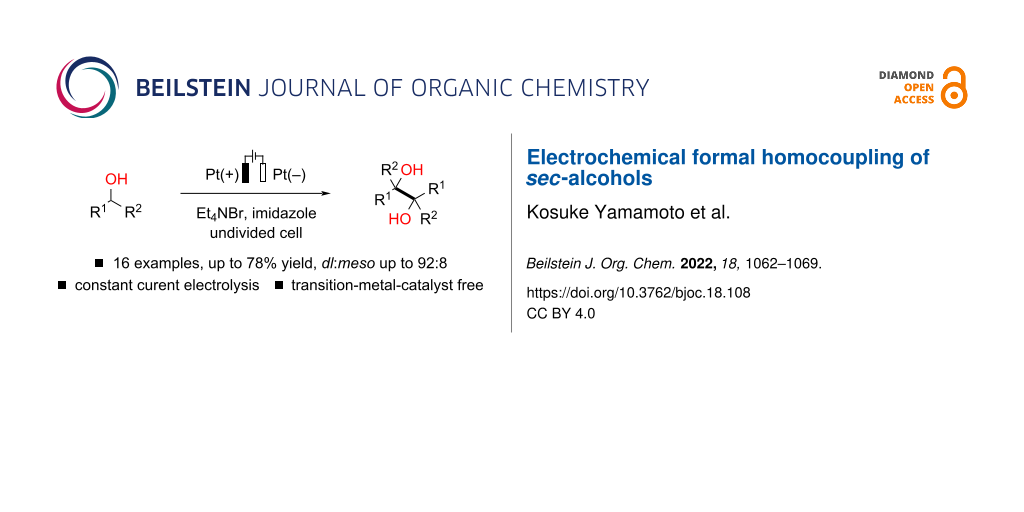
Abstract
Electrochemical pinacol coupling of carbonyl compounds in an undivided cell with a sacrificial anode would be a promising approach toward synthetically valuable vic-1,2-diol scaffolds without using low-valent metal reductants. However, sacrificial anodes produce an equimolar amount of metal waste, which may be a major issue in terms of sustainable chemistry. Herein, we report a sacrificial anode-free electrochemical protocol for the synthesis of pinacol-type vic-1,2-diols from sec-alcohols, namely benzyl alcohol derivatives and ethyl lactate. The corresponding vic-1,2-diols are obtained in moderate to good yields, and good to high levels of stereoselectivity are observed for sec-benzyl alcohol derivatives. The present transformations smoothly proceed in a simple undivided cell under constant current conditions without the use of external chemical oxidants/reductants, and transition-metal catalysts.

Graphical Abstract
Introduction
Carbon–carbon bond formation is one of the most fundamental and important reactions in synthetic organic chemistry. Reductive coupling of carbonyl compounds known as pinacol coupling would be a powerful method to construct vic-1,2-diol scaffolds through C–C bond formation [1,2]. Such scaffolds are widely utilized as versatile building blocks in the synthesis of biologically active compounds [3-7], chiral auxiliaries [8,9], and chiral ligands [10-13]. Traditional pinacol coupling reactions are performed with a stoichiometric or even excess amount of low-valent metal reductants, such as Al, Ti, V, Zn, and Sm (Scheme 1a). Although these protocols have proven to be a reliable strategy to access vic-1,2-diols, producing a large amount of metal waste may be a major drawback especially in a large-scale synthesis. Thus, the improved procedures using a catalytic amount of transition-metal reductants have been developed, but stoichiometric silicon electrophiles and co-reductants such as Zn were commonly required to complete the catalytic cycle [14]. More recently, visible light-mediated pinacol coupling reactions have been disclosed by several groups [15-18]. In addition to the reductive coupling of carbonyl compounds, oxidative homocoupling reactions of benzyl alcohols under transition metal- or semiconductor-based photoredox catalysis have been demonstrated as attractive approaches to access vic-1,2-diols [19-23].
![[1860-5397-18-108-i1]](/bjoc/content/inline/1860-5397-18-108-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Strategies for the synthesis of vic-1,2-diols.
Scheme 1: Strategies for the synthesis of vic-1,2-diols.
