Abstract
A series of spiro[isoindole-1,5-isoxazolidin]-3(2H)-ones has been synthesized by 1,3-dipolar cycloaddition of N-benzylnitrone with isoindolin-3-methylene-1-ones. The regio- and stereoselectivity of the process have been rationalized by computational methods. The obtained compounds show cytotoxic properties and antiproliferative activity in the range of 9–22 μM. Biological tests suggest that the antitumor activity could be linked to the inhibition of the protein–protein p53-MDM2 interaction. Docking measurements support the biological data.
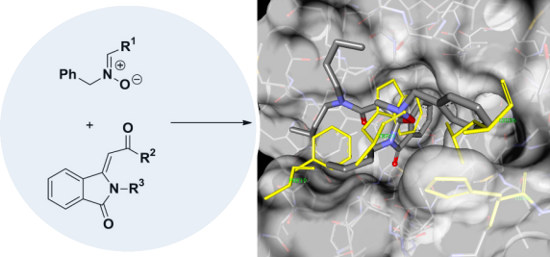
Graphical Abstract
Introduction
The p53 tumor suppressor protein is a transcriptional factor that plays a key role in the regulation of several cellular processes, including apoptosis, DNA repair, and angiogenesis [1-4]. The murine double minute 2 (MDM2) protein is the primary cellular inhibitor of p53, functioning through direct interaction with p53 [5]: tumoral cells show an overexpression of MDM2 which suppresses the functions of the p53 protein [5-8].
The design of non-peptide, small-molecule inhibitors that block the MDM2-p53 interaction has been sought as an attractive strategy to activate p53 for the treatment of cancer and other human diseases [9-11]. Major advances have been made in the design of lipophilic small-molecule inhibitors of the MDM2-p53 interaction in recent years, and several compounds have moved into advanced preclinical development or clinical trials [12-14]. Potent MDM2-p53 inhibitors, such as Nutlin-3 [12] and the spirooxindoles, for example MI-63 and MI-219, [15-18] have demonstrated cellular activity consistent with inhibition of MDM2-p53 binding and have shown in vivo antitumor activity [15,19] (Figure 1).
![[1860-5397-12-278-1]](/bjoc/content/figures/1860-5397-12-278-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some relevant MDM2-p53 interaction inhibitors.
Figure 1: Some relevant MDM2-p53 interaction inhibitors.
Isoindolinones, belonging to the alkaloids family, are found in many natural products such as vitedoamine A, chilenine, lennoxamine, magallanesine and nuevamine [20-25]. These compounds possess a lot of pharmacological activities such as anxiolytic/anticonvulsant, TNFα-inhibitory, antiangiogenic, 5-HT antagonistic/antidepressant [26-30], PARP-1-inhibitory [31], histone deacetylase inhibitory [32] and cytotoxic activity.
Recently, MDM2-p53 inhibitors based on an isoindolinone scaffold [33,34] have been reported. These latter results demonstrate the versatility of the isoindolinone scaffold as MDM2-p53 inhibitor and show that significant improvements in potency may be gained by modest structural modifications.
Introducing structural diversity into the isoindolinone scaffold can represent an important approach towards the design of new chemotherapeutics.
On the basis of these considerations, we have designed a route towards a new class of potential MDM2-p53 inhibitors 3, through the construction of the spiro[isoxazolidin-isoindolinone] system. The synthetic scheme (Figure 2) exploits the strategy of the 1,3-dipolar cycloaddition of nitrones on the substrate isoindolin-3-methylene-1-one 2, obtained by a recent methodology of a PdI2 catalyzed aminocarbonylation-N-heterocyclization of 2-ethynylbenzamides 1 [35].
![[1860-5397-12-278-2]](/bjoc/content/figures/1860-5397-12-278-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic route to spiro[isoindole-1,5-isoxazolidin]-3(2H)-ones 3.
Figure 2: Retrosynthetic route to spiro[isoindole-1,5-isoxazolidin]-3(2H)-ones 3.
The rationale of our choice is based on molecular docking data. Using the published structure of the MDM2–p53 binding site, we have employed computational methods and focused library synthesis based on the isoindolinone template, to develop compounds with inhibitory activity. These studies have resulted in the identification of a number of potential MDM2-p53 interaction inhibitors. Biological tests confirm our initial hypothesis indicating for compounds 3 an antiproliferative activity in the range of 9–22 μM: the antitumor activity appears to be linked to the inhibition of the protein–protein p53-MDM2 interaction.
Results and Discussion
Chemistry
The synthetic scheme towards the construction of the spiro[isoxazolidin-isoindolinone] system 3 starts from isoindolinones 2 which have been synthesized, as reported [35], by Pd-catalyzed aminocarbonylation-N-heterocyclization of 2-ethynylbenzamides 1. The latter are easily accessible through ethynylation of N-substituted 2-iodobenzamides, with secondary amines (Scheme 1).
![[1860-5397-12-278-i1]](/bjoc/content/inline/1860-5397-12-278-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Compounds 2a–e, as E/Z mixtures, have been reacted with nitrone 4. Different experimental conditions have been exploited: by reacting the nitrone and dipolarophile in toluene solution under reflux, the formation of products was not observed, even after extension of the reaction time to 72 h. Under these conditions the process led only to decomposition of the nitrone with the recovery of unaltered isoindolinone. The best results have been obtained by performing the 1,3-dipolar cycloaddition in toluene at 110 °C for 4 h, under microwave irradiation: cycloadducts 6a–e have been isolated in 38–60% yield, as major isomers, together with isomers 7 as minor adducts and unreacted isoindolinones 2a–e mainly in E configuration (Scheme 2, Table 1).
![[1860-5397-12-278-i2]](/bjoc/content/inline/1860-5397-12-278-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 6a–f by 1,3-dipolar cycloaddition.
Scheme 2: Synthesis of 6a–f by 1,3-dipolar cycloaddition.
Table 1: Synthesis of 6a–f by 1,3-dipolar cycloaddition.
| Entry | Nitrones | Dipolarofile | (Z/E ratio) | Product | Ratio | Yield %a |
|---|---|---|---|---|---|---|
| 1 | 4 |
2a R2 = N(C4H8)O,
R3 = PhCH2 |
2.2 | 6a/7a | 85:15 | 60 |
| 2 | 4 |
2b R2 = N(C4H8),
R3 = Bu |
1.8 | 6b/7bb | 95:5 | 55 |
| 3 | 4 |
2c R2 = N(C4H8)O,
R3 = Bu |
2 | 6c/7cb | 94:6 | 40 |
| 4 | 4 |
2d R2 = N(C5H10),
R3 = Bu |
1 | 6d/7db | 99:1 | 35 |
| 5 | 4 |
2e R2 = N(Bu)2,
R3 = Bu |
1 | 6e/7eb | 99:1 | 38 |
| 6 | 4 |
2f R2 = N(C4H8)O,
R3 = Ph |
only Z isomer | 6f/7f | 100:0 | 65 |
| 7 | 4 |
2g R2 = N(C4H8)O,
R3 = N-t-Bu |
only E isomer | 6g/7g | 0:100 | 10 |
| 8 | 5 |
2a R2 = N(C4H8)O,
R3 = PhCH2 |
2.2 | – | – | – |
aIsomeric mixture; bnot isolated.
The cycloaddition reaction of 2f, obtained only as Z isomer in the aminocarbonylation procedure, produces only 6f in 65 % yield, while the reaction performed with 2g, which is present only as E isomer, leads to adduct 7g even if in low yield (10%). These set of experiments indicate that the Z isomers are more reactive than E derivatives and that the Z compounds give only cycloadducts 6, while E lead only to adducts 7.
The 1H NMR spectrum of the crude reaction mixture shows the stereoisomers 6a–f as the main products, while stereoisomers 7a–f are present as minor components or only in traces.
The cycloaddition reaction showed complete regioselectivity and a high stereoselectivity in favour of the [1(RS),4’(RS)]2,2’-dibenzyl-4’-substituted spiro[isoindole-1,5’-isoxazolidin]-3(2H)-ones 6a–f.
The structure of adducts 6 and 7 has been elucidated by 1H NMR and 13C NMR spectroscopies and MS spectrometry. In particular, the 1H NMR spectrum of 6a, chosen as model compound, shows the diagnostic resonance of the H9 proton at 3.83 ppm, while the methylene protons at C8 resonate at 3.71 and 3.41 ppm. In compound 7a, H9 proton resonates at 2.70 ppm, while the methylene protons at C8 resonate at 3.84 and 4.08 ppm. The detailed long-range coupling analysis observed in the 1H,13C-HBMC confirms the attributions; thus, the long-range coupling between H9 and C7 (66.34 ppm) of the benzyl substituent at the nitrogen atom of the isoxazolidine ring, observed in compound 6a, is in agreement with the proposed structure. Conversely, for 7a, a long-range coupling was detected between H9 and C18 (43.40 ppm), the carbon atom of the benzyl group at N2.
