Abstract
The homogeneous titanium- and dye-catalyzed as well as the heterogeneous semiconductor particle-catalyzed photohydroxymethylation of ketones by methanol were investigated in order to evaluate the most active photocatalyst system. Dialkoxytitanium dichlorides are the most efficient species for chemoselective hydroxymethylation of acetophenone as well as other aromatic and aliphatic ketones. Pinacol coupling is the dominant process for semiconductor catalysis and ketone reduction dominates the Ti(OiPr)4/methanol or isopropanol systems. Application of dilution effects on the TiO2 catalysis leads to an increase in hydroxymethylation at the expense of the pinacol coupling.

Graphical Abstract
Introduction
Stimulated by the principles of sustainable chemical synthesis and the progress in our understanding of catalytic and photoinduced electron-transfer processes, in recent years photoredox catalysis emerged as a new and powerful area for advanced synthesis [1-10]. There are numerous features that characterize an effective photoredox catalytic cycle: light absorption, charge separation, charge transport and annihilation as well as the use of appropriate sacrificial compounds such as electron and hole donors, elements that also appear in the natural photosynthesis, the role model for all applications. Catalysts that can function as light-absorbing and as redox-activating species must combine several features: redox-inactivity in the electronic ground states, optimal absorption properties in the near UV or visible region and appropriate redox activity in the excited states. Many potent photoredox catalysts with sufficient long-term stability are transition metal complexes with excited MLCT states that can be generated in the visible. Another important group of photocatalytic active compounds are semiconductor particles that absorb in the UV-A and near visible reagion. The widely used TiO2 has found numerous applications in photochemical water detoxification or surface purification because of its favourable excited-state redox properties [11]. In synthetic applications of semiconductor photocatalysis two clearly distinguishable reaction protocols were designated as type A and B by Kisch and co-workers [12-15]. In the type A process, two different products are formed from the initially formed electron–hole pair, one from the substrate radical cation that is formed from electron transfer to the semiconductor valence band hole, the other from the substrate radical anion that is formed from reduction by the semiconductor conduction band electron. Mostly, one of these steps consumes a sacrificial electron/hole donor. In type B photocatalysis, combination of the radical ions leads to a new product without the need of sacrificial components. The latter process proceeds with a high degree of atom economy [16]. We have recently demonstrated this for the azido-hydroperoxidation of alkenes, a convenient method for the synthesis of 1,2-amino alcohols [17,18]. In the field of C–C coupling reactions, the direct hydroxyalkylation of carbonyl compounds and carbonyl analogs is a demanding task because the α-CH activation of alcohols must occur in the presence of the acidic and nucleophilic hydroxy group. Thus, protection and deactivation of this group is necessary for thermal processes. In contrast to that, photochemical redox activation is possible in the presence of titanium(IV) catalysts [19-22]. As shown in a series of papers by Sato and coworkers, carbonyl compounds 1 as well as imines couple with methanol to give the 1,2-diols or 1,2-amino alcohols, respectively, when irradiated in the presence of stoichiometric or sub-stoichiometric amounts of titanium tetrachloride (Scheme 1). In order to run these reactions to completion, not less than 0.5 equivalents of TiCl4 were necessary which accounts for severe catalyst consumption. Furthermore, the addition of TiCl4 to methanol solutions is cumbersome and it is unclear what species is catalytically active. These processes have thermal counterparts in reduced titanium-mediated chemistry, e.g., the Ti(III)/t-BuOOH-mediated hydroxymethylation of imines [23,24].
![[1860-5397-10-114-i1]](/bjoc/content/inline/1860-5397-10-114-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photohydroxymethylation of carbonyl compounds and imines.
Scheme 1: Photohydroxymethylation of carbonyl compounds and imines.
In order to evaluate the nature of the active catalytic species in the photochemical homogenous titanium-catalyzed hydroxymethylation and to develop a truly catalytic process, we used a model reaction for catalyst screening (acetophenone/methanol) and applied the optimal homogenous reaction conditions involving titanium catalysis to other ketones and keto esters.
Results
Nature of the homogeneous catalytic titanium species
The original protocol for photocatalytic hydroxymethylation involves titanium tetrachloride in methanol as the reactive catalyst/donor mixture and carbonyl compounds as the acceptor components. During the exothermic dissolution process of TiCl4 in methanol with formation of gaseous HCl, a slightly yellowish solution is formed that, after irradiation with UV-A light, turns into a bluish solution indicating the formation of reduced titanium species. Obviously, ligand exchange reactions lead to a series of chloro- and methoxy-titanium complexes that have different catalytic activities. In order to simulate the different complex stages, we applied different monomeric titanium complexes of the type TiCln(OiPr)4−n (n = 0, 1, 2, 3) [25-27] in the model process, the irradiation of a solution of acetophenone (3) in methanol (Scheme 2). In the absence of any titanium species, photolysis at 254 and 300 nm, respectively, led only to trace amounts of the hydroxymethylation product 4 via a (triplet carbonyl) excited-state hydrogen-transfer process, obviously a sluggish process under these conditions (Table 1). In the presence of titanium complexes TiCln(OiPr)4−n, coupling and reduction products 4 and 6 were formed without pinacol 5 formation (Scheme 2).