Electroorganic chemistry has been recognized as an environmentally benign and powerful strategy to promote redox reactions using electricity as a traceless oxidant or reductant [24-28]. Electrochemical pinacol coupling would be a promising alternative to avoid the use of low-valent metal reductants. The reported methods commonly carried out in a divided cell [29-34] or an undivided cell with sacrificial anodes [35], such as Al, Mg, and Sn, to prevent undesired oxidative reactions (Scheme 1b) [36-39]. While sacrificial anodes enable the reactions to be performed with a simple and user-friendly undivided cell set-up, consuming the anode material with generating stoichiometric metal waste may be a serious issue in terms of green and sustainable chemistry. Thus, the development of a sacrificial anode-free process such as paired electrolysis would be highly desirable [40-44]. The group of Wang recently reported the sacrificial anode-free electroreduction of benzophenone derivatives to afford vic-1,2-diols using over-stoichiometric NaN3 under acidic conditions, but appropriate precautions should be taken for in situ-generated explosive and toxic HN3 [45]. Kim et al. reported the formation of vic-1,2-diols in the sacrificial anode-free electrocarboxylation of 1-phenylethanol and benzyl alcohol which involves tetramethylpiperidine-1-oxyl-mediated alcohol oxidation as an anodic event [46]. However, vic-1,2-diols were obtained only as minor products and formal homocoupling of benzhydrol did not occur under Kim’s reaction conditions. Thus, the development of an environmentally benign and efficient electrochemical protocol to access vic-1,2-diols would be still highly desirable. Herein, we report the sacrificial anode-free electrochemical synthesis of vic-1,2-diols through the formal homocoupling of sec-alcohols using platinum electrodes in an undivided cell (Scheme 1c).
Results and Discussion
We commenced the optimization study for the electrochemical formal homocoupling of sec-alcohols by using 1-phenylethanol (1a) as a model substrate. The results are summarized in Table 1. The electrolysis was carried out using an undivided cell in the presence of Et4NBr as an electrolyte with a mixed solvent of MeCN and H2O under air atmosphere. When 4 F/mol of electricity was passed through the reaction mixture using two platinum electrodes at 0 ºC, the corresponding pinacol-type product 2a was obtained in 58% yield with an 89:11 ratio of dl and meso isomers (Table 1, entry 1). Acetophenone (3a) was also formed in 32% yield under the reaction conditions described in entry 1. Using different electrode materials such as Ni, Zn, and graphite as cathode did not improve the yield of 2a (Table 1, entries 2–4). The present reaction proceeded in the presence of quaternary ammonium salts with different counter anions including the BF4 anion, and Et4NBr was found to be the preferable electrolyte among them (Table 1, entry 1 vs entries 5–7). Next, we examined the effect of acidic and basic additives on the reaction outcome. While the use of Mg(OTf)2, HCO2H, or 2,6-lutidine resulted in reduced reaction efficiency, imidazole exhibited the positive effect on the product yield, providing 2a in 72% yield (Table 1, entries 8–11). Addition of H2O was crucial to obtain 2a in a high yield, and we chose 125 μL of H2O as the optimal volume for the present transformation (Table 1, entries 11–13). The reaction under inert atmosphere did not improve the yield of 2a (Table 1, entry 14).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-108-i7.svg?max-width=637&scale=1.0)
|
||||||
| entry | (+)-(−) | electrolyte | additive | yield (%)b | dl:meso for 2ac | |
| 2a | 3a | |||||
| 1 | Pt-Pt | Et4NBr | – | 58 | 32 | 89:11 |
| 2 | Pt-Ni | Et4NBr | – | 5 | 49 | 90:10 |
| 3 | Pt-Zn | Et4NBr | – | 24 | 20 | 89:11 |
| 4 | Pt-C | Et4NBr | – | 28 | 40 | 89:11 |
| 5 | Pt-Pt | Et4NCl | – | 39 | 30 | 90:10 |
| 6 | Pt-Pt | Et4NI | – | 10 | 5 | 90:10 |
| 7 | Pt-Pt | Et4NBF4 | – | 46 | 32 | 90:10 |
| 8 | Pt-Pt | Et4NBr | Mg(OTf)2 | 26 | 64 | 90:10 |
| 9 | Pt-Pt | Et4NBr | HCO2H | 39 | 33 | 89:11 |
| 10 | Pt-Pt | Et4NBr | 2,6-lutidine | 24 | 41 | 89:11 |
| 11 | Pt-Pt | Et4NBr | imidazole | 72 | 24 | 90:10 |
| 12d | Pt-Pt | Et4NBr | imidazole | 78 (78) | 8 | 90:10 |
| 13e | Pt-Pt | Et4NBr | imidazole | 39 | 11 | 77:23 |
| 14d,f | Pt-Pt | Et4NBr | imidazole | 77 | 12 | 90:10 |
aReaction conditions: 1a (1.0 mmol), electrolyte (0.1 equiv), additive (0.05 equiv), MeCN (5 mL), H2O (250 μL), 50 mA constant current (cc), 4 F/mol, 0 °C, under air. bDetermined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. The number in parentheses refers to the isolated yield. cDetermined by 1H NMR analysis. dH2O (125 μL). eWithout H2O. fUnder Ar.