NOE experiments support the assigned stereochemical relationships. In agreement with the internuclear distance values, obtained from computational data (see Supporting Information File 1), irradiation of H9, in compound 6a, induces a positive NOE effect for methylene protons at C7 (4.25 and 4.06 ppm), while, for 7a, a NOE enhancement was observed for methylene protons at C18 (5.06 and 4.82 ppm) (Figure 3).
![[1860-5397-12-278-3]](/bjoc/content/figures/1860-5397-12-278-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected NOESY observed for compounds 6a and 7a.
Figure 3: Selected NOESY observed for compounds 6a and 7a.
Furthermore, X-ray diffraction measurements confirm the structural assignment. Unfortunately, it was possible only to obtain a single crystal for the minor isomer 7 and the relative configuration, as RS/SR, at C10 and C9, respectively, is reported in Figure 4 [36].
![[1860-5397-12-278-4]](/bjoc/content/figures/1860-5397-12-278-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP drawing of the X-ray crystal structure of 7a showing the model atomic numbering scheme (C10 and C9 correspond to the chiral centres C1 and C4’, respectively). Crystal packing is a racemate due to the centrosymmetric symmetry and in the picture the choice of the enantiomer is arbitrary. C: light blue, H: white, O: red, N: magenta. Probability of the ORTEP ellipsoids is set to 50%, whereas H size is arbitrary.
Figure 4: ORTEP drawing of the X-ray crystal structure of 7a showing the model atomic numbering scheme (C10 a...
In order to further evaluate the regio- and the stereochemical outcome of the cycloaddition reaction and to extend the potentiality of the synthetic process, dipolarophile 2a was reacted with nitrones 5 under the same experimental conditions. However, only the starting isoindolinone was recovered. Also the use of more drastic reaction conditions, such as the use of o-xylene as a solvent and higher temperatures up to 140 °C failed, leading to the degradation of the nitrone and the isolation of the unaltered dipolarophile, so clearly indicating that the steric factors play a crucial role in the cycloaddition process (see computational data).
Theoretical calculations
The obtained results and stereochemical outcome of the 1,3-dipolar cycloaddition process have been rationalized through a mechanistic study on the basis of our expertise in the study of cycloaddition reactions and heterocyclic compounds [37-42]. Calculations were performed using the Gaussian09 program package, [43] through optimizations with the Thrular’s functional M06 [44] and 6-31+G(d,p) basis set. A simplified model, able to correctly mimic the system, was considered and the reaction between dipolarophile 8 (Z and E) and nitrone 4 (Scheme 3) was studied.
![[1860-5397-12-278-i3]](/bjoc/content/inline/1860-5397-12-278-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The reaction pathway was considered and the transition states leading to the (R,R)-9 or (R,S)-10 adducts were modeled. Figure 5 shows the TSs (named endo, N or exo, X) for reaction of the two isomeric dipolarophiles (Z)-8 and (E)-8 with nitrone 4, respectively. All the possible degrees of conformational freedom were considered, in particular the different orientations of the benzylic moiety. The possibility, in the endo TSs, of stacking interactions between aromatic rings, as hypothesized in the literature [45], was taken into account, but the corresponding geometries are too high in energy and evolve to the TSs reported in Figure 5.
![[1860-5397-12-278-5]](/bjoc/content/figures/1860-5397-12-278-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Three-dimensional plots of TSs of reaction of dipolarophiles (Z)-8 and (E)-8 with nitrone 4. The labels endo N and exo E refer to the location of the nitrone N-benzyl in the formed five-membered ring on the same or opposite side of the phenyl ring of dipolarophiles. Distances between atoms involved in the forming bonds (Å) are reported in the 3D plots.
Figure 5: Three-dimensional plots of TSs of reaction of dipolarophiles (Z)-8 and (E)-8 with nitrone 4. The la...
The profiles of the four reactions are shown in Figure 6 and the percentages of the adducts derived from the TSs at 408 K are given in Table 2. The cycloadducts 9 and 10 are formed with a ratio of 92:8.
![[1860-5397-12-278-6]](/bjoc/content/figures/1860-5397-12-278-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Free energy profiles for the cycloaddition reaction of the two isomers Z (A) or E (B) of dipolarophile 8 with nitrone 4, considering both the endo (N, black) or exo (X, red) path.
Figure 6: Free energy profiles for the cycloaddition reaction of the two isomers Z (A) or E (B) of dipolaroph...
Calculations supported the experimental data, showing that the adduct RR in the racemic mixture is the mainly obtained product. It is worthy pointing out that increasing the steric hindrance on the carbon atom of nitrone 5, such as replacing one hydrogen atom with a methyl group, the energy barriers become significantly higher (about 30 kcal/mol), so the reaction is expected to be difficult.
Biological tests
Cellular viability and proliferation
The synthesized compounds were assayed for their biological activity on three human cancer cell lines (the neuroblastoma SH-SY5Y, the HT-29 colorectal adenocarcinoma and the HepG2 hepatocellular carcinoma cells) treated for 24–72 h with the tested compounds. The MTS assay [46,47] showed a significant reduction in cellular viability in all cancer cell lines treated with compounds 6a–f at concentrations ranging from 1 to 100 µM, when compared with respective controls. No significant effect in cellular viability in all cancer cell lines was found when the cells were exposed to the synthesized compounds for 24 and 48 h (data not shown). In particular, compound 6e showed to be the most active derivative and displayed the greatest activity in the range of 9.41 to 21.58 µM. Furthermore, SH-SY5Y cell lines were more susceptible to treatment with 6e, than the HT-29 and HepG2 cells. Thus, the other experiments have been performed using 6e as model compound.
In general, all the synthesized compounds showed a certain degree of antiproliferative effect against all the examined cancer cells with a similar trend (see Supporting Information File 1, Figure S1). Noteworthy, compound 6e exhibited superior activity with respect to other derivatives. As shown in Figure 7a, treatment of SH-SY5Y, HT-29 and HepG2 cells with 6e ranging from 1 µM to 100 µM, for 24–72 h, reduced cell growth in all cancer cell lines. In particular, the maximal growth inhibitory effect of 6e was reached after 72 h of incubation with the 100 µM concentration, corresponding to 72% in HepG2 (IC50 10.50 µM), 83% and 84% in HT-29 (IC50 21.58 µM) and SH-SY5Y (IC50 9.41 µM) cell lines, respectively (P < 0.001 vs control). Significant reduction of cell proliferation was also observed when the cultures were exposed to 6e for 24 hours (P < 0.01 vs control) and 48 (P < 0.001 vs control). Lesser, but still significant, an antiproliferative effect was also found treating the cells with 6e at concentrations of 50, 10 and 5 µM for all time of exposure, while a concentration of 1 µM did not exert a significant antiproliferative effect.
![[1860-5397-12-278-7]](/bjoc/content/figures/1860-5397-12-278-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Compound 6e reduces cancer cell proliferation. Treatment of SH-SY5Y, HT-29 and HepG2 cells with 6e in a range from 1 to 100 µM for 24, 48 and 72 h reduced the growth rate in a time and concentration-dependent manner. The experiments were performed by the MTS assay (a) and BrdU test (b). Results are expressed as percentages of growth rates of treated cells compared to untreated cultures, and are the means ± SEM from of independent experiments performed in eightplicate (MTS assay) or in triplicate (BrdU test). *P < 0.05, **P < 0.01 and ***P < 0.001 vs untreated cells.
Figure 7: Compound 6e reduces cancer cell proliferation. Treatment of SH-SY5Y, HT-29 and HepG2 cells with 6e ...
Assessment of cell proliferation was also performed cytofluorimetrically by the BrdU assay, [48] obtaining results that reflect data from MTS test (Figure 7b).
Cytotoxic effect
The cytotoxic effect induced by 6a–f was evaluated by an LDH assay [49], revealing that significant cytotoxicity was exerted only at the higher concentrations (50 and 100 µM; see Supporting Information File 1). Figure 8a shows that 6e caused a significant increase of LDH release at 10, 50 and 100 µM concentration in all cell lines used in this study (P < 0.01 and P < 0.001 for SH-SY5Y cells and P < 0.05 and P < 0.01 for HT-29 and HepG2 cells). The LDH release was accompanied by a significant increase in cell death, as detected by flow cytometry through a propidium iodide assay (Figure 8b) [50,51].
![[1860-5397-12-278-8]](/bjoc/content/figures/1860-5397-12-278-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Cytotoxic effect of 6e. The cytotoxic activity of 6e was assessed in terms of both LDH release (a) and cell death (b). LDH levels are extrapolated as the values detected in untreated cells, which are arbitrarily expressed as 1. Cell death was reported as the percentage of dead cells vs untreated cultures point to 0. Data, expressed as mean ± S.E.M., represent the values obtained in three different sets of experiments made in triplicate. *P < 0.05, **P < 0.01 and ***P < 0.001 vs untreated cells.
Figure 8: Cytotoxic effect of 6e. The cytotoxic activity of 6e was assessed in terms of both LDH release (a) ...