![[1860-5397-10-114-i2]](/bjoc/content/inline/1860-5397-10-114-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Model process: photocatalyzed acetophenone/methanol reaction.
Scheme 2: Model process: photocatalyzed acetophenone/methanol reaction.
Table 1: Homogeneous sensitizer variation for the acetophenone model reaction.
| catalysta | irrad. wave-length (nm) | yield 4 (%)b | yield 5 (%)b | yield 6 (%)b |
|---|---|---|---|---|
| none |
300
254 |
<5c
<5c |
–
– |
–
– |
| TiCl4 |
300
254 |
33
34 |
–
– |
–
– |
| TiCl3OiPr |
300
254 |
31
47 |
–
– |
–
– |
| TiCl2(OiPr)2 |
300
254 |
0
60 |
–
– |
–
– |
| TiCl(OiPr)3 |
300
254 |
<5d
<5d |
–
– |
–
– |
| Ti(OiPr)4 |
300
254 |
–
– |
–
– |
–
46 |
|
Ti(OiPr)4/iPrOHe
Ti(OiPr)4/BF3d Ti(OiPr)4/AlCl3d |
254
254 254 |
–
45 49 |
–
– – |
43
– – |
a0.375 mmol catalyst in methanol (6 mL), 0.75 mmol acetophenone, irradiation time 72 h, rt; bisolated yields ; ctrace amounts detected in 1H NMR; d0.75 mmol added in methanol; e0.375 mmol cat. in isopropanol.
In the presence of the tetraalkoxide Ti(OiPr)4, only the reduction product 6 was detected which demonstrates that chlorotitanium complexes are crucial for the desired reaction path. The results from TiCl4 and TiCl3OiPr were nearly identical at both wavelengths whereas for TiCl2(OiPr)2 catalytic activity was preserved only for the 254 nm irradiation. These results show that different catalytically active species must exist that can be excited in different wavelength regions. TiCl(OiPr)3 showed no activity at all, meaning that no further ligand exchange to a tetraalkoxide did occur from this complex. The optimal results concerning reaction time and yields were observed for the TiCl2(OiPr)2 species. The apparently different catalytic activity of titanium tetraalkoxide complexes could be switched back to hydroxymethylation in the presence of additional strong Lewis acids such as AlCl3 or BF3. These compounds alone did not show catalytic activity in methanol, only in combination with Ti(OiPr)4. With the optimal homogenous reaction conditions in hand, we applied several other aromatic and aliphatic open chain and cyclic ketones as substrates (Scheme 3). Even benzophenone, a notorious pinacol forming substrate, gave moderate yields of the hydroxymethylation product 7. Among the aromatic ketones, para-fluoroacetophenone was the most reactive ketone. Excellent yields were obtained for 2-pentanone where the product 18 was isolated without purification after extraction. For comparison, the results from the Ti(OiPr)4 catalysis are included in Table 2. Additonally, the comparision with the heterogeneous TiO2 photolyses demonstrates that under semiconductor conditions pinacolization becomes the major path, but only for aromatic ketones. Aliphatic ketones did not show conversion under TiO2 photolyses.
![[1860-5397-10-114-i3]](/bjoc/content/inline/1860-5397-10-114-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Photocatalyzed acetophenone/methanol reaction: types I–III.
Scheme 3: Photocatalyzed acetophenone/methanol reaction: types I–III.
Table 2: Substrate variation under optimized conditions.
| entry | Substrate | TiCl2(OiPr)2a | TiO2 P25b | Ti(OiPr)4a | |
|---|---|---|---|---|---|
|
yield (%)c
type I |
yield (%)c
type I |
yield (%)c
type II |
yield (%)c
type III |
||
| 1 | Ph(CO)CH3 | 60 (4 [28]) | 1 (4) | 82 (5 [29]) | 46 (6 [30]) |
| 2 | Ph(CO)Ph | 28 (7 [31]) | 20 (7) | 71 (8 [24]) | 50 (9 [32]) |
| 3 | 2’-F-Ph((CO)CH3 | 56 (10) | – | 54 (11) | 40 (12 [33]) |
| 4 | 4’-MeO-Ph(CO)CH3 | – | – | 23 (13 [34]) | – |
| 5 | 4’-NO2-Ph(CO)CH3 | 19 (14 [35]) | – | – | |
| 6 | 4’-Me-Ph(CO)CH3 | 27 (15 [36]) | 6 (15) | 54 (16 [34]) | 16 (17 [37]) |
| 7 | C3H7(CO)CH3 | 92 (18 [38]) | – | – | |
| 8 | cyclohexanone | 38 (19 [39])d | – | – | |
a0.75 mmol ketone in methanol (6 mL), 0.5 equiv cat., irradiation time 72 h, λ = 254 nm (TiCl2(OiPr)2) and 300 nm (Ti(OiPr)4), rt; b0.75 mmol ketone in methanol (6 mL), 1.4 wt % TiO2 P25, irradiation time 48 h irradiation, λ = 350 nm, rt; cisolated yields; dmixture consisting of 9% 1,2-diol and 29% acetalization product with cyclohexanone (20).
