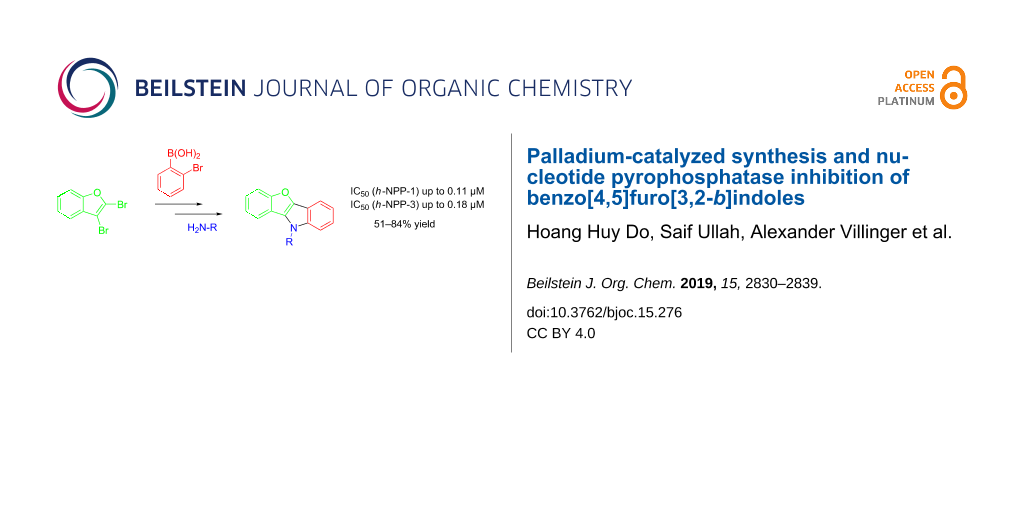
Abstract
A two-step palladium-catalyzed procedure based on Suzuki–Miyaura cross coupling, followed by a double Buchwald–Hartwig reaction, allows for the synthesis of pharmaceutically relevant benzo[4,5]furo[3,2-b]indoles in moderate to very good yield. The synthesized compounds have been analyzed with regard to their inhibitory activity (IC50) of nucleotide pyrophosphatases h-NPP1 and h-NPP3. The activity lies in the nanomolar range. The results were rationalized based on docking studies.

Graphical Abstract
Introduction
Furoindoles and their derivatives have received a lot of attention based on their versatile pharmaceutical activities. Furoindols were reported to show potent antiallergic [1-4], anticancer [5,6], analgesic and anti-inflammatory activity [7] (Figure 1). Similarly, benzofuroindol has been studied intensively as a pharmacophore of calcium-activated potassium channel (BKCa) opening activity [8-13]. Hence, benzofuroindols have been discussed as potential drug candidates for antispasmodic activity and thus therapeutic treatment of, e.g., urge urinary incontinence [8,9,11]. In addition, derivatives might be applied for therapeutic treatment of stroke, asthma, hypertensions, convulsion, traumatic brain injury [10] or treatment of erectile dysfunction [12,13]. Other derivatives show selective activity as estrogen, androgen and/or progestrin receptor modulators [14].
![[1860-5397-15-276-1]](/bjoc/content/figures/1860-5397-15-276-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Pharmacologically relevant furoindoles.
Figure 1: Pharmacologically relevant furoindoles.