Involvement of p53 in the pharmacological activity
Tumor suppressor p53 plays an important role in conserving genome stability by preventing its mutation. Normally, p53 is found at low levels because of its continuous proteasomal degradation promoted by MDM2. MDM2 inhibits p53 activity in two ways: i) by targeting p53 into the region of interaction with CBP/p300, thus preventing its transcriptional activity, and ii) by exporting p53 from the nucleus to the cytosol, acting as E3 ubiquitin ligase that marks p53 for degradation by the proteasome. Following DNA damage, p53 rises and becomes phosphorylated, thus translocates to nucleus where it binds DNA and activates expression of several proteins that arrest cell proliferation until the damage is repaired. If the damage is too severe, p53 trigs apoptosis which allows the elimination of damaged cells, also by its translocation in the mitochondria where it inhibits the activity of anti-apoptotic proteins. Many tumors overproduce MDM2 to impair p53 function, thus promoting cancerogenesis.
In order to evaluate the possible involvement of p53 in the antiproliferative and cytotoxic effect of 6e, we assessed the levels of p53, MDM2 and p21 by Western blot analysis [52]. We have chosen the SH-SY5Y cells because of their greatest sensitivity to this molecule in comparison to the other cultures employed in this study. The cells were treated for 72 h with 6e at concentrations that do not induce any cytotoxic effects (1–10 µM). As shown in Figure 9, incubation with 5 and 10 µM concentration decreased the levels of p53 in the cytosol (P < 0.01 vs untreated cells) and increased those in the nucleus (P < 0.01 and P < 0.001 vs untreated cells), demonstrating its involvement in the anti-cancer effect elicited by 6e.
![[1860-5397-12-278-9]](/bjoc/content/figures/1860-5397-12-278-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: 6e modulate the levels of p53 in SH-SY5Y cells. (a) The SH-SY5Y cells were treated for 24 h with the indicated concentration of 6e, and then, both cytosolic and nuclear proteins were analyzed by Western blot for p53 protein. A representative immunoblot of three independent experiments is shown. (b) A densitometric analysis of autoradiographic bands collected from three separate experiments is shown. Levels of nuclear or cytosolic protein were normalized for laminin or β-actin, respectively. **P < 0.01 and ***P < 0.001 vs untreated cells.
Figure 9: 6e modulate the levels of p53 in SH-SY5Y cells. (a) The SH-SY5Y cells were treated for 24 h with th...
Treatment of the cells with 6e leads to increased p53 protein expression, a compensatory increase in MDM2 expression, and activates p53-mediated apoptosis with an increase in p21 expression, when compared with the control. The effects appeared lower than it can be found in cells treared with Nutlin-3, a known MDM2-p53 antagonist [12] (Figure 10).
![[1860-5397-12-278-10]](/bjoc/content/figures/1860-5397-12-278-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: (A) Representative Western Blots and (B) semiquantitative analyses of p53, MDM2, p21 expression levels in total cellular lysates of SH-SY5Y cells non-exposed (a), exposed at 72 h to 10 μM 6e (b) or 10 μM Nutlin-3 (c), performed after normalization with β-tubulin. Blots shown are representative ones of a Western Blot analysis of four experiments in duplicate. Results are expressed as the mean ± S.D. of the values of four experiments in duplicate. *P < 0.05, significant differences vs controls.
Figure 10: (A) Representative Western Blots and (B) semiquantitative analyses of p53, MDM2, p21 expression lev...
Effect of 6e on apoptotic pathway activation
To elucidate whether 6e might be related to apoptotic pathway, we studied by Western Blot analysis, caspase-3 and PARP cleavage, in SH-SY5Y cell line cultures.
A significant activation of caspase-3 and PARP cleavage in 10 µM SH-SY5Y-treated cells was found (Figure 11b), when compared with the untreated ones (Figure 11a), even if its effect is lower than found in Nutlin-3-treated cells (Figure 11c).
![[1860-5397-12-278-11]](/bjoc/content/figures/1860-5397-12-278-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: (A) Representative Western Blots and (B) densitometric analysis of caspase-3 and PARP cleavage in total cellular lysates of SH-SY5Y cells non-exposed (a), exposed at 72 h to 10 μM 6e (b) or 10 μM Nutlin-3 (c), performed after normalization with β-tubulin. Blots shown are representative of Western blot analysis of four experiments in duplicate. Results are expressed as the mean ± SD of the values of four experiments in duplicate. *P < 0.05, significant differences vs controls.
Figure 11: (A) Representative Western Blots and (B) densitometric analysis of caspase-3 and PARP cleavage in t...
These set of experiments demonstrate that the exposure of SH-SY5Y cancer cell lines to 10 µM 6e for 72 h was able to activate the apoptotic pathway.
Docking studies
To support the suggested interaction of synthesized compounds with MDM2, docking studies were applied, starting from the X-ray coordinates of the complex of the MI63-analogue with MDM2 [53,54]. The protein structure PDB ID 3LBL was chosen as the reference receptor because its ligand had high binding affinity and high resolution (1.6 Å). Docking studies were performed using AutoDock4.2 and both enantiomers of compounds 6a–f were docked into the MDM2 binding site.
The docking protocol starts with the redocking of the MI63 analogue in the binding site to determine the lowest RMSD relative to the crystallographic pose. The ligand was successfully redocked with a RMSD of 0.59 Å. Determination of the single binding mode of spiro-isoxazolidin isoindolinone scaffold in the receptor/ligand complex was difficult because of the open and lipophilic nature of the p53 binding site on MDM2. Therefore, prediction of the possible binding mode was based on two energy types, i.e., the lowest binding energy of the largest cluster and the intermolecular energy (Table 3).
Through the docking results analysis, we found out that spiro[isoindolin-isoxazolidin] derivatives efficiently bind to the surface of MDM2 only by hydrophobic interaction. In particular, the best docking results were obtained for the (S,S)-enantiomers and, in agreement with the biological evaluation, the compound (S,S)-6e has shown an intermolecular energy value comparable with that of the co-crystallized ligand (MI63 analogue).
In addition to a lowest binding energy and intermolecular energy of docking success, we have also performed a visual inspection to confirm that all the chosen docking poses reproduced the p53 residue (Phe19, Trp23 and Leu26) and the ligand moieties occupied the three main hydrophobic pockets of MDM2.
As shown in Figure 12, compounds 6a and 6c bind to MDM2 with a similar pose. Specifically, the isoindolinone moiety occupies the Trp23 pocket, the isoxazolidine ring projects its benzyl and morpholinamide groups into the Phe19 and Leu26 pockets respectively. Likewise compounds 6b, 6d and 6g, did show a similar binding mode.
![[1860-5397-12-278-12]](/bjoc/content/figures/1860-5397-12-278-12.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: Compounds 6a (green) and 6c (blue) docked into MDM2 structure (PDB-ID: 3LBL) superimposed on the key amino acid side chain residues of the p53/MDM2 (yellow sticks, PDB-ID: 1YCR).
Figure 12: Compounds 6a (green) and 6c (blue) docked into MDM2 structure (PDB-ID: 3LBL) superimposed on the ke...
Conversely, for spiro[isoxazolidin-isoindolinone] 6e, the presence of the acyclic amide group on the isoxazolidine ring produces a 180° rotation of the isoindolinone core such that the benzyl group and dialkylamide occupy the Leu26 and Phe19 pockets, respectively. The reoriented binding mode, similar to the MI63 analogue, takes advantage of the π–π stacking interaction with His96 of MDM2 and the benzyl moiety of compound 6e, and could be related to the greater stability of the complex ligand/MDM2 (Figure 13) and to the better biological activity.
![[1860-5397-12-278-13]](/bjoc/content/figures/1860-5397-12-278-13.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 13: A) Compounds 6e (green) docked into MDM2 structure (PDB-ID: 3LBL); His96 and p53 residue (yellow sticks, PDB-ID 1YCR) are included for reference. B) Co-crystal structure of MI63-analogue in MDM2 and redocked MI63-analogue (green sticks) superimposed on the key amino acid side chain residue of the p53/MDM2 (yellow sticks, PDB-ID: 1YCR).
Figure 13: A) Compounds 6e (green) docked into MDM2 structure (PDB-ID: 3LBL); His96 and p53 residue (yellow st...
Conclusion
Spiro[isoindole-1,5-isoxazolidin]-3(2H)-ones 6a–f, synthesized by 1,3-dipolar cycloaddition of N-benzylnitrone with isoindolin-3-methylene-1-ones, have shown interesting cytotoxic and antiproliferative activity on three human cancer cell lines, the neuroblastoma SH-SY5Y, the HT-29 colorectal adenocarcinoma and the HepG2 hepatocellular carcinoma cells. In particular, the most active compound 6e shows an IC50 in the range of 9–22 μM: biological tests suggest that the antitumor activity could be linked to the inhibition of the protein–protein p53-MDM2 interaction. Docking measurements support the biological data. Further studies are needed to better clarify the role played by the new synthesized compounds in SH-SY5Y cancer cell lines as inhibitor of MDM2-p53 interaction.