These conditions were applied to keto ester substrates that feature an additional trapping site for the primary hydroxy group. We envisaged the formation of lactones from the corresponding hydroxymethylation products (Scheme 4). Methyl benzoylformate (21) gave a mixture of pinacol and the 1,2-diol without lactone formation. The higher homologs (Table 3, entries 2–4) resulted in the corresponding lactones with ring sizes of 5 and 6, respectively, with the δ-keto ester (Table 3, entry 4) leading to the δ-pentyrolactone and not the corresponding seven-membered lactone. The primary product from hydroxymethylation was also isolated from the reaction of the ε-keto ester 25 [40].
![[1860-5397-10-114-i4]](/bjoc/content/inline/1860-5397-10-114-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Photohydroxymethylation and subsequent lactonization of keto esters.
Scheme 4: Photohydroxymethylation and subsequent lactonization of keto esters.
Table 3: Photohydroxymethylation of keto esters: 1,2-diol and lactone formation.
a0.75 mmol keto ester in methanol (6 mL), 0.5 equiv TiCl2(OiPr)2, irradiation time 72 h, λ = 254 nm, rt; bisolated yields; cadditionally 11% of the corresponding pinacol 27 was formed.
Heterogeneous and dye-sensitized photocatalysis
The results with the semiconductor particle TiO2 P25 under low catalyst loading conditions appear in Table 2 for a series of ketone substrates. In order to explore the catalyst profile we tested other reaction conditions for the TiO2 catalysis, other metal-containing heterogeneous and homogeneous catalysts as well as the classical organic PET catalyst 9,10-dicyanoanthracene (DCA). The results are summarized in Table 4 for the model reaction of acetophenone in methanol (Scheme 5). Except for the Ru(bpy)3Cl2 system, all catalysts enabled a high degree of conversion and high yields of the pinacol 5 were detected. The hydroxymethylation product 4 was detected only in few experiments with a maximum yield of 6% from one TiO2 experiment. The best results were obtained for TiO2 P25 catalysis in the presence of molecular sieves (Table 4, entry 3). In all cases, the pinacol diastereoisomers were formed in nearly equal amounts. Also the change in irradiation wavelength did not lead to substantial changes in conversion and chemoselectivity. The formation of formaldehyde as the final oxidation product was proven qualitatively (colorless precipitation of polyformaldehyde was observed in most experiments) and by a GC–MS online detection of monomeric formaldehyde.
![[1860-5397-10-114-i5]](/bjoc/content/inline/1860-5397-10-114-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Model reaction for heterogeneous and dye-sensitized catalysis.
Scheme 5: Model reaction for heterogeneous and dye-sensitized catalysis.
Table 4: Heterogeneous sensitizer variation for model reaction in comparison with optimized homogeneous conditions for model process.
| entrya | catalyst | loading | conversion (%)b | yield 4 (%)b | yield 5 (%)b |
|---|---|---|---|---|---|
| 1 | TiO2 P25 | 2.8 wt % | 77 | – | 66 |
| 2 | 2.8 wt %c | 76 | – | 58 | |
| 3 | 2.8 wt %d | 96 | 3 | 83 | |
| 4 | 2.8 wt %e | 71 | – | 48 | |
| 5 | 2.2 wt % | 89 | 6 | 79 | |
| 6 | 1.4 wt % | 95 | 1 | 82 | |
| 7 | 3.3 wt % | 81 | 2 | 74 | |
| 8 | TiO2-pigment | 2.8 wt % | 89 | – | 69 |
| 9 | zinc white | 2.8 wt % | 79 | – | 69 |
| 10 | WO3, <100 nm | 2.8 wt % | 98 | – | 70 |
| 11 | 1.4 wt % | 95 | 1 | 75 | |
| 12 | Fe2O3, <50 nm | 2.8 wt % | 92 | – | 75 |
| 13 | 1.4 wt % | 95 | – | 80 | |
| 14 | ZnO, 6% Al doped, <50 nm | 2.8 wt % | 63 | – | 46 |
| 15 | InSnO, <50 nm | 2.8 wt % | 65 | – | 45 |
| 16 | Ir(ppy)3 | 2.5 mol % | 91 | – | 50 |
| 17 | 2.5 mol %f | 65 | – | 55 | |
| 18 | 0.5 mol % | 89 | – | 82 | |
| 19 | Ru(bpy)3Cl2 | 2.5 mol % | 27 | – | 1 |
| 20 | DCA | 2.5 mol % | 95 | 1 | 94 |
| 21 | 0.5 mol % | 96 | 1 | 92 | |
| 22 | none | – | <5% | – | – |
a0.75 mmol acetophenone in methanol (6 mL), irradiation time 24 h, λ = 350 nm, rt; bdetermined by GC; cλ = 300 nm; d600 mg molecular sieves were added; e100 µL H2O was added; fIr(ppy)3 regained from entry 16.