With the optimized conditions in hand, the substrate scope of the present transformation was investigated as shown in Scheme 2. Various 1-arylethanol derivatives were firstly examined. Substrates bearing p-methyl (1b) or p-tert-butyl (1c) groups afforded the desired products 2b and 2c in moderate yields. Halogen substituents such as fluorine (1d) and chlorine (1e) atoms were tolerated under the present reaction conditions providing 2d and 2e in 70% and 57% yields, respectively, with high diastereoselectivities. Substrates having electron-withdrawing groups such as ester (1f) and trifluoromethyl (1g) on the para-position of the aryl moiety afforded the desired products in good yields (2f and 2g). On the other hand, the reaction of 1-(4-cyanophenyl)ethanol (1h) resulted in a decrease in both the yield and the dl:meso ratio. While steric hindrance of substituents on the meta-position of the aryl moiety did not impede the present transformation (2i and 2j), the ortho-substituted substrate 1k gave 2k in a less satisfactory yield but with good diastereoselectivity. 1-Phenyl-1-propanol (1l) was successfully transformed into the desired product 2l in a moderate yield. In addition, ethyl lactate (1m) provided the corresponding vic-1,2-diol 2m in 60% yield but with low diastereoselectivity [47]. Benzhydrol derivatives (1n–p) were found to be good substrates for the present reaction, affording the corresponding benzopinacols (2n–p) in good yields after the passage of 8 F/mol in a mixed solvent of MeCN/MeOH.
![[1860-5397-18-108-i2]](/bjoc/content/inline/1860-5397-18-108-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope. Reaction conditions: 1 (1.0 mmol), Et4NBr (0.1 equiv), imidazole (0.05 equiv), MeCN (5 mL), H2O (125 μL), 50 mA cc, 4 F/mol, 0 °C, under air. a100 mA cc. b6 F/mol, imidazole (0.075 equiv). c6 F/mol. d8 F/mol, imidazole (0.1 equiv) e8 F/mol, MeCN/MeOH (4:1, 5 mL) without H2O.
Scheme 2: Substrate scope. Reaction conditions: 1 (1.0 mmol), Et4NBr (0.1 equiv), imidazole (0.05 equiv), MeC...
Next, we examined the possibility to extend the present process to the cross-coupling reaction of two different benzyl alcohols (Scheme 3). Pleasingly, the reaction using a 1:1 mixture of 1a and 1f under the standard reaction conditions provided the cross-coupling product 2af (dr = 94:6) together with the homocoupling products 2a and 2f.
![[1860-5397-18-108-i3]](/bjoc/content/inline/1860-5397-18-108-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Investigation of cross-coupling reaction.
Scheme 3: Investigation of cross-coupling reaction.
To demonstrate the scalability of the present electrochemical transformation, a large-scale experiment was performed as shown in Scheme 4. The formal homocoupling of 1a smoothly proceeded on a 10 mmol scale to provide the desired product in 72% yield under slightly modified reaction conditions.
![[1860-5397-18-108-i4]](/bjoc/content/inline/1860-5397-18-108-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
In order to gain insight into the present reaction, several control experiments were conducted as shown in Scheme 5. When acetophenone (3a) was used as a starting material under the standard reaction conditions, vic-1,2-diol 2a and 3a were obtained in 39% and 52% yields, respectively, and 1-phenylethanol (1a) was not observed in this reaction (Scheme 5a). The dl:meso ratio of 2a was identical compared with that observed in the reaction using 1a as the starting material. This observation indicated that ketone 3a would be the intermediate in the present transformation. The reaction in the absence of imidazole also proceeded to afford 2a in a somewhat lower yield with a high diastereoselectivity. In both cases, the reaction proceeded with the good mass balance of 2a and 3a. On the other hand, the reaction without adding water resulted in a decrease in the dl:meso ratio of 2a, and ketone 3a was transformed into unidentified byproducts. When dl-2a was subjected to the present reaction conditions, oxidative C–C bond cleavage of dl-2a proceeded to give the corresponding ketone 3a (Scheme 5b) [48]. Recovered 2a was found to be a mixture of dl and meso isomers, indicating that homocoupling of in situ-generated ketone 3a occurred under the reaction conditions. While ketone 3a was obtained in a higher yield when the reaction was performed in the absence of imidazole, a lower yield of 3a and a poor mass balance were observed in the reaction without adding water. These results indicate that imidazole may suppress the formation of the ketone from the corresponding vic-1,2-diol. Water may play a role as a proton source to facilitate the formation of the protonated ketyl radical through a concerted proton-electron transfer toward the ketone or smooth protonation of the radical anion species, which readily dimerize to vic-1,2-diol 2a [46,49]. The addition of water may be also important to achieve high diastereoselectivity in the present reaction.