Experimental
General: Solvents and reagents are commercial. Microwave assisted synthesis was performed in a CEM Discover microwave oven using sealed reaction vessels. The temperature was monitored using a vertically focused IR temperature sensor. In order to have a homogenous system all the batches were started with a ramp time of 120 seconds and when the temperature program was completed a cooling period of 10 minutes was included. ESI-HRMS were determined with a Thermo Fischer Scientific LTQ Orbitrap XL. NMR spectra (1H NMR at 500 MHz, 13C NMR at 126 MHz) were recorded with Varian instruments and are reported in ppm relative to CDCl3 (7.26 ppm). Merck silica gel 60-F254 precoated aluminum plates have been used for thin-layer chromatographic separations. Flash chromatography was performed on Merck silica gel (200–400 mesh). Preparative separations were carried out by a MPLC Büchi C-601 by using Merck silica gel 0.040–0.063 mm. X-ray crystal structure determination of compound 6a was performed at room temperature on a suitable single crystal, obtained by recrystallization from ether/dichloromethane 2:1, by a Bruker AXS K Apex II CCD diffractometer with Mo Kα radiation (λ = 0.71073 Å). The structure was solved with the SIR2011 [55] structure solution program using Direct Methods and were refined with the ShelXL [56] refinement package using Weighted Least Squares minimization. All compounds were determined to havea a purity >95% using a Shimadzu LC/MS/MS-8040 system (C18 column; eluting gradient 10–90% acetonitrile in water).
Materials: Nitrones 4, 5 and dipolarophiles 2a–g have been prepared according to known procedures [35,57-59].
General 1,3-dipolar cycloaddition procedure. A solution of 2a (0.287 mmol) and nitrone 4 (0.287 mmol) in toluene (5 mL) was put in a sealed tube and irradiated under microwave conditions at 200 W, 110 °C, for 4 h (CEM Discover Microwave reactor). The removal of the solvent in vacuo afforded a crude material which, after flash chromatography purification by using as eluent a mixture of chloroform/cyclohexane/ethyl acetate 5:3:2, gave compounds 6a and 7a, as white amorphous mass in 60% yield. The 1H NMR spectrum shows the presence of (R,R)/(S,S)- and (R,S)/(S,R)-isomers respectively in 85:15 ratio. A single crystal of 7a suitable for X-ray diffraction studies was obtained by slow diffusion of diethyl ether into a hot dichloromethane solution of isomeric mixture, followed by filtration.
(1RS,4'RS)-2,2'-Dibenzyl-4'-(morpholine-4-carbonyl)spiro[isoindoline-1,5'-isoxazolidin]-3-one (6a): White solid; mp 197–199 °C (Yield: 71 mg, 51%); IR (KBr) νmax: 1685, 1638 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.82–7.78 (m, 1H), 7.62–7.47 (m, 5H), 7.43 (d, J = 7.1 Hz, 2H), 7.36 (t, J = 7.3 Hz, 2H), 7.33–7.27 (m, 4H), 5.21 (d, J = 14.7 Hz, 1H), 4.90 (d, J = 14.7 Hz, 1H), 4.25 (d, J = 13.0 Hz, 1H), 4.08 (d, J = 13.0 Hz, 1H), 3.87–3.79 (m, 1H), 3.76–3.67 (m, 1H), 3.53–3.44 (m, 1H), 3.30 (dt, J = 11.5, 4.5 Hz, 1H), 3.10–3.00 (m, 3H), 2.82–2.74 (m, 1H), 2.62–2.54 (m, 1H), 2.53–2.46 (m, 1H), 2.38–2.30 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 167.8, 166.5, 141.19, 138.7, 136.4, 132.4, 131.2, 130.5, 129.5, 129.1, 128.8, 128.7, 128.0, 127.9, 124.7, 123.2, 97.3, 66.3, 65.8, 63.1, 60.0, 51.7, 44.9, 43.7, 42.2; HRMS–ESI (m/z): [M + H]+ calcd for C29H30N3O4, 484.2236; found, 484.2262.
(1RS,4'SR)-2,2'-Dibenzyl-4'-(morpholine-4-carbonyl)spiro[isoindoline-1,5'-isoxazolidin]-3-one (7a): White crystals, mp 185–187 °C (Yield: 12 mg, 9%); IR (KBr) νmax: 1680, 1642 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 6.7 Hz, 1H), 7.62–7.46 (m, 4H), 7.45–7.20 (m, 10H), 5.05 (d, J = 11.6 Hz, 1H), 4.83 (d, J = 11.6 Hz, 1H), 4.25 (d, J = 13.0 Hz, 1H), 4.12–3.98 (m, 2H), 3.86–3.80 (m, 1H), 3.42–3.37 (m, 1H), 3.33–3.27 (m, 2H), 3.08–3.04 (m, 1H), 2.80–2.72 (m, 1H), 2.74–2.64 (m, 1H), 2.61–2.52 (m, 1H), 2.54–2.46 (m, 1H), 2.39–2.31 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 167.6, 162.4, 132.1, 130.3, 129.2, 129.0, 128.9, 128.5, 128.4, 128.2, 127.8, 127.6, 127.3, 126.8, 125.0, 124.4, 123.0, 100.0, 66.1, 65.5, 62.9, 59.7, 53.5, 44. 7, 43.4, 41.9; HRMS–ESI (m/z): [M + H]+ calcd for C29H30N3O4, 484.2236; found, 484.2271.
(1RS,4'RS)-2'-Benzyl-2-butyl-4'-(pyrrolidine-1-carbonyl)spiro[isoindoline-1,5'-isoxazolidin]-3-one (6b): Yellow sticky oil (Yield: 69 mg, 48 %); IR (neat) νmax: 1685, 1640 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.72–7.68 (m, 1H), 7.62–7.53 (m, 1H), 7.53–7.49 (m, 1H), 7.47–7.42 (m, 1H), 7.41–7.34 (m, 2H), 7.34–7.27 (m, 3H), 4.20 (d, J = 12.7 Hz, 1H), 4.00 (d, J = 12.7 Hz, 1H), 3.92–3.83 (m, 1H), 3.75–3.66 (m, 1H), 3.64–3.54 (m, 1H), 3.25–3.17 (m, 1H), 3.16–3.08 (m, 1H), 2.73–2.61 (m, 2H), 1.75–1.60 (m, 4H), 1.59–1.51 (m, 2H), 1.40–1.24 (m, 4H), 0.90 (t, J = 7.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.3, 166.3, 136.4, 131.9, 130.1, 129.4, 129.2, 128.5, 127.8, 124.7, 122.6, 97.6, 63.1, 59.5, 53.9, 46.1, 46.0, 40.6, 31.4, 25.9, 23.8, 20.7, 13.9; HRMS–ESI (m/z): [M + H]+ calcd for C26H32N3O3, 434.2444; found, 434.2471.
(1RS,4'RS)-2'-Benzyl-2-butyl-4'-(morpholine-4-carbonyl)spiro[isoindoline-1,5'-isoxazolidin]-3-one (6c): White solid, mp 122–124 °C (Yield: 54 mg, 38%); IR (KBr) νmax: 1687, 1632 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.77–7.74 (m, 1H), 7.57–7.48 (m, 3H), 7.38–7.27 (m, 5H), 4.19 (d, J = 12.7 Hz, 1H), 4.00 (d, J = 12.7 Hz, 1H), 3.84 (s, 1H), 3.78–3.70 (m, 1H), 3.64–3.55 (m, 1H), 3.47–3.39 (m, 2H), 3.36–3.28 (m, 1H), 3.10–3.02 (m, 2H), 2.83–2.73 (m, 2H), 2.71–2.65 (m, 1H), 1.76–1.66 (m, 2H), 1.39–1.29 (m, 2H), 1.24–1.17 (m, 2H), 0.92–0.86 (m, 3H); 13C NMR (126 MHz, CDCl3) δ 167.1, 166.5, 141.1, 136.1, 132.0, 130.3, 129.2, 129.1, 128.3, 127.7, 124.5, 122.8, 97.1, 66.2, 65.8 62.9, 59.9, 51.6, 45.2, 42.1, 40.3, 31.1, 29.7, 20.6, 13.7; HRMS–ESI (m/z): [M + H]+ calcd for C26H32N3O4, 450.2393; found, 450.2422.