From these results, we reasoned that methanol oxidation is the primary event (e.g., from the DCA results) and acetophenone reduction follows resulting in the corresponding hydroxybenzyl radicals that couple to give the pinacol 5. Under this assumption, a decrease in ketone concentration should favour radical combination of the hydroxybenzyl and the (more reactive and easier oxidizable) hydroxymethyl radicals. As shown in Table 5, this is actually the case for the semiconductor particle TiO2 P25 catalysis. If acetophenone is added to the photolysis solution constantly over a period of 24 h, the absolute amount of hydroxymethylation product 4 can be increased to 40%.
Table 5: Chemoselectivity modification by application of a dilution effect.
| entry | drop rate | TiO2 in methanol (30 mL) | reaction time | conversion (%) | yield 4 (%) | yield 5 (%) |
|---|---|---|---|---|---|---|
| 1a | 0.28 mmol/h | 15 mg | 18.7 h | 92 | 36 | 52 |
| 2a,b | 0.28 mmol/h | 15 mg | 18.2 h | 82 | 25 | 38 |
| 3a | 0.21 mmol/h | 15 mg | 24 h | 95 | 33 | 55 |
| 4a | 0.21 mmol/h | 10 mg | 24 h | 89 | 13 | 60 |
| 5c | 0.16 mmol/h | 10 mg | 24 h | 96 | 40 | 50 |
a5 mmol acetophenone dissolved in methanol (10 mL) was slowly added to a TiO2 P25 (15 mg/ 10 mg) suspension in methanol (30 mL) irradiated (300 nm) at 15 °C; bTiO2 P25 suspension was cooled to −5 °C; c3.72 mmol acetophenone dissolved in methanol (10 mL) was slowly added to a TiO2 P25 (10 mg) suspension in methanol (30 mL) irradiated (300 nm) at 15 °C.
Discussion
Three product-forming routes can be assumed for the three classes of products observed in the study (Scheme 6): hydroxymethylation (route I), pinacolization (route II) and reduction/hydrogenation (route III). The crucial primary step for all processes is methanol oxidation [44]. By using appropriate reaction conditions, every route can be switched on exclusively. Hydroxymethylation is favoured if both hydroxyalkyl radicals are generated in close proximity by a coupled electron transfer/back transfer process. According to this expectation, the optimal conditions for route I are fulfilled for TiCl2(OR)2, a species that is capable of oxidizing methanol in the excited state and simultaneously acting as a ground-state Lewis acid that complexes the carbonyl compound. A much weaker Lewis acid such as Ti(OR)4 is capable of methanol oxidation but prefers hydrogen transfer at the first or second oxidation event. The pinacolization route II is favoured for heterogeneous and dye-catalyzed conditions. Interestingly, the combination of TiO2 P25 with an organic dye prefers largely the hydrogenation route III [45].
![[1860-5397-10-114-i6]](/bjoc/content/inline/1860-5397-10-114-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Product forming routes I to III for photoredox catalysis of methanol/carbonyl compounds.
Scheme 6: Product forming routes I to III for photoredox catalysis of methanol/carbonyl compounds.