![[1860-5397-18-108-i5]](/bjoc/content/inline/1860-5397-18-108-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Control experiments. aDetermined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. bDetermined by 1H NMR analysis.
Scheme 5: Control experiments. aDetermined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. b...
On the basis of the results of the control experiments, a plausible reaction mechanism is depicted in Scheme 6. Initially, sec-alcohol 1 is oxidized by an anodically generated Br+ species to provide the corresponding ketone 3. Then, ketone 3 undergoes electrochemical pinacol coupling to form vic-1,2-diol 2. Overoxidation of compound 2 could proceed under the reaction conditions to reproduce ketone 3, which could be transformed again into 2. Initial screening of electrolytes indicated that direct oxidation of sec-alcohol 1 to ketone 3 could also proceed under the present reaction conditions.
![[1860-5397-18-108-i6]](/bjoc/content/inline/1860-5397-18-108-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have developed the sacrificial anode-free electrochemical protocol for the synthesis of vic-1,2-diols from sec-alcohols without external chemical oxidants or reductants. The present reaction smoothly proceeded in a simple undivided cell with platinum electrodes under constant current conditions, affording pinacol-type products in moderate to good yields with good to high diastereoselectivities. The successful large-scale experiment showed the potential synthetic utility of this transformation. Further investigations of the reaction mechanism are currently underway in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental procedure, characterization data, and copies of NMR spectra of the products. | ||
| Format: PDF | Size: 2.6 MB | Download |
Funding
This work was supported by the Japan Society of Promotion for the Sciences (JSPS) Grant-in-Aid for Scientific Research (19K05459, 19K16317, and 18K06582). The spectral data were collected with the research equipment shared in the MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting introduction of the new sharing system JPMXS0422500320).
References
-
Suzuki, K.; Tamiya, M. Pinacol Coupling Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 3, pp 580–620. doi:10.1016/b978-0-08-097742-3.00316-5
Return to citation in text: [1] -
Chatterjee, A.; Joshi, N. N. Tetrahedron 2006, 62, 12137–12158. doi:10.1016/j.tet.2006.09.002
Return to citation in text: [1] -
Ogata, M.; Matsumoto, H.; Takahashi, K.; Shimizu, S.; Kida, S.; Murabayashi, A.; Shiro, M.; Tawara, K. J. Med. Chem. 1987, 30, 1054–1068. doi:10.1021/jm00389a016
Return to citation in text: [1] -
Nicolaou, K. C.; Yang, Z.; Liu, J. J.; Ueno, H.; Nantermet, P. G.; Guy, R. K.; Claiborne, C. F.; Renaud, J.; Couladouros, E. A.; Paulvannan, K.; Sorensen, E. J. Nature 1994, 367, 630–634. doi:10.1038/367630a0
Return to citation in text: [1] -
Mateo, C.; Álvarez, R.; Pérez-Melero, C.; Peláez, R.; Medarde, M. Bioorg. Med. Chem. Lett. 2007, 17, 6316–6320. doi:10.1016/j.bmcl.2007.08.075
Return to citation in text: [1] -
Singh, P.; Mittal, A.; Kaur, S.; Holzer, W.; Kumar, S. Org. Biomol. Chem. 2008, 6, 2706–2712. doi:10.1039/b803608j
Return to citation in text: [1] -
Werner, B.; Kalesse, M. Org. Lett. 2017, 19, 1524–1526. doi:10.1021/acs.orglett.7b00288
Return to citation in text: [1] -