(1RS,4'RS)-2'-Benzyl-2-butyl-4'-(piperidine-1-carbonyl)spiro[isoindoline-1,5'-isoxazolidin]-3-one (6d): White sticky oil (Yield: 45 mg, 35%); IR (neat) νmax: 1684, 1631 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.73–7.69 (m, 1H), 7.54–7.43 (m, 3H), 7.37–7.34 (m, 2H), 7.33–7.26 (m, 3H), 4.18 (d, J = 12.7 Hz, 1H), 4.13–4.06 (m, 1H), 3.99 (d, J = 12.7 Hz, 1H), 3.90–3.81 (m, 1H), 3.78–3.70 (m, 1H), 3.61–3.55 (m, 1H), 3.51–3.43 (m, 1H), 3.18–3.10 (m, 1H), 3.08–2.96 (m, 2H), 2.76–2.67 (m, 1H), 1.78–1.69 (m, 2H), 1.39–1.23 (m, 8H), 0.89 (t, J = 7.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.4, 166.1, 141.4, 136.4, 131.9, 130.1, 129.4, 128.4, 127.8, 124.6, 122.6, 97.4, 63.1, 60.4, 51.6, 45.9, 42.9, 40.4, 31.2, 25.8, 25.2, 24.1, 20.7, 13.9; HRMS–ESI (m/z): [M + H]+ calcd for C27H34N3O3, 448.2600; found, 448.2629.
(1R,4'R)-2'-Benzyl-N,N,2-tributyl-3-oxospiro[isoindoline-1,5'-isoxazolidine]-4'-carboxamide (6e): Yellow oil (Yield: 52 mg, 38%); IR (neat) νmax: 1705, 1686, 1628 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.71–7.67 (m, 1H), 7.62–7.26 (m, 8H), 4.17 (d, J = 12.7 Hz, 1H), 4.10–4.03 (m, 1H), 3.99 (d, J = 12.7 Hz, 1H), 3.88–3.79 (m, 1H), 3.74–3.66 (m, 1H), 3.62–3.54 (m, 1H), 3.54–3.40 (m, 2H), 3.39–3.34 (m, 1H), 3.19–3.14 (m, 1H), 3.03–2.96 (m, 1H), 2.48–2.30 (m, 2H), 1.68–1.54 (m, 4H), 1.37–1.20 (m, 6H), 0.95–0.85 (m, 6H), 0.67 (t, J = 7.2 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.3, 166.8, 136.3, 131.9, 130.1, 129.5, 129.3, 128.5, 127.8, 127.7, 125.2, 122.6, 97.6, 63.1, 60.5, 51.6, 47.9, 47.1, 46.4, 45.3, 43.7, 40.1, 31.3, 30.8, 29.6, 28.9, 20.7, 20.2, 19.9, 14.0; HRMS–ESI (m/z): [M + H]+ calcd for C30H42N3O3, 492.3226; found, 492.3255.
(1RS,4'RS)-2'-Benzyl-4'-(morpholine-4-carbonyl)-2-phenylspiro[isoindoline-1,5'-isoxazolidin]-3-one (6f): Yellow oil (Yield: 91 mg, 65%); IR (neat) νmax: 1683, 1636 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.88–7.82 (m, 1H), 7.66–7.60 (m, 2H), 7.58–7.44 (m, 3H), 7.44–7.28 (m, 8H), 4.18 (d, J = 13.0 Hz, 1H), 4.10–3.96 (m, 1H), 3.78–3.72 (m, 1H), 3.62–3.57 (m, 1H), 3.56–3.49 (m, 1H), 3.49–3.44 (m, 1H), 3.44–3.39 (m, 1H), 3.39–3.32 (m, 2H), 3.24–3.05 (m, 3H), 2.86–2.79 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 166.9, 166.5, 136.2, 134.7, 133.1, 132.9, 131.0, 130.5, 129.9, 129.4, 129.0, 128.8, 128.4, 128.0, 127.8, 125.0, 123.6, 101.5, 66.9, 66.5, 66.0, 45.4, 42.45; HRMS–ESI (m/z): [M + H]+ calcd for C28H28N3O4, 470.2080; found, 470.2052.
(1RS,4'SR) 2'-Benzyl-2-tert-butyl-4'-(morpholine-4-carbonyl)spiro[isoindoline-1,5'-isoxazolidin]-3-one (7g): Yellow oil (Yield: 14 mg, 10%); IR (neat) νmax: 1681, 1629 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 7.4 Hz, 1H), 7.53–7.35 (m, 5H), 7.34–7.23 (m, 3H), 4.30–4.11 (m, 4H), 3.53–3.39 (m, 2H), 3.33–3.17 (m, 4H), 3.05–2.95 (m, 1H), 2.95–2.86 (m, 1H), 2.73–2.65 (m, 1H), 1.70 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 169.76, 136.12, 132.16, 129.71, 129.10, 128.46, 127.65, 122.20, 100.19, 66.49, 65.91, 57.79, 53.10, 45.81, 42.45, 29.68; HRMS–ESI (m/z): [M + H]+ calcd for C26H32N3O4, 450,2393; found, 450.2371.
Supporting Information
| Supporting Information File 1: Biological tests, 1H and 13C NMR spectra of all new compounds, computational methods and X-ray data. | ||
| Format: PDF | Size: 1.7 MB | Download |
Acknowledgements
We gratefully acknowledge the Italian Ministry of Education, Universities, and Research (MIUR: PRIN 2010-2011, 20109Z2XRJ_007), the Universities of Messina, Catania and Calabria, Interuniversity Research Centre on Pericyclic Reactions and Synthesis of Hetero and Carbocycles Systems and the Interuniversity Consortium for Innovative Methodologies and Processes for Synthesis (CINMPIS) for partial financial support. The authors are very grateful to Professor Pierluigi Caramella for helpful discussions and suggestions.
References
-
Teodoro, J. G.; Evans, S. K.; Green, M. R. J. Mol. Med. 2007, 85, 1175–1186. doi:10.1007/s00109-007-0221-2
Return to citation in text: [1] -
Fridman, J. S.; Lowe, S. W. Oncogene 2003, 22, 9030–9040. doi:10.1038/sj.onc.1207116
Return to citation in text: [1] -
Vazquez, A.; Bond, E. E.; Levine, A. J.; Bond, G. L. Nat. Rev. Drug Discovery 2008, 7, 979–987. doi:10.1038/nrd2656
Return to citation in text: [1] -
Vogelstein, B.; Lane, D.; Levine, A. J. Nature 2000, 408, 307–310. doi:10.1038/35042675
Return to citation in text: [1] -
Chène, P. Nat. Rev. Cancer 2003, 3, 102–109. doi:10.1038/nrc991
Return to citation in text: [1] [2] -
Momand, J.; Zambetti, G. P.; Olson, D. C.; George, D.; Levine, A. J. Cell 1992, 69, 1237–1245. doi:10.1016/0092-8674(92)90644-R
Return to citation in text: [1] -
Toledo, F.; Wahl, G. M. Nat. Rev. Cancer 2006, 6, 909–923. doi:10.1038/nrc2012
Return to citation in text: [1] -
Fuchs, S. Y.; Adler, V.; Buschmann, T.; Wu, X.; Ronai, Z. Oncogene 1998, 17, 2543–2547. doi:10.1038/sj.onc.1202200
Return to citation in text: [1] -
Millard, M.; Pathania, D.; Grande, F.; Xu, S.; Neamati, N. Curr. Pharm. Des. 2011, 17, 536–559. doi:10.2174/138161211795222649
Return to citation in text: [1] -
Fischer, P. Int. J. Pept. Res. Ther. 2006, 12, 3–19. doi:10.1007/s10989-006-9016-5
Return to citation in text: [1] -
Weber, L. Expert Opin. Ther. Pat. 2010, 20, 179–191.
