 20, 175–180. doi:10.1038/s41563-020-0797-2 |
| 2. | Song, X.; Lu, G.; Man, Y.; Zhang, J.; Chen, S.; Han, C.; Xu, H. Angew. Chem., Int. Ed. 2023, 62, e202300980. doi:10.1002/anie.202300980 |
| 3. | Hu, Y.; Cai, W.; Ying, L.; Chen, D.; Yang, X.; Jiang, X.-F.; Su, S.; Huang, F.; Cao, Y. J. Mater. Chem. C 2018, 6, 2690–2695. doi:10.1039/c7tc04064d |
| 4. | Cai, S.; Shi, H.; Zhang, Z.; Wang, X.; Ma, H.; Gan, N.; Wu, Q.; Cheng, Z.; Ling, K.; Gu, M.; Ma, C.; Gu, L.; An, Z.; Huang, W. Angew. Chem., Int. Ed. 2018, 57, 4005–4009. doi:10.1002/anie.201800697 |
| 5. | Cai, S.; Shi, H.; Li, J.; Gu, L.; Ni, Y.; Cheng, Z.; Wang, S.; Xiong, W.-W.; Li, L.; An, Z.; Huang, W. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1701244. doi:10.1002/adma.201701244 |
| 18. | Li, E.; Xu, X.; Li, H.; Zhang, H.; Xu, X.; Yuan, X.; Li, Y. Tetrahedron 2009, 65, 8961–8968. doi:10.1016/j.tet.2009.08.075 |
| 19. | Liao, Q.; Zhang, L.; Wang, F.; Li, S.; Xi, C. Eur. J. Org. Chem. 2010, 5426–5431. doi:10.1002/ejoc.201000665 |
| 29. | Damrath, M.; Caspers, L. D.; Duvinage, D.; Nachtsheim, B. J. Org. Lett. 2022, 24, 2562–2566. doi:10.1021/acs.orglett.2c00691 |
| 15. | Kajiyama, D.; Inoue, K.; Ishikawa, Y.; Nishiyama, S. Tetrahedron 2010, 66, 9779–9784. doi:10.1016/j.tet.2010.11.015 |
| 16. | Wang, Q.; Zhang, X.; Wang, P.; Gao, X.; Zhang, H.; Lei, A. Chin. J. Chem. 2021, 39, 143–148. doi:10.1002/cjoc.202000407 |
| 17. | Antonchick, A. P.; Samanta, R.; Kulikov, K.; Lategahn, J. Angew. Chem., Int. Ed. 2011, 50, 8605–8608. doi:10.1002/anie.201102984 |
| 36. | Kuczmera, T. J.; Dietz, A.; Boelke, A.; Nachtsheim, B. J. Beilstein J. Org. Chem. 2023, 19, 317–324. doi:10.3762/bjoc.19.27 |
| 13. | Umehara, A.; Shimizu, S.; Sasaki, M. ChemCatChem 2023, 15, e202201596. doi:10.1002/cctc.202201596 |
| 14. | Umehara, A.; Shimizu, S.; Sasaki, M. Adv. Synth. Catal. 2023, 365, 2367–2376. doi:10.1002/adsc.202300487 |
| 34. | Peng, X.; Li, L.; Ren, Y.; Xue, H.; Liu, J.; Wen, S.; Chen, J. Adv. Synth. Catal. 2020, 362, 2030–2038. doi:10.1002/adsc.201901460 |
| 6. | Liu, Y.; Guo, Y.; Ji, F.; Gao, D.; Song, C.; Chang, J. J. Org. Chem. 2016, 81, 4310–4315. doi:10.1021/acs.joc.6b00729 |
| 7. | Utaipan, T.; Athipornchai, A.; Suksamrarn, A.; Jirachotikoon, C.; Yuan, X.; Lertcanawanichakul, M.; Chunglok, W. J. Nat. Med. 2017, 71, 158–169. doi:10.1007/s11418-016-1045-6 |
| 8. | Nishiyama, T.; Matsuoka, A.; Honda, R.; Kitamura, T.; Hatae, N.; Choshi, T. Tetrahedron 2020, 76, 131110. doi:10.1016/j.tet.2020.131110 |
| 9. | Crabtree, R. H. ACS Sustainable Chem. Eng. 2017, 5, 4491–4498. doi:10.1021/acssuschemeng.7b00983 |
| 10. | You, X.; Zhu, D.; Lu, W.; Sun, Y.; Qiao, S.; Luo, B.; Du, Y.; Pi, R.; Hu, Y.; Huang, P.; Wen, S. RSC Adv. 2018, 8, 17183–17190. doi:10.1039/c8ra02939c |
| 11. | Kim, Y.; Yeom, M.; Lee, S.; Tae, J.; Kim, H. J.; Rhim, H.; Seong, J.; Choi, K. I.; Min, S.-J.; Choo, H. Bull. Korean Chem. Soc. 2018, 39, 1083–1089. doi:10.1002/bkcs.11555 |