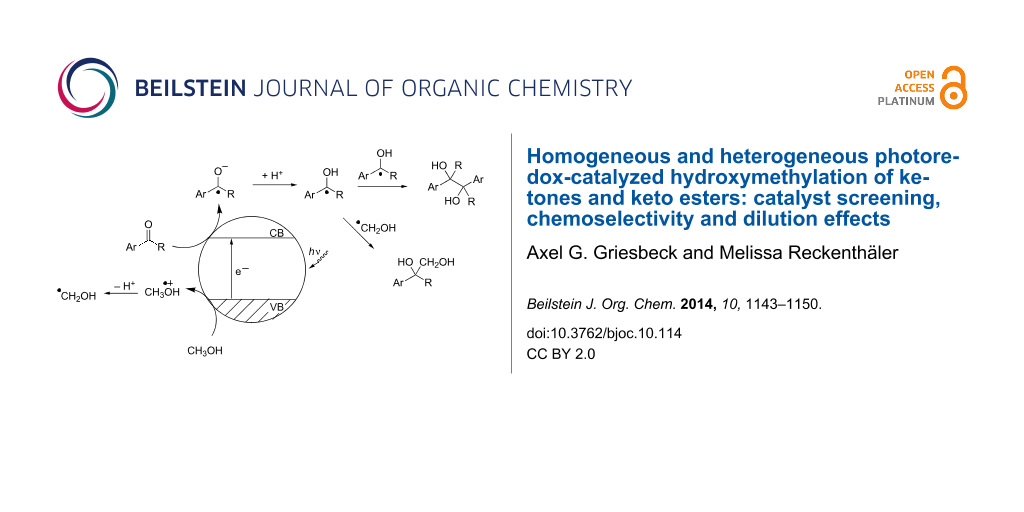
On the surface of the relatively large semiconductor particles, combination events are rare between the hydroxymethyl radical from methanol oxidation and the hydroxyalkyl radical from ketone reduction. Thus, further oxidation of the hydroxymethyl species to give methanol and another hydroxyalkyl radical is feasible. The combination of two hydroxyalkyl radicals is then dictated by diffusion kinetics (Scheme 7). The dilution experiments described in Table 5 indicate that the probability for pinacol formation is reduced by reducing the stationary concentration of the aromatic ketone. It was shown that hydroxymethyl radicals are formed from methanol during the photolysis of TiO2 in the absence of additional acceptor compounds with formation of hydrogen and eventually formation of formaldehyde [46]. Both hydrogen and formaldehyde were also detected in our experiments by gas-phase analysis. Thus, higher amounts of hydroxymethyl radicals can be produced under lower concentration of the acceptor ketone and the probability of hydroxybenzyl radical dimerization (i.e., route II) is disfavoured under these conditions.
![[1860-5397-10-114-i7]](/bjoc/content/inline/1860-5397-10-114-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Photoredox initiated steps on semiconductor particle surfaces, CB, VB = conduction and valence band.
Scheme 7: Photoredox initiated steps on semiconductor particle surfaces, CB, VB = conduction and valence band....
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 238.1 KB | Download |
References
-
Reckenthäler, M.; Griesbeck, A. G. Adv. Synth. Catal. 2013, 355, 2727–2744. doi:10.1002/adsc.201300751
Return to citation in text: [1] -
Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387–2403. doi:10.1039/c3ob40137e
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Zou, Y.-Q.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2013, 52, 11701–11703. doi:10.1002/anie.201307206
Return to citation in text: [1] -
König, B., Ed. Photoredox Catalysis; De Gruyter: Berlin/Boston, 2013.
Return to citation in text: [1] -
Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234
Return to citation in text: [1] -
Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x
Return to citation in text: [1] -
Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223
Return to citation in text: [1] -
Narayanam, J. M. R.; Jagan, M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] -
Niwa, T. J. Synth. Org. Chem., Jpn. 2010, 68, 1307–1308.
Return to citation in text: [1] -
Fujishima, A.; Rao, T. N.; Tryk, D. A. J. Photochem. Photobiol., C 2000, 1, 1–21. doi:10.1016/S1389-5567(00)00002-2
Return to citation in text: [1] -
Keck, H.; Schindler, W.; Knoch, F.; Kisch, H. Chem.–Eur. J. 1997, 3, 1638–1645. doi:10.1002/chem.19970031013
Return to citation in text: [1] -
Hörner, G.; Johne, P.; Künneth, R.; Twardzik, G.; Roth, H.; Clark, T.; Kisch, H. Chem.–Eur. J. 1999, 5, 208–217. doi:10.1002/(SICI)1521-3765(19990104)5:1<208::AID-CHEM208>3.0.CO;2-0
Return to citation in text: [1] -
Hopfner, M.; Weiß, H.; Meissner, D.; Heinemann, F. W.; Kisch, H. Photochem. Photobiol. Sci. 2002, 1, 696–703. doi:10.1039/b204569a
Return to citation in text: [1] -
Kisch, H. Angew. Chem., Int. Ed. 2013, 52, 812–847. doi:10.1002/anie.201201200
Return to citation in text: [1] -
Trost, B. M. Acc. Chem. Res. 2002, 35, 695–705. doi:10.1021/ar010068z
Return to citation in text: [1] -
Griesbeck, A. G.; Reckenthäler, M.; Uhlig, J. Photochem. Photobiol. Sci. 2010, 9, 775–778. doi:10.1039/c0pp00033g
Return to citation in text: [1] -