Return to citation in text: [1] -
Vassilev, L. T.; Vu, B. T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E. A. Science 2004, 303, 844–848. doi:10.1126/science.1092472
Return to citation in text: [1] [2] [3] -
Rew, Y.; Sun, D.; Gonzalez-Lopez De Turiso, F.; Bartberger, M. D.; Beck, H. P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B. M.; Gustin, D.; Huang, X.; Jiang, M.; Jiao, X.; Jin, L.; Kayser, F.; Kopecky, D. J.; Li, Y.; Lo, M.-C.; Long, A. M.; Michelsen, K.; Oliner, J. D.; Osgood, T.; Ragains, M.; Saiki, A. Y.; Schneider, S.; Toteva, M.; Yakowec, P.; Yan, X.; Ye, Q.; Yu, D.; Zhao, X.; Zhou, J.; Medina, J. C.; Olson, S. H. J. Med. Chem. 2012, 55, 4936–4954. doi:10.1021/jm300354j
Return to citation in text: [1] -
Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Tomita, Y.; Parrish, D. A.; Deschamps, J. R.; Wang, S. J. Am. Chem. Soc. 2005, 127, 10130–10131. doi:10.1021/ja051147z
Return to citation in text: [1] -
Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R. S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; Bernard, D.; Zhang, J.; Lu, Y.; Gu, Q.; Shah, R. B.; Pienta, K. J.; Ling, X.; Kang, S.; Guo, M.; Sun, Y.; Yang, D.; Wang, S. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3933–3938. doi:10.1073/pnas.0708917105
Return to citation in text: [1] [2] -
Popowicz, G. M.; Czarna, A.; Wolf, S.; Wang, K.; Wang, W.; Dömling, A.; Holak, T. A. Cell Cycle 2010, 9, 1104–1111. doi:10.4161/cc.9.6.10956
Return to citation in text: [1] -
Yu, S.; Qin, D.; Shangary, S.; Chen, J.; Wang, G.; Ding, K.; McEachern, D.; Qiu, S.; Nikolovska-Coleska, Z.; Miller, R.; Kang, S.; Yang, D.; Wang, S. J. Med. Chem. 2009, 52, 7970–7973. doi:10.1021/jm901400z
Return to citation in text: [1] -
Yong, S. R.; Ung, A. T.; Pyne, S. G.; Skelton, P. W.; White, A. H. Tetrahedron 2007, 63, 5579–5586. doi:10.1016/j.tet.2007.04.028
Return to citation in text: [1] -
Mohammad, R. M.; Wu, J.; Azmi, A. S.; Aboukameel, A.; Sosin, A.; Wu, S.; Yang, D.; Wang, S.; Al-Katib, A. M. Mol. Cancer 2009, 8, No. 115. doi:10.1186/1476-4598-8-115
Return to citation in text: [1] -
Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 2007, 63, 2664–2669. doi:10.1016/j.tet.2007.01.021
Return to citation in text: [1] -
Daïch, A.; Marchalin, S.; Pigeon, P.; Decroix, B. Tetrahedron Lett. 1998, 39, 9187–9190. doi:10.1016/S0040-4039(98)02075-9
Return to citation in text: [1] -
Taniguchi, T.; Iwasaki, K.; Uchiyama, M.; Tamura, O.; Ishibashi, H. Org. Lett. 2005, 7, 4389–4390. doi:10.1021/ol051563o
Return to citation in text: [1] -
Fang, F. G.; Feigelson, G. B.; Danishefsky, S. J. Tetrahedron Lett. 1989, 30, 2743–2746. doi:10.1016/S0040-4039(00)99114-7
Return to citation in text: [1] -
Alonso, R.; Castedo, L.; Dominguez, D. Tetrahedron Lett. 1985, 26, 2925–2928. doi:10.1016/S0040-4039(00)98873-7
Return to citation in text: [1] -
Kim, G.; Jung, P.; Tuan, L. A. Tetrahedron Lett. 2008, 49, 2391–2392. doi:10.1016/j.tetlet.2008.02.057
Return to citation in text: [1] -
Stuk, T. L.; Assink, B. K.; Bates, R. C., Jr.; Erdman, D. T.; Fedij, V.; Jennings, S. M.; Lassig, J. A.; Smith, R. J.; Smith, T. L. Org. Process Res. Dev. 2003, 7, 851–855. doi:10.1021/op034060b
Return to citation in text: [1] -
Luzzio, F. A.; Zacherl, D. P.; Figg, W. D. Tetrahedron Lett. 1999, 40, 2087–2090. doi:10.1016/S0040-4039(99)00152-5
Return to citation in text: [1] -
Luzzio, F. A.; Mayorov, A. V.; Ng, S. S. W.; Kruger, E. A.; Figg, W. D. J. Med. Chem. 2003, 46, 3793–3799. doi:10.1021/jm020079d
Return to citation in text: [1] -
Norman, M. H.; Minick, D. J.; Rigdon, G. C. J. Med. Chem. 1996, 39, 149–157. doi:10.1021/jm9502201
Return to citation in text: [1] -
Luci, D. K.; Lawson, E. C.; Ghosh, S.; Kinney, W. A.; Smith, C. E.; Qi, J.; Wang, Y.; Minor, L. K.; Maryanoff, B. E. Tetrahedron Lett. 2009, 50, 4958–4961. doi:10.1016/j.tetlet.2009.06.025
Return to citation in text: [1] -
Papeo, G. M. E.; Krasavin, M. Y.; Orsini, P.; Scolaro, A. 4-Carboxamido-isoindolinone derivatives as selective parp-1 inhibitors. PCT Pat. Appl. WO2014064149A1, May 1, 2014.
Return to citation in text: [1] -
Lee, S.; Shinji, C.; Ogura, K.; Shimizu, M.; Maeda, S.; Sato, M.; Yoshida, M.; Hashimoto, Y.; Miyachi, H. Bioorg. Med. Chem. Lett. 2007, 17, 4895–4900. doi:10.1016/j.bmcl.2007.06.038
Return to citation in text: [1] -
Hardcastle, I. R.; Ahmed, S. U.; Atkins, H.; Farnie, G.; Golding, B. T.; Griffin, R. J.; Guyenne, S.; Hutton, C.; Källblad, P.; Kemp, S. J.; Kitching, M. S.; Newell, D. R.; Norbedo, S.; Northen, J. S.; Reid, R. J.; Saravanan, K.; Willems, H. M. G.; Lunec, J. J. Med. Chem. 2006, 49, 6209–6221. doi:10.1021/jm0601194
Return to citation in text: [1] -
Hardcastle, I. R.; Liu, J.; Valeur, E.; Watson, A.; Ahmed, S. U.; Blackburn, T. J.; Bennaceur, K.; Clegg, W.; Drummond, C.; Endicott, J. A.; Golding, B. T.; Griffin, R. J.; Gruber, J.; Haggerty, K.; Harrington, R. W.; Hutton, C.; Kemp, S.; Lu, X.; McDonnell, J. M.; Newell, D. R.; Noble, M. E. M.; Payne, S. L.; Revill, C. H.; Riedinger, C.; Xu, Q.; Lunec, J. J. Med. Chem. 2011, 54, 1233–1243. doi:10.1021/jm1011929
Return to citation in text: [1] -
Mancuso, R.; Ziccarelli, I.; Armentano, D.; Marino, N.; Giofrè, S. V.; Gabriele, B. J. Org. Chem. 2014, 79, 3506–3518. doi:10.1021/jo500281h
Return to citation in text: [1] [2] [3] -
Crystallographic data (excluding structure factors) for the structure 6a in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1479267. Copies of the data can be obtained, free of charge, via http://www.ccdc.cam.ac.uk/conts/retrieving.html or on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Return to citation in text: [1] -
Legnani, L.; Lunghi, C.; Marinone Albini, F.; Nativi, C.; Richichi, B.; Toma, L. Eur. J. Org. Chem. 2007, 3547–3554. doi:10.1002/ejoc.200700112
Return to citation in text: [1] -
Iannazzo, D.; Brunaccini, E.; Giofrè, S. V.; Piperno, A.; Romeo, G.; Ronsisvalle, S.; Chiacchio, M. A.; Lanza, G.; Chiacchio, U. Eur. J. Org. Chem. 2010, 5897–5905. doi:10.1002/ejoc.201000579
Return to citation in text: [1] -
Luparia, M.; Legnani, L.; Porta, A.; Zanoni, G.; Toma, L.; Vidari, G. J. Org. Chem. 2009, 74, 7100–7110. doi:10.1021/jo9014936
Return to citation in text: [1] -
Quadrelli, P.; Mella, M.; Legnani, L.; Al-Saad, D. Eur. J. Org. Chem. 2013, 4655–4665. doi:10.1002/ejoc.201300119
Return to citation in text: [1] -
Savion, M.; Memeo, M. G.; Bovio, B.; Grazioso, G.; Legnani, L.; Quadrelli, P. Tetrahedron 2012, 68, 1845–1852. doi:10.1016/j.tet.2011.12.086
Return to citation in text: [1] -
Masciocchi, D.; Villa, S.; Meneghetti, F.; Pedretti, A.; Barlocco, D.; Legnani, L.; Toma, L.; Kwon, B.-M.; Nakano, S.; Asai, A.; Gelain, A. Med. Chem. Commun. 2012, 3, 592–599. doi:10.1039/c2md20018j
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2010.