Alexakis, A.; Mangeney, P. Tetrahedron: Asymmetry 1990, 1, 477–511. doi:10.1016/s0957-4166(00)80536-4
Return to citation in text: [1] -
Fujioka, H.; Kita, Y. Asymmetric Synthesis Using Chiral Acetals from C2-Symmetric Diols and Its Application to Natural Product Synthesis. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, Netherlands, 1994; Vol. 14, pp 469–516. doi:10.1016/b978-0-444-81780-8.50018-1
Return to citation in text: [1] -
Fleischer, R.; Braun, M. Synlett 1998, 1441–1443. doi:10.1055/s-1998-1953
Return to citation in text: [1] -
Stodulski, M.; Jaźwiński, J.; Mlynarski, J. Eur. J. Org. Chem. 2008, 5553–5562. doi:10.1002/ejoc.200800726
Return to citation in text: [1] -
Hashimoto, T.; Gálvez, A. O.; Maruoka, K. J. Am. Chem. Soc. 2013, 135, 17667–17670. doi:10.1021/ja407764u
Return to citation in text: [1] -
Gök, Y.; Karayiğit, İ. Ü.; Gök, H. Z. J. Organomet. Chem. 2017, 846, 44–50. doi:10.1016/j.jorganchem.2017.05.059
Return to citation in text: [1] -
Hirao, T. Catalytic Reductive Coupling of Carbonyl Compounds – The Pinacol Coupling Reaction and Beyond. In Metal Catalyzed Reductive C–C Bond Formation; Krische, M. J., Ed.; Topics in Current Chemistry, Vol. 279; Springer: Berlin, Germany, 2007; pp 53–75. doi:10.1007/128_2007_120
Return to citation in text: [1] -
Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. doi:10.1002/anie.201501556
Return to citation in text: [1] -
Liu, M.; Tan, L.; Rashid, R. T.; Cen, Y.; Cheng, S.; Botton, G.; Mi, Z.; Li, C.-J. Chem. Sci. 2020, 11, 7864–7870. doi:10.1039/d0sc02718a
Return to citation in text: [1] -
Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Feng, C.-W.; Qin, Y.; Shen, Y.-M.; Pu, C.; Peng, X. J. Org. Chem. 2021, 86, 2474–2488. doi:10.1021/acs.joc.0c02627
Return to citation in text: [1] -
Wang, H.; Qu, J.-P.; Kang, Y.-B. Org. Lett. 2021, 23, 2900–2903. doi:10.1021/acs.orglett.1c00537
Return to citation in text: [1] -
Mitkina, T.; Stanglmair, C.; Setzer, W.; Gruber, M.; Kisch, H.; König, B. Org. Biomol. Chem. 2012, 10, 3556–3561. doi:10.1039/c2ob07053g
Return to citation in text: [1] -
McClelland, K. P.; Weiss, E. A. ACS Appl. Energy Mater. 2019, 2, 92–96. doi:10.1021/acsaem.8b01652
Return to citation in text: [1] -
Wang, J.; Feng, Y.-X.; Zhang, M.; Zhang, C.; Li, M.; Li, S.-J.; Zhang, W.; Lu, T.-B. CCS Chem. 2020, 2, 81–88. doi:10.31635/ccschem.020.201900093
Return to citation in text: [1] -
Fujita, M.; Kobayashi, F.; Ide, T.; Egami, H.; Hamashima, Y. Eur. J. Org. Chem. 2020, 7151–7155. doi:10.1002/ejoc.202001329
Return to citation in text: [1] -
Ishida, N.; Son, M.; Kawasaki, T.; Ito, M.; Murakami, M. Synlett 2021, 32, 2067–2070. doi:10.1055/a-1644-4876
Return to citation in text: [1] -
Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d
Return to citation in text: [1] -
Schäfer, H. J. C. R. Chim. 2011, 14, 745–765. doi:10.1016/j.crci.2011.01.002
Return to citation in text: [1] -
Meyer, T. H.; Finger, L. H.; Gandeepan, P.; Ackermann, L. Trends Chem. 2019, 1, 63–76. doi:10.1016/j.trechm.2019.01.011
Return to citation in text: [1] -
Cembellín, S.; Batanero, B. Chem. Rec. 2021, 21, 2453–2471. doi:10.1002/tcr.202100128
Return to citation in text: [1] -
Claraz, A.; Masson, G. ACS Org. Inorg. Au 2022, 2, 126–147. doi:10.1021/acsorginorgau.1c00037
Return to citation in text: [1] -
Stocker, J. H.; Jenevein, R. M. J. Org. Chem. 1968, 33, 294–297. doi:10.1021/jo01265a058