Griesbeck, A. G.; Steinwascher, J.; Reckenthäler, M.; Uhlig, J. Res. Chem. Intermed. 2013, 39, 33–42. doi:10.1007/s11164-012-0629-3
Return to citation in text: [1] -
Sato, T.; Izumi, G.; Imamura, T. J. Chem. Soc., Perkin Trans. 1 1976, 788–791. doi:10.1039/p19760000788
Return to citation in text: [1] -
Sato, T.; Yoshiie, S.; Imamura, T.; Hasegawa, K.; Miyahara, M.; Yamamura, S.; Ito, O. Bull. Chem. Soc. Jpn. 1977, 50, 2714–2730. doi:10.1246/bcsj.50.2714
Return to citation in text: [1] -
Sato, T.; Yamaguchi, S.-i.; Kaneko, H. Tetrahedron Lett. 1979, 20, 1863–1864. doi:10.1016/S0040-4039(01)86861-1
Return to citation in text: [1] -
Sato, T.; Kaneko, H.; Yamaguchi, S. J. Org. Chem. 1980, 45, 3778–3782. doi:10.1021/jo01307a011
Return to citation in text: [1] -
Rossi, B.; Prosperini, S.; Pastori, N.; Clerici, A.; Punta, C. Molecules 2012, 17, 14700–14732. doi:10.3390/molecules171214700
Return to citation in text: [1] -
Clerici, A.; Ghilardi, A.; Pastori, N.; Punta, C.; Porta, O. Org. Lett. 2012, 10, 5063–5066. doi:10.1021/ol802244n
Return to citation in text: [1] [2] -
Kamigaito, M.; Sawamoto, M.; Higashimura, T. Macromolecules 1995, 28, 5671–5675. doi:10.1021/ma00120a037
Return to citation in text: [1] -
Birse, E. F.; McKenzie, A.; Murray, A. W. J. Chem. Soc., Perkin Trans. 1 1988, 1039–1046. doi:10.1039/p19880001039
Return to citation in text: [1] -
Reetz, M. T.; Westermann, J.; Steinbach, R.; Wenderoth, B.; Peter, R.; Ostarek, R.; Maus, S. Chem. Ber. 1985, 118, 1421–1440. doi:10.1002/cber.19851180412
Return to citation in text: [1] -
Wang, A.; Jiang, H. J. Org. Chem. 2010, 75, 2321–2326. doi:10.1021/jo100125q
Return to citation in text: [1] -
Stocker, J. H.; Kern, D. H. J. Org. Chem. 1968, 33, 291–294. doi:10.1021/jo01265a057
Return to citation in text: [1] -
Lee, J. M.; Park, E. J.; Cho, S. H.; Chang, S. J. Am. Chem. Soc. 2008, 130, 7824–7825. doi:10.1021/ja8031218
Return to citation in text: [1] -
Ortiz, J.; Guijarro, A.; Yus, M. Eur. J. Org. Chem. 1999, 3005–3012. doi:10.1002/(SICI)1099-0690(199911)1999:11<3005::AID-EJOC3005>3.0.CO;2-7
Return to citation in text: [1] -
Karthikeyan, J.; Jeganmohan, M.; Cheng, C.-H. Chem.–Eur. J. 2010, 16, 8989–8992. doi:10.1002/chem.201001160
Return to citation in text: [1] -
Johnson, T. C.; Totty, W. G.; Wills, M. Org. Lett. 2012, 14, 5230–5233. doi:10.1021/ol302354z
Return to citation in text: [1] -
Uchiyama, M.; Matsumoto, Y.; Nakamura, S.; Ohwada, T.; Kobayashi, N.; Yamashita, N.; Matsumiya, A.; Sakamoto, T. J. Am. Chem. Soc. 2004, 126, 8755–8759. doi:10.1021/ja039674a
Return to citation in text: [1] [2] -
Cleij, M.; Archelas, A.; Furstoss, R. J. Org. Chem. 1999, 64, 5029–5035. doi:10.1021/jo982101+
Return to citation in text: [1] -
Chavan, S. P.; Khatod, H. S. Tetrahedron: Asymmetry 2012, 23, 1410–1415. doi:10.1016/j.tetasy.2012.09.008
Return to citation in text: [1] -
Li, J.; Wang, C.; Xue, D.; Wei, Y.; Xiao, J. Green Chem. 2013, 15, 2685–2689. doi:10.1039/c3gc41133h
Return to citation in text: [1] -
DeGoey, D. A.; Chen, H.-J.; Flosi, W. J.; Grampovnik, D. J.; Yeung, C. M.; Klein, L. L.; Kempf, D. J. J. Org. Chem. 2002, 67, 5445–5453. doi:10.1021/jo0162890
Return to citation in text: [1] -
Itami, K.; Kamei, T.; Mitsudo, K.; Nokami, T.; Yoshida, J.-i. J. Org. Chem. 2001, 66, 3970–3976. doi:10.1021/jo015528g
Return to citation in text: [1] -
Ito, S.; Matsumoto, M. J. Org. Chem. 1983, 48, 1133–1135. doi:10.1021/jo00155a051
Return to citation in text: [1] -
Wang, Z.-M.; Sharpless, K. B. Synlett 1993, 603–604. doi:10.1055/s-1993-22547
Return to citation in text: [1] -
Eliel, E. L.; Bai, X.; Ohwa, M. J. Chin. Chem. Soc. 2000, 47, 63–70.