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] -
Rigolet, S.; Goncalo, P.; Mélot, J. M.; Vébrel, J. J. Chem. Res., Synop. 1998, 686–687. doi:10.1039/a803841d
Return to citation in text: [1] -
Romeo, R.; Giofrè, S. V.; Carnovale, C.; Chiacchio, M. A.; Campisi, A.; Mancuso, R.; Cirmi, S.; Navarra, M. Eur. J. Org. Chem. 2014, 5442–5447. doi:10.1002/ejoc.201402106
Return to citation in text: [1] -
Romeo, R.; Giofrè, S. V.; Garozzo, A.; Bisignano, B.; Corsaro, A.; Chiacchio, M. A. Bioorg. Med. Chem. 2013, 21, 5688–5693. doi:10.1016/j.bmc.2013.07.031
Return to citation in text: [1] -
Visalli, G.; Ferlazzo, N.; Cirmi, S.; Campiglia, P.; Gangemi, S.; Di Pietro, A.; Calapai, G.; Navarra, M. Anti-Cancer Agents Med. Chem. 2014, 14, 1402–1413. doi:10.2174/1871520614666140829120530
Return to citation in text: [1] -
Giofrè, S. V.; Romeo, R.; Carnovale, C.; Mancuso, R.; Cirmi, S.; Navarra, M.; Garozzo, A.; Chiacchio, M. A. Molecules 2015, 20, 5260–5275. doi:10.3390/molecules20045260
Return to citation in text: [1] -
Ferlazzo, N.; Visalli, G.; Smeriglio, A.; Cirmi, S.; Lombardo, G. E.; Campiglia, P.; Di Pietro, A.; Navarra, M. Evidence-Based Complementary Altern. Med. 2015, No. 957031. doi:10.1155/2015/957031
Return to citation in text: [1] -
Romeo, R.; Navarra, M.; Giofrè, S. V.; Carnovale, C.; Cirmi, S.; Lanza, G.; Chiacchio, M. A. Bioorg. Med. Chem. 2014, 22, 3379–3385. doi:10.1016/j.bmc.2014.04.047
Return to citation in text: [1] -
Ferlazzo, N.; Cirmi, S.; Russo, M.; Ursino, M. R.; Trapasso, E.; Lombardo, G. E.; Gangemi, S.; Calapai, G.; Navarra, M. Life Sci. 2016, 146, 81–91. doi:10.1016/j.lfs.2015.12.040
Return to citation in text: [1] -
Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Wang, S. J. Med. Chem. 2006, 49, 3432–3435. doi:10.1021/jm051122a
Return to citation in text: [1] -
Estrada-Ortiz, N.; Neochoritis, C. G.; Dömling, A. ChemMedChem 2016, 11, 757–772. doi:10.1002/cmdc.201500487
Return to citation in text: [1] -
Burla, M. C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. J. Appl. Crystallogr. 2007, 40, 609–613. doi:10.1107/S0021889807010941
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/S2053229614024218
Return to citation in text: [1] -
Aschwanden, P.; Kværnø, L.; Geisser, R. W.; Kleinbeck, F.; Carreira, E. M. Org. Lett. 2005, 7, 5741–5742. doi:10.1021/ol052540c
Return to citation in text: [1] -
Romeo, R.; Giofrè, S. V.; Carnovale, C.; Campisi, A.; Parenti, R.; Bandini, L.; Chiacchio, M. A. Bioorg. Med. Chem. 2013, 21, 7929–7937. doi:10.1016/j.bmc.2013.10.001
Return to citation in text: [1] -
Romeo, R.; Carnovale, C.; Giofrè, S. V.; Chiacchio, M. A.; Garozzo, A.; Amata, E.; Romeo, G.; Chiacchio, U. Beilstein J. Org. Chem. 2015, 11, 328–334. doi:10.3762/bjoc.11.38
Return to citation in text: [1]
| 55. | Burla, M. C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G. L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. J. Appl. Crystallogr. 2007, 40, 609–613. doi:10.1107/S0021889807010941 |
| 56. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/S2053229614024218 |
| 35. | Mancuso, R.; Ziccarelli, I.; Armentano, D.; Marino, N.; Giofrè, S. V.; Gabriele, B. J. Org. Chem. 2014, 79, 3506–3518. doi:10.1021/jo500281h |
| 57. | Aschwanden, P.; Kværnø, L.; Geisser, R. W.; Kleinbeck, F.; Carreira, E. M. Org. Lett. 2005, 7, 5741–5742. doi:10.1021/ol052540c |
| 58. | Romeo, R.; Giofrè, S. V.; Carnovale, C.; Campisi, A.; Parenti, R.; Bandini, L.; Chiacchio, M. A. Bioorg. Med. Chem. 2013, 21, 7929–7937. doi:10.1016/j.bmc.2013.10.001 |
| 59. | Romeo, R.; Carnovale, C.; Giofrè, S. V.; Chiacchio, M. A.; Garozzo, A.; Amata, E.; Romeo, G.; Chiacchio, U. Beilstein J. Org. Chem. 2015, 11, 328–334. doi:10.3762/bjoc.11.38 |
| 1. | Teodoro, J. G.; Evans, S. K.; Green, M. R. J. Mol. Med. 2007, 85, 1175–1186. doi:10.1007/s00109-007-0221-2 |
| 2. | Fridman, J. S.; Lowe, S. W. Oncogene 2003, 22, 9030–9040. doi:10.1038/sj.onc.1207116 |
| 3. | Vazquez, A.; Bond, E. E.; Levine, A. J.; Bond, G. L. Nat. Rev. Drug Discovery 2008, 7, 979–987. doi:10.1038/nrd2656 |
| 4. | Vogelstein, B.; Lane, D.; Levine, A. J. Nature 2000, 408, 307–310. doi:10.1038/35042675 |
| 12. | Vassilev, L. T.; Vu, B. T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E. A. Science 2004, 303, 844–848. doi:10.1126/science.1092472 |
| 13. | Rew, Y.; Sun, D.; Gonzalez-Lopez De Turiso, F.; Bartberger, M. D.; Beck, H. P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B. M.; Gustin, D.; Huang, X.; Jiang, M.; Jiao, X.; Jin, L.; Kayser, F.; Kopecky, D. J.; Li, Y.; Lo, M.-C.; Long, A. M.; Michelsen, K.; Oliner, J. D.; Osgood, T.; Ragains, M.; Saiki, A. Y.; Schneider, S.; Toteva, M.; Yakowec, P.; Yan, X.; Ye, Q.; Yu, D.; Zhao, X.; Zhou, J.; Medina, J. C.; Olson, S. H. J. Med. Chem. 2012, 55, 4936–4954. doi:10.1021/jm300354j |
| 14. | Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Tomita, Y.; Parrish, D. A.; Deschamps, J. R.; Wang, S. J. Am. Chem. Soc. 2005, 127, 10130–10131. doi:10.1021/ja051147z |
| 35. | Mancuso, R.; Ziccarelli, I.; Armentano, D.; Marino, N.; Giofrè, S. V.; Gabriele, B. J. Org. Chem. 2014, 79, 3506–3518. doi:10.1021/jo500281h |
| 9. | Millard, M.; Pathania, D.; Grande, F.; Xu, S.; Neamati, N. Curr. Pharm. Des. 2011, 17, 536–559. doi:10.2174/138161211795222649 |
| 10. | Fischer, P. Int. J. Pept. Res. Ther. 2006, 12, 3–19. doi:10.1007/s10989-006-9016-5 |
| 11. | Weber, L. Expert Opin. Ther. Pat. 2010, 20, 179–191. |
| 36. | Crystallographic data (excluding structure factors) for the structure 6a in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1479267. Copies of the data can be obtained, free of charge, via http://www.ccdc.cam.ac.uk/conts/retrieving.html or on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk). |
| 5. | Chène, P. Nat. Rev. Cancer 2003, 3, 102–109. doi:10.1038/nrc991 |
| 6. | Momand, J.; Zambetti, G. P.; Olson, D. C.; George, D.; Levine, A. J. Cell 1992, 69, 1237–1245. doi:10.1016/0092-8674(92)90644-R |
| 7. | Toledo, F.; Wahl, G. M. Nat. Rev. Cancer 2006, 6, 909–923. doi:10.1038/nrc2012 |
| 8. | Fuchs, S. Y.; Adler, V.; Buschmann, T.; Wu, X.; Ronai, Z. Oncogene 1998, 17, 2543–2547. doi:10.1038/sj.onc.1202200 |
| 33. | Hardcastle, I. R.; Ahmed, S. U.; Atkins, H.; Farnie, G.; Golding, B. T.; Griffin, R. J.; Guyenne, S.; Hutton, C.; Källblad, P.; Kemp, S. J.; Kitching, M. S.; Newell, D. R.; Norbedo, S.; Northen, J. S.; Reid, R. J.; Saravanan, K.; Willems, H. M. G.; Lunec, J. J. Med. Chem. 2006, 49, 6209–6221. doi:10.1021/jm0601194 |
| 34. | Hardcastle, I. R.; Liu, J.; Valeur, E.; Watson, A.; Ahmed, S. U.; Blackburn, T. J.; Bennaceur, K.; Clegg, W.; Drummond, C.; Endicott, J. A.; Golding, B. T.; Griffin, R. J.; Gruber, J.; Haggerty, K.; Harrington, R. W.; Hutton, C.; Kemp, S.; Lu, X.; McDonnell, J. M.; Newell, D. R.; Noble, M. E. M.; Payne, S. L.; Revill, C. H.; Riedinger, C.; Xu, Q.; Lunec, J. J. Med. Chem. 2011, 54, 1233–1243. doi:10.1021/jm1011929 |