Return to citation in text: [1] -
Maurice, C.; Schöllhorn, B.; Canet, I.; Mousset, G.; Mousty, C.; Guilbot, J.; Plusquellec, D. Eur. J. Org. Chem. 2000, 813–821. doi:10.1002/(sici)1099-0690(200003)2000:5<813::aid-ejoc813>3.0.co;2-q
Return to citation in text: [1] -
Lagrost, C.; Hapiot, P.; Vaultier, M. Green Chem. 2005, 7, 468–474. doi:10.1039/b500839e
Return to citation in text: [1] -
Kise, N.; Isemoto, S.; Sakurai, T. Tetrahedron 2012, 68, 8805–8816. doi:10.1016/j.tet.2012.07.094
Return to citation in text: [1] -
Nakahara, K.; Naba, K.; Saitoh, T.; Sugai, T.; Obata, R.; Nishiyama, S.; Einaga, Y.; Yamamoto, T. ChemElectroChem 2019, 6, 4153–4157. doi:10.1002/celc.201900202
Return to citation in text: [1] -
Liu, C.; Li, R.; Zhou, W.; Liang, Y.; Shi, Y.; Li, R.-L.; Ling, Y.; Yu, Y.; Li, J.; Zhang, B. ACS Catal. 2021, 11, 8958–8967. doi:10.1021/acscatal.1c01382
Return to citation in text: [1] -
Chaussard, J.; Folest, J.-C.; Nedelec, J.-Y.; Perichon, J.; Sibille, S.; Troupel, M. Synthesis 1990, 369–381. doi:10.1055/s-1990-26880
Return to citation in text: [1] -
Léonard, E.; Duñach, E.; Périchon, J. J. Chem. Soc., Chem. Commun. 1989, 276–277. doi:10.1039/c39890000276
Return to citation in text: [1] -
Thomas, H. G.; Littmann, K. Synlett 1990, 757–758. doi:10.1055/s-1990-21241
Return to citation in text: [1] -
Sahloul, K.; Sun, L.; Requet, A.; Chahine, Y.; Mellah, M. Chem. – Eur. J. 2012, 18, 11205–11209. doi:10.1002/chem.201201390
Return to citation in text: [1] -
Kronenwetter, H.; Husek, J.; Etz, B.; Jones, A.; Manchanayakage, R. Green Chem. 2014, 16, 1489–1495. doi:10.1039/c3gc41641k
Return to citation in text: [1] -
Hilt, G. ChemElectroChem 2020, 7, 395–405. doi:10.1002/celc.201901799
Return to citation in text: [1] -
Sbei, N.; Hardwick, T.; Ahmed, N. ACS Sustainable Chem. Eng. 2021, 9, 6148–6169. doi:10.1021/acssuschemeng.1c00665
Return to citation in text: [1] -
Senboku, H.; Nagakura, K.; Fukuhara, T.; Hara, S. Tetrahedron 2015, 71, 3850–3856. doi:10.1016/j.tet.2015.04.020
Return to citation in text: [1] -
Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. ChemElectroChem 2015, 2, 73–76. doi:10.1002/celc.201402299
Return to citation in text: [1] -
Medvedev, J. J.; Steksova, Y. P.; Medvedeva, X. V.; Pivovarova, Y.; Krivoshapkina, E. F.; Klinkova, A. J. Electrochem. Soc. 2020, 167, 155521. doi:10.1149/1945-7111/abb839
Return to citation in text: [1] -
Wang, Y.; Zhao, J.; Qiao, T.; Zhang, J.; Chen, G. Chin. J. Chem. 2021, 39, 3297–3302. doi:10.1002/cjoc.202100508
Return to citation in text: [1] -
Muchez, L.; De Vos, D. E.; Kim, M. ACS Sustainable Chem. Eng. 2019, 7, 15860–15864. doi:10.1021/acssuschemeng.9b04612
Return to citation in text: [1] [2] -
The reaction using 4-phenylbutan-2-ol did not gave the corresponding vic-1,2-diol, and the staring alcohol and the corresponding ketone were detected in 53% and 1% NMR yields, respectively.
Return to citation in text: [1] -
Shono, T.; Matsumura, Y.; Hashimoto, T.; Hibino, K.; Hamaguchi, H.; Aoki, T. J. Am. Chem. Soc. 1975, 97, 2546–2548. doi:10.1021/ja00842a044
Return to citation in text: [1] -
Wang, S.; Singh, P. S.; Evans, D. H. J. Phys. Chem. C 2009, 113, 16686–16693. doi:10.1021/jp904976v
Return to citation in text: [1]
| 46. | Muchez, L.; De Vos, D. E.; Kim, M. ACS Sustainable Chem. Eng. 2019, 7, 15860–15864. doi:10.1021/acssuschemeng.9b04612 |