| 12. | Kang, B.; Yasuno, Y.; Okamura, H.; Sakai, A.; Satoh, T.; Kuse, M.; Shinada, T. Bull. Chem. Soc. Jpn. 2020, 93, 993–999. doi:10.1246/bcsj.20200116 |
| 29. | Damrath, M.; Caspers, L. D.; Duvinage, D.; Nachtsheim, B. J. Org. Lett. 2022, 24, 2562–2566. doi:10.1021/acs.orglett.2c00691 |
| 33. | Zhu, D.; Liu, Q.; Luo, B.; Chen, M.; Pi, R.; Huang, P.; Wen, S. Adv. Synth. Catal. 2013, 355, 2172–2178. doi:10.1002/adsc.201300271 |
| 34. | Peng, X.; Li, L.; Ren, Y.; Xue, H.; Liu, J.; Wen, S.; Chen, J. Adv. Synth. Catal. 2020, 362, 2030–2038. doi:10.1002/adsc.201901460 |
| 35. | Wu, B.; Yoshikai, N. Angew. Chem., Int. Ed. 2015, 54, 8736–8739. doi:10.1002/anie.201503134 |
| 32. | Riedmüller, S.; Nachtsheim, B. J. Beilstein J. Org. Chem. 2013, 9, 1202–1209. doi:10.3762/bjoc.9.136 |
| 5. | Cai, S.; Shi, H.; Li, J.; Gu, L.; Ni, Y.; Cheng, Z.; Wang, S.; Xiong, W.-W.; Li, L.; An, Z.; Huang, W. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1701244. doi:10.1002/adma.201701244 |
| 29. | Damrath, M.; Caspers, L. D.; Duvinage, D.; Nachtsheim, B. J. Org. Lett. 2022, 24, 2562–2566. doi:10.1021/acs.orglett.2c00691 |
| 31. | Boelke, A.; Kuczmera, T. J.; Caspers, L. D.; Lork, E.; Nachtsheim, B. J. Org. Lett. 2020, 22, 7261–7266. doi:10.1021/acs.orglett.0c02593 |
| 20. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 21. | Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521–2523. doi:10.1039/b701864a |
| 22. | Grushin, V. V. Chem. Soc. Rev. 2000, 29, 315–324. doi:10.1039/a909041j |
| 23. | Liu, Z.; Luo, B.; Liu, X.; Hu, Y.; Wu, B.; Huang, P.; Wen, S. Eur. J. Org. Chem. 2016, 1110–1118. doi:10.1002/ejoc.201501544 |
| 24. | Zhu, D.; Wu, Z.; Liang, L.; Sun, Y.; Luo, B.; Huang, P.; Wen, S. RSC Adv. 2019, 9, 33170–33179. doi:10.1039/c9ra07288h |
| 25. | Zhu, D.; Wu, Z.; Luo, B.; Du, Y.; Liu, P.; Chen, Y.; Hu, Y.; Huang, P.; Wen, S. Org. Lett. 2018, 20, 4815–4818. doi:10.1021/acs.orglett.8b01969 |
| 26. | Vlasenko, Y. A.; Postnikov, P. S.; Trusova, M. E.; Shafir, A.; Zhdankin, V. V.; Yoshimura, A.; Yusubov, M. S. J. Org. Chem. 2018, 83, 12056–12070. doi:10.1021/acs.joc.8b01995 |
| 27. | Wang, M.; Fan, Q.; Jiang, X. Org. Lett. 2018, 20, 216–219. doi:10.1021/acs.orglett.7b03564 |
| 28. | Chatterjee, N.; Goswami, A. Eur. J. Org. Chem. 2017, 3023–3032. doi:10.1002/ejoc.201601651 |
| 29. | Damrath, M.; Caspers, L. D.; Duvinage, D.; Nachtsheim, B. J. Org. Lett. 2022, 24, 2562–2566. doi:10.1021/acs.orglett.2c00691 |
| 30. | Peng, X.; Rahim, A.; Peng, W.; Jiang, F.; Gu, Z.; Wen, S. Chem. Rev. 2023, 123, 1364–1416. doi:10.1021/acs.chemrev.2c00591 |
| 18. | Li, E.; Xu, X.; Li, H.; Zhang, H.; Xu, X.; Yuan, X.; Li, Y. Tetrahedron 2009, 65, 8961–8968. doi:10.1016/j.tet.2009.08.075 |
© 2024 Clamor et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.