| 35. | Mancuso, R.; Ziccarelli, I.; Armentano, D.; Marino, N.; Giofrè, S. V.; Gabriele, B. J. Org. Chem. 2014, 79, 3506–3518. doi:10.1021/jo500281h |
| 20. | Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 2007, 63, 2664–2669. doi:10.1016/j.tet.2007.01.021 |
| 21. | Daïch, A.; Marchalin, S.; Pigeon, P.; Decroix, B. Tetrahedron Lett. 1998, 39, 9187–9190. doi:10.1016/S0040-4039(98)02075-9 |
| 22. | Taniguchi, T.; Iwasaki, K.; Uchiyama, M.; Tamura, O.; Ishibashi, H. Org. Lett. 2005, 7, 4389–4390. doi:10.1021/ol051563o |
| 23. | Fang, F. G.; Feigelson, G. B.; Danishefsky, S. J. Tetrahedron Lett. 1989, 30, 2743–2746. doi:10.1016/S0040-4039(00)99114-7 |
| 24. | Alonso, R.; Castedo, L.; Dominguez, D. Tetrahedron Lett. 1985, 26, 2925–2928. doi:10.1016/S0040-4039(00)98873-7 |
| 25. | Kim, G.; Jung, P.; Tuan, L. A. Tetrahedron Lett. 2008, 49, 2391–2392. doi:10.1016/j.tetlet.2008.02.057 |
| 31. | Papeo, G. M. E.; Krasavin, M. Y.; Orsini, P.; Scolaro, A. 4-Carboxamido-isoindolinone derivatives as selective parp-1 inhibitors. PCT Pat. Appl. WO2014064149A1, May 1, 2014. |
| 15. | Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R. S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; Bernard, D.; Zhang, J.; Lu, Y.; Gu, Q.; Shah, R. B.; Pienta, K. J.; Ling, X.; Kang, S.; Guo, M.; Sun, Y.; Yang, D.; Wang, S. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3933–3938. doi:10.1073/pnas.0708917105 |
| 19. | Mohammad, R. M.; Wu, J.; Azmi, A. S.; Aboukameel, A.; Sosin, A.; Wu, S.; Yang, D.; Wang, S.; Al-Katib, A. M. Mol. Cancer 2009, 8, No. 115. doi:10.1186/1476-4598-8-115 |
| 32. | Lee, S.; Shinji, C.; Ogura, K.; Shimizu, M.; Maeda, S.; Sato, M.; Yoshida, M.; Hashimoto, Y.; Miyachi, H. Bioorg. Med. Chem. Lett. 2007, 17, 4895–4900. doi:10.1016/j.bmcl.2007.06.038 |
| 15. | Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R. S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; Bernard, D.; Zhang, J.; Lu, Y.; Gu, Q.; Shah, R. B.; Pienta, K. J.; Ling, X.; Kang, S.; Guo, M.; Sun, Y.; Yang, D.; Wang, S. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3933–3938. doi:10.1073/pnas.0708917105 |
| 16. | Popowicz, G. M.; Czarna, A.; Wolf, S.; Wang, K.; Wang, W.; Dömling, A.; Holak, T. A. Cell Cycle 2010, 9, 1104–1111. doi:10.4161/cc.9.6.10956 |
| 17. | Yu, S.; Qin, D.; Shangary, S.; Chen, J.; Wang, G.; Ding, K.; McEachern, D.; Qiu, S.; Nikolovska-Coleska, Z.; Miller, R.; Kang, S.; Yang, D.; Wang, S. J. Med. Chem. 2009, 52, 7970–7973. doi:10.1021/jm901400z |
| 18. | Yong, S. R.; Ung, A. T.; Pyne, S. G.; Skelton, P. W.; White, A. H. Tetrahedron 2007, 63, 5579–5586. doi:10.1016/j.tet.2007.04.028 |
| 12. | Vassilev, L. T.; Vu, B. T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E. A. Science 2004, 303, 844–848. doi:10.1126/science.1092472 |
| 26. | Stuk, T. L.; Assink, B. K.; Bates, R. C., Jr.; Erdman, D. T.; Fedij, V.; Jennings, S. M.; Lassig, J. A.; Smith, R. J.; Smith, T. L. Org. Process Res. Dev. 2003, 7, 851–855. doi:10.1021/op034060b |
| 27. | Luzzio, F. A.; Zacherl, D. P.; Figg, W. D. Tetrahedron Lett. 1999, 40, 2087–2090. doi:10.1016/S0040-4039(99)00152-5 |
| 28. | Luzzio, F. A.; Mayorov, A. V.; Ng, S. S. W.; Kruger, E. A.; Figg, W. D. J. Med. Chem. 2003, 46, 3793–3799. doi:10.1021/jm020079d |
| 29. | Norman, M. H.; Minick, D. J.; Rigdon, G. C. J. Med. Chem. 1996, 39, 149–157. doi:10.1021/jm9502201 |
| 30. | Luci, D. K.; Lawson, E. C.; Ghosh, S.; Kinney, W. A.; Smith, C. E.; Qi, J.; Wang, Y.; Minor, L. K.; Maryanoff, B. E. Tetrahedron Lett. 2009, 50, 4958–4961. doi:10.1016/j.tetlet.2009.06.025 |
| 44. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 37. | Legnani, L.; Lunghi, C.; Marinone Albini, F.; Nativi, C.; Richichi, B.; Toma, L. Eur. J. Org. Chem. 2007, 3547–3554. doi:10.1002/ejoc.200700112 |
| 38. | Iannazzo, D.; Brunaccini, E.; Giofrè, S. V.; Piperno, A.; Romeo, G.; Ronsisvalle, S.; Chiacchio, M. A.; Lanza, G.; Chiacchio, U. Eur. J. Org. Chem. 2010, 5897–5905. doi:10.1002/ejoc.201000579 |
| 39. | Luparia, M.; Legnani, L.; Porta, A.; Zanoni, G.; Toma, L.; Vidari, G. J. Org. Chem. 2009, 74, 7100–7110. doi:10.1021/jo9014936 |
| 40. | Quadrelli, P.; Mella, M.; Legnani, L.; Al-Saad, D. Eur. J. Org. Chem. 2013, 4655–4665. doi:10.1002/ejoc.201300119 |
| 41. | Savion, M.; Memeo, M. G.; Bovio, B.; Grazioso, G.; Legnani, L.; Quadrelli, P. Tetrahedron 2012, 68, 1845–1852. doi:10.1016/j.tet.2011.12.086 |
| 42. | Masciocchi, D.; Villa, S.; Meneghetti, F.; Pedretti, A.; Barlocco, D.; Legnani, L.; Toma, L.; Kwon, B.-M.; Nakano, S.; Asai, A.; Gelain, A. Med. Chem. Commun. 2012, 3, 592–599. doi:10.1039/c2md20018j |
| 12. | Vassilev, L. T.; Vu, B. T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E. A. Science 2004, 303, 844–848. doi:10.1126/science.1092472 |
| 53. | Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Wang, S. J. Med. Chem. 2006, 49, 3432–3435. doi:10.1021/jm051122a |
| 54. | Estrada-Ortiz, N.; Neochoritis, C. G.; Dömling, A. ChemMedChem 2016, 11, 757–772. doi:10.1002/cmdc.201500487 |
| 50. | Ferlazzo, N.; Visalli, G.; Smeriglio, A.; Cirmi, S.; Lombardo, G. E.; Campiglia, P.; Di Pietro, A.; Navarra, M. Evidence-Based Complementary Altern. Med. 2015, No. 957031. doi:10.1155/2015/957031 |
| 51. | Romeo, R.; Navarra, M.; Giofrè, S. V.; Carnovale, C.; Cirmi, S.; Lanza, G.; Chiacchio, M. A. Bioorg. Med. Chem. 2014, 22, 3379–3385. doi:10.1016/j.bmc.2014.04.047 |
| 52. | Ferlazzo, N.; Cirmi, S.; Russo, M.; Ursino, M. R.; Trapasso, E.; Lombardo, G. E.; Gangemi, S.; Calapai, G.; Navarra, M. Life Sci. 2016, 146, 81–91. doi:10.1016/j.lfs.2015.12.040 |
| 48. | Visalli, G.; Ferlazzo, N.; Cirmi, S.; Campiglia, P.; Gangemi, S.; Di Pietro, A.; Calapai, G.; Navarra, M. Anti-Cancer Agents Med. Chem. 2014, 14, 1402–1413. doi:10.2174/1871520614666140829120530 |
| 49. | Giofrè, S. V.; Romeo, R.; Carnovale, C.; Mancuso, R.; Cirmi, S.; Navarra, M.; Garozzo, A.; Chiacchio, M. A. Molecules 2015, 20, 5260–5275. doi:10.3390/molecules20045260 |
| 45. | Rigolet, S.; Goncalo, P.; Mélot, J. M.; Vébrel, J. J. Chem. Res., Synop. 1998, 686–687. doi:10.1039/a803841d |
| 46. | Romeo, R.; Giofrè, S. V.; Carnovale, C.; Chiacchio, M. A.; Campisi, A.; Mancuso, R.; Cirmi, S.; Navarra, M. Eur. J. Org. Chem. 2014, 5442–5447. doi:10.1002/ejoc.201402106 |
| 47. | Romeo, R.; Giofrè, S. V.; Garozzo, A.; Bisignano, B.; Corsaro, A.; Chiacchio, M. A. Bioorg. Med. Chem. 2013, 21, 5688–5693. doi:10.1016/j.bmc.2013.07.031 |
© 2016 Giofrè et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)