| 49. | Wang, S.; Singh, P. S.; Evans, D. H. J. Phys. Chem. C 2009, 113, 16686–16693. doi:10.1021/jp904976v |
| 1. | Suzuki, K.; Tamiya, M. Pinacol Coupling Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 3, pp 580–620. doi:10.1016/b978-0-08-097742-3.00316-5 |
| 2. | Chatterjee, A.; Joshi, N. N. Tetrahedron 2006, 62, 12137–12158. doi:10.1016/j.tet.2006.09.002 |
| 14. | Hirao, T. Catalytic Reductive Coupling of Carbonyl Compounds – The Pinacol Coupling Reaction and Beyond. In Metal Catalyzed Reductive C–C Bond Formation; Krische, M. J., Ed.; Topics in Current Chemistry, Vol. 279; Springer: Berlin, Germany, 2007; pp 53–75. doi:10.1007/128_2007_120 |
| 47. | The reaction using 4-phenylbutan-2-ol did not gave the corresponding vic-1,2-diol, and the staring alcohol and the corresponding ketone were detected in 53% and 1% NMR yields, respectively. |
| 10. | Fleischer, R.; Braun, M. Synlett 1998, 1441–1443. doi:10.1055/s-1998-1953 |
| 11. | Stodulski, M.; Jaźwiński, J.; Mlynarski, J. Eur. J. Org. Chem. 2008, 5553–5562. doi:10.1002/ejoc.200800726 |
| 12. | Hashimoto, T.; Gálvez, A. O.; Maruoka, K. J. Am. Chem. Soc. 2013, 135, 17667–17670. doi:10.1021/ja407764u |
| 13. | Gök, Y.; Karayiğit, İ. Ü.; Gök, H. Z. J. Organomet. Chem. 2017, 846, 44–50. doi:10.1016/j.jorganchem.2017.05.059 |
| 48. | Shono, T.; Matsumura, Y.; Hashimoto, T.; Hibino, K.; Hamaguchi, H.; Aoki, T. J. Am. Chem. Soc. 1975, 97, 2546–2548. doi:10.1021/ja00842a044 |
| 8. | Alexakis, A.; Mangeney, P. Tetrahedron: Asymmetry 1990, 1, 477–511. doi:10.1016/s0957-4166(00)80536-4 |
| 9. | Fujioka, H.; Kita, Y. Asymmetric Synthesis Using Chiral Acetals from C2-Symmetric Diols and Its Application to Natural Product Synthesis. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, Netherlands, 1994; Vol. 14, pp 469–516. doi:10.1016/b978-0-444-81780-8.50018-1 |
| 45. | Wang, Y.; Zhao, J.; Qiao, T.; Zhang, J.; Chen, G. Chin. J. Chem. 2021, 39, 3297–3302. doi:10.1002/cjoc.202100508 |
| 3. | Ogata, M.; Matsumoto, H.; Takahashi, K.; Shimizu, S.; Kida, S.; Murabayashi, A.; Shiro, M.; Tawara, K. J. Med. Chem. 1987, 30, 1054–1068. doi:10.1021/jm00389a016 |
| 4. | Nicolaou, K. C.; Yang, Z.; Liu, J. J.; Ueno, H.; Nantermet, P. G.; Guy, R. K.; Claiborne, C. F.; Renaud, J.; Couladouros, E. A.; Paulvannan, K.; Sorensen, E. J. Nature 1994, 367, 630–634. doi:10.1038/367630a0 |
| 5. | Mateo, C.; Álvarez, R.; Pérez-Melero, C.; Peláez, R.; Medarde, M. Bioorg. Med. Chem. Lett. 2007, 17, 6316–6320. doi:10.1016/j.bmcl.2007.08.075 |
| 6. | Singh, P.; Mittal, A.; Kaur, S.; Holzer, W.; Kumar, S. Org. Biomol. Chem. 2008, 6, 2706–2712. doi:10.1039/b803608j |
| 7. | Werner, B.; Kalesse, M. Org. Lett. 2017, 19, 1524–1526. doi:10.1021/acs.orglett.7b00288 |
| 46. | Muchez, L.; De Vos, D. E.; Kim, M. ACS Sustainable Chem. Eng. 2019, 7, 15860–15864. doi:10.1021/acssuschemeng.9b04612 |
| 29. | Stocker, J. H.; Jenevein, R. M. J. Org. Chem. 1968, 33, 294–297. doi:10.1021/jo01265a058 |
| 30. | Maurice, C.; Schöllhorn, B.; Canet, I.; Mousset, G.; Mousty, C.; Guilbot, J.; Plusquellec, D. Eur. J. Org. Chem. 2000, 813–821. doi:10.1002/(sici)1099-0690(200003)2000:5<813::aid-ejoc813>3.0.co;2-q |
| 31. | Lagrost, C.; Hapiot, P.; Vaultier, M. Green Chem. 2005, 7, 468–474. doi:10.1039/b500839e |
| 32. | Kise, N.; Isemoto, S.; Sakurai, T. Tetrahedron 2012, 68, 8805–8816. doi:10.1016/j.tet.2012.07.094 |