Return to citation in text: [1] -
Sato, T.; Maeno, H.; Noro, T.; Fujisawa, T. Chem. Lett. 1988, 17, 1739–1742. doi:10.1246/cl.1988.1739
Return to citation in text: [1] -
Bowker, M. Green Chem. 2011, 13, 2235–2246. doi:10.1039/c1gc00022e
Return to citation in text: [1] -
Kohtani, S.; Nishioka, S.; Yoshioka, E.; Miyabe, H. Catal. Commun. 2014, 43, 61–65. doi:10.1016/j.catcom.2013.09.006
Return to citation in text: [1] -
Micic, O. I.; Zhang, Y.; Cromack, K. R.; Trifunac, A. D.; Thurnauer, M. C. J. Phys. Chem. 1993, 97, 13284–13288. doi:10.1021/j100152a036
Return to citation in text: [1]
| 45. | Kohtani, S.; Nishioka, S.; Yoshioka, E.; Miyabe, H. Catal. Commun. 2014, 43, 61–65. doi:10.1016/j.catcom.2013.09.006 |
| 46. | Micic, O. I.; Zhang, Y.; Cromack, K. R.; Trifunac, A. D.; Thurnauer, M. C. J. Phys. Chem. 1993, 97, 13284–13288. doi:10.1021/j100152a036 |
| 1. | Reckenthäler, M.; Griesbeck, A. G. Adv. Synth. Catal. 2013, 355, 2727–2744. doi:10.1002/adsc.201300751 |
| 2. | Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387–2403. doi:10.1039/c3ob40137e |
| 3. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 4. | Zou, Y.-Q.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2013, 52, 11701–11703. doi:10.1002/anie.201307206 |
| 5. | König, B., Ed. Photoredox Catalysis; De Gruyter: Berlin/Boston, 2013. |
| 6. | Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234 |
| 7. | Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x |
| 8. | Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223 |
| 9. | Narayanam, J. M. R.; Jagan, M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 10. | Niwa, T. J. Synth. Org. Chem., Jpn. 2010, 68, 1307–1308. |
| 17. | Griesbeck, A. G.; Reckenthäler, M.; Uhlig, J. Photochem. Photobiol. Sci. 2010, 9, 775–778. doi:10.1039/c0pp00033g |
| 18. | Griesbeck, A. G.; Steinwascher, J.; Reckenthäler, M.; Uhlig, J. Res. Chem. Intermed. 2013, 39, 33–42. doi:10.1007/s11164-012-0629-3 |
| 33. | Johnson, T. C.; Totty, W. G.; Wills, M. Org. Lett. 2012, 14, 5230–5233. doi:10.1021/ol302354z |
| 34. | Uchiyama, M.; Matsumoto, Y.; Nakamura, S.; Ohwada, T.; Kobayashi, N.; Yamashita, N.; Matsumiya, A.; Sakamoto, T. J. Am. Chem. Soc. 2004, 126, 8755–8759. doi:10.1021/ja039674a |
| 12. | Keck, H.; Schindler, W.; Knoch, F.; Kisch, H. Chem.–Eur. J. 1997, 3, 1638–1645. doi:10.1002/chem.19970031013 |
| 13. | Hörner, G.; Johne, P.; Künneth, R.; Twardzik, G.; Roth, H.; Clark, T.; Kisch, H. Chem.–Eur. J. 1999, 5, 208–217. doi:10.1002/(SICI)1521-3765(19990104)5:1<208::AID-CHEM208>3.0.CO;2-0 |
| 14. | Hopfner, M.; Weiß, H.; Meissner, D.; Heinemann, F. W.; Kisch, H. Photochem. Photobiol. Sci. 2002, 1, 696–703. doi:10.1039/b204569a |
| 15. | Kisch, H. Angew. Chem., Int. Ed. 2013, 52, 812–847. doi:10.1002/anie.201201200 |
| 24. | Clerici, A.; Ghilardi, A.; Pastori, N.; Punta, C.; Porta, O. Org. Lett. 2012, 10, 5063–5066. doi:10.1021/ol802244n |
| 11. | Fujishima, A.; Rao, T. N.; Tryk, D. A. J. Photochem. Photobiol., C 2000, 1, 1–21. doi:10.1016/S1389-5567(00)00002-2 |
| 32. | Karthikeyan, J.; Jeganmohan, M.; Cheng, C.-H. Chem.–Eur. J. 2010, 16, 8989–8992. doi:10.1002/chem.201001160 |
| 30. | Lee, J. M.; Park, E. J.; Cho, S. H.; Chang, S. J. Am. Chem. Soc. 2008, 130, 7824–7825. doi:10.1021/ja8031218 |
| 25. | Kamigaito, M.; Sawamoto, M.; Higashimura, T. Macromolecules 1995, 28, 5671–5675. doi:10.1021/ma00120a037 |
| 26. | Birse, E. F.; McKenzie, A.; Murray, A. W. J. Chem. Soc., Perkin Trans. 1 1988, 1039–1046. doi:10.1039/p19880001039 |
| 27. | Reetz, M. T.; Westermann, J.; Steinbach, R.; Wenderoth, B.; Peter, R.; Ostarek, R.; Maus, S. Chem. Ber. 1985, 118, 1421–1440. doi:10.1002/cber.19851180412 |
| 31. | Ortiz, J.; Guijarro, A.; Yus, M. Eur. J. Org. Chem. 1999, 3005–3012. doi:10.1002/(SICI)1099-0690(199911)1999:11<3005::AID-EJOC3005>3.0.CO;2-7 |
| 23. | Rossi, B.; Prosperini, S.; Pastori, N.; Clerici, A.; Punta, C. Molecules 2012, 17, 14700–14732. doi:10.3390/molecules171214700 |
| 24. | Clerici, A.; Ghilardi, A.; Pastori, N.; Punta, C.; Porta, O. Org. Lett. 2012, 10, 5063–5066. doi:10.1021/ol802244n |
| 19. | Sato, T.; Izumi, G.; Imamura, T. J. Chem. Soc., Perkin Trans. 1 1976, 788–791. doi:10.1039/p19760000788 |
| 20. | Sato, T.; Yoshiie, S.; Imamura, T.; Hasegawa, K.; Miyahara, M.; Yamamura, S.; Ito, O. Bull. Chem. Soc. Jpn. 1977, 50, 2714–2730. doi:10.1246/bcsj.50.2714 |
| 21. | Sato, T.; Yamaguchi, S.-i.; Kaneko, H. Tetrahedron Lett. 1979, 20, 1863–1864. doi:10.1016/S0040-4039(01)86861-1 |
| 22. | Sato, T.; Kaneko, H.; Yamaguchi, S. J. Org. Chem. 1980, 45, 3778–3782. doi:10.1021/jo01307a011 |
| 29. | Stocker, J. H.; Kern, D. H. J. Org. Chem. 1968, 33, 291–294. doi:10.1021/jo01265a057 |
| 34. | Uchiyama, M.; Matsumoto, Y.; Nakamura, S.; Ohwada, T.; Kobayashi, N.; Yamashita, N.; Matsumiya, A.; Sakamoto, T. J. Am. Chem. Soc. 2004, 126, 8755–8759. doi:10.1021/ja039674a |
| 35. | Cleij, M.; Archelas, A.; Furstoss, R. J. Org. Chem. 1999, 64, 5029–5035. doi:10.1021/jo982101+ |
| 36. | Chavan, S. P.; Khatod, H. S. Tetrahedron: Asymmetry 2012, 23, 1410–1415. doi:10.1016/j.tetasy.2012.09.008 |
| 43. | Sato, T.; Maeno, H.; Noro, T.; Fujisawa, T. Chem. Lett. 1988, 17, 1739–1742. doi:10.1246/cl.1988.1739 |
| 41. | Wang, Z.-M.; Sharpless, K. B. Synlett 1993, 603–604. doi:10.1055/s-1993-22547 |
| 39. | Itami, K.; Kamei, T.; Mitsudo, K.; Nokami, T.; Yoshida, J.-i. J. Org. Chem. 2001, 66, 3970–3976. doi:10.1021/jo015528g |
| 40. | Ito, S.; Matsumoto, M. J. Org. Chem. 1983, 48, 1133–1135. doi:10.1021/jo00155a051 |
| 37. | Li, J.; Wang, C.; Xue, D.; Wei, Y.; Xiao, J. Green Chem. 2013, 15, 2685–2689. doi:10.1039/c3gc41133h |
| 38. | DeGoey, D. A.; Chen, H.-J.; Flosi, W. J.; Grampovnik, D. J.; Yeung, C. M.; Klein, L. L.; Kempf, D. J. J. Org. Chem. 2002, 67, 5445–5453. doi:10.1021/jo0162890 |
© 2014 Griesbeck and Reckenthäler; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)