| 33. | Nakahara, K.; Naba, K.; Saitoh, T.; Sugai, T.; Obata, R.; Nishiyama, S.; Einaga, Y.; Yamamoto, T. ChemElectroChem 2019, 6, 4153–4157. doi:10.1002/celc.201900202 |
| 34. | Liu, C.; Li, R.; Zhou, W.; Liang, Y.; Shi, Y.; Li, R.-L.; Ling, Y.; Yu, Y.; Li, J.; Zhang, B. ACS Catal. 2021, 11, 8958–8967. doi:10.1021/acscatal.1c01382 |
| 36. | Léonard, E.; Duñach, E.; Périchon, J. J. Chem. Soc., Chem. Commun. 1989, 276–277. doi:10.1039/c39890000276 |
| 37. | Thomas, H. G.; Littmann, K. Synlett 1990, 757–758. doi:10.1055/s-1990-21241 |
| 38. | Sahloul, K.; Sun, L.; Requet, A.; Chahine, Y.; Mellah, M. Chem. – Eur. J. 2012, 18, 11205–11209. doi:10.1002/chem.201201390 |
| 39. | Kronenwetter, H.; Husek, J.; Etz, B.; Jones, A.; Manchanayakage, R. Green Chem. 2014, 16, 1489–1495. doi:10.1039/c3gc41641k |
| 24. | Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d |
| 25. | Schäfer, H. J. C. R. Chim. 2011, 14, 745–765. doi:10.1016/j.crci.2011.01.002 |
| 26. | Meyer, T. H.; Finger, L. H.; Gandeepan, P.; Ackermann, L. Trends Chem. 2019, 1, 63–76. doi:10.1016/j.trechm.2019.01.011 |
| 27. | Cembellín, S.; Batanero, B. Chem. Rec. 2021, 21, 2453–2471. doi:10.1002/tcr.202100128 |
| 28. | Claraz, A.; Masson, G. ACS Org. Inorg. Au 2022, 2, 126–147. doi:10.1021/acsorginorgau.1c00037 |
| 40. | Hilt, G. ChemElectroChem 2020, 7, 395–405. doi:10.1002/celc.201901799 |
| 41. | Sbei, N.; Hardwick, T.; Ahmed, N. ACS Sustainable Chem. Eng. 2021, 9, 6148–6169. doi:10.1021/acssuschemeng.1c00665 |
| 42. | Senboku, H.; Nagakura, K.; Fukuhara, T.; Hara, S. Tetrahedron 2015, 71, 3850–3856. doi:10.1016/j.tet.2015.04.020 |
| 43. | Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. ChemElectroChem 2015, 2, 73–76. doi:10.1002/celc.201402299 |
| 44. | Medvedev, J. J.; Steksova, Y. P.; Medvedeva, X. V.; Pivovarova, Y.; Krivoshapkina, E. F.; Klinkova, A. J. Electrochem. Soc. 2020, 167, 155521. doi:10.1149/1945-7111/abb839 |
| 19. | Mitkina, T.; Stanglmair, C.; Setzer, W.; Gruber, M.; Kisch, H.; König, B. Org. Biomol. Chem. 2012, 10, 3556–3561. doi:10.1039/c2ob07053g |
| 20. | McClelland, K. P.; Weiss, E. A. ACS Appl. Energy Mater. 2019, 2, 92–96. doi:10.1021/acsaem.8b01652 |
| 21. | Wang, J.; Feng, Y.-X.; Zhang, M.; Zhang, C.; Li, M.; Li, S.-J.; Zhang, W.; Lu, T.-B. CCS Chem. 2020, 2, 81–88. doi:10.31635/ccschem.020.201900093 |
| 22. | Fujita, M.; Kobayashi, F.; Ide, T.; Egami, H.; Hamashima, Y. Eur. J. Org. Chem. 2020, 7151–7155. doi:10.1002/ejoc.202001329 |
| 23. | Ishida, N.; Son, M.; Kawasaki, T.; Ito, M.; Murakami, M. Synlett 2021, 32, 2067–2070. doi:10.1055/a-1644-4876 |
| 15. | Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. doi:10.1002/anie.201501556 |
| 16. | Liu, M.; Tan, L.; Rashid, R. T.; Cen, Y.; Cheng, S.; Botton, G.; Mi, Z.; Li, C.-J. Chem. Sci. 2020, 11, 7864–7870. doi:10.1039/d0sc02718a |
| 17. | Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Feng, C.-W.; Qin, Y.; Shen, Y.-M.; Pu, C.; Peng, X. J. Org. Chem. 2021, 86, 2474–2488. doi:10.1021/acs.joc.0c02627 |
| 18. | Wang, H.; Qu, J.-P.; Kang, Y.-B. Org. Lett. 2021, 23, 2900–2903. doi:10.1021/acs.orglett.1c00537 |
| 35. | Chaussard, J.; Folest, J.-C.; Nedelec, J.-Y.; Perichon, J.; Sibille, S.; Troupel, M. Synthesis 1990, 369–381. doi:10.1055/s-1990-26880 |
© 2022 Yamamoto et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.