Hence, the synthesis of furoindoles has been intensively studied in recent years [8,10,15-21]. We and others extensively studied double Buchwald–Hartwig reactions as the key step for the synthesis of heterocycles. For example, the cyclization of 2,2’-dibromobiaryls with amines allows for a convenient synthesis of carbazole derivatives [22-30]. Recently, we reported the synthesis of diindolofurans by regioselective Suzuki–Miyaura couplings of tetrabromofuran and subsequent cyclization by tetrafold Buchwald–Hartwig reaction [31]. We also studied the synthesis of benzo[4,5]furo[3,2-b]indoles by a similar concept. However, while performing our studies, Truong et al. reported the synthesis of these target molecules by a related strategy. The cyclization of 2-alkynylphenols with iodinde gave a 2-(2-bromophenyl)-3-iodobenzo[b]furan which could be cyclized by Buchwald–Hartwig reactions [32]. Altogether, the synthesis of four derivatives was reported. Herein, we wish to report the synthesis of ten benzo[4,5]furo[3,2-b]indole derivatives based on regioselective Suzuki–Miyaura reaction of 2,3-dibromobenzofuran with 2-bromophenylboronic acid and subsequent cyclization. The difference between the work of Truong and our approach mainly lies in the synthesis of the cyclization precursor. The method of Truong and our approach are equally efficient in this regard. However, the use of 2-alkynylphenols, as reported by Truong, requires one or two additional synthetic steps. The Buchwald–Hartwig reaction was individually optimized by both groups. While Truong and co-workers exclusively used anilines as reagents, we also successfully employed alkyl- and benzylamines which required an additional optimization of the conditions and the employment of different ligands. Therefore, we feel that our approach is more general and merits publication. In addition to the synthetic work, we report, for the first time, a study related to the activity of the products as nucleotide pyrophosphatase inhibitors. In this context, we also studied the biological activity of previously synthesized diindolofurans and the results are compared with those of benzofuroindoles.
Results and Discussion
Following a literature procedure, 2,3-dibromobenzofuran (1) was synthesized by bromination of benzofuran [33]. The Suzuki–Miyaura reaction of 1 with 2-bromophenylboronic acid (2), carried out under standard conditions using Pd(PPh3)4, afforded the desired product 3 in 84% yield and with very good regioselectivity. The synthesis of benzo[4,5]furo[3,2-b]indoles by double Buchwald–Hartwig reaction was studied next. The conditions were optimized for the reaction of 3 with p-toluidine (4b, Scheme 1, Table 1). The amount of ligand and palladium precursor was optimized using different solvents (dioxane, toluene, and DMF). Monodentate ligands, like XPhos, SPhos, DavePhos, RuPhos, or P(t-Bu)3·HBF4, were not effective in the reaction and gave product 5b in low yields. Bidentate phosphine ligands, such as BINAP, XantPhos, dppe, or dppf (Table 1), worked very well and allowed to improve the yield of 5b up to 75% (Table 1, entry 4). As compared to Pd2(dba)3, the use of Pd(OAc)2 as the Pd source resulted in a decrease of the yield (52%). Performing the reaction in dioxane or DMF gave lower yields as well. In summary, up to 75% yield of 5b could be achieved using BINAP and Pd2(dba)3 as the catalytic system.
![[1860-5397-15-276-i1]](/bjoc/content/inline/1860-5397-15-276-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of benzo[4,5]furo[3,2-b]indoles 5a–j. Conditions: (i) 1.2 equiv 2-bromophenylboronic acid (2), 5 mol % Pd(PPh3)4, 3.0 equiv K3PO4, 1,4-dioxane, H2O, 100 ºC, 8 h. (ii) 1.5 equiv 4, 3.0 equiv NaOt-Bu, 5 mol % Pd2(dba)3, 10 mol % ligand, toluene, 110 ºC, 12 h.
Scheme 1: Synthesis of benzo[4,5]furo[3,2-b]indoles 5a–j. Conditions: (i) 1.2 equiv 2-bromophenylboronic acid...
Table 1: Optimization for the Synthesis of 5b.
| Entry | Pd precursor | Ligand | Yield (%)a |
| 1 | Pd2(dba)3 | dppf | 57 |
| 2 | Pd2(dba)3 | XantPhos | 54 |
| 3 | Pd2(dba)3 | dppe | 62 |
| 4 | Pd2(dba)3 | BINAP | 75 |
| 5 | Pd2(dba)3 | XPhos | 36 |
| 6 | Pd2(dba)3 | SPhos | 44 |
| 7 | Pd2(dba)3 | DavePhos | 15 |
| 8 | Pd2(dba)3 | RuPhos | 45 |
| 9 | Pd2(dba)3 | Pt-Bu3·HBF4 | 41 |
| 10 | Pd(OAc)2 | BINAP | 52 |
| 11b | Pd2(dba)3 | BINAP | 61 |
| 12c | Pd2(dba)3 | BINAP | 14 |
aYield calculated by 1H NMR of the crude product using 1,4-dioxane as an internal standard. bDioxane. cDMF.
Subsequently, the scope of the reaction of 3 was studied using different amines. The reaction of 3 with various anilines afforded products 5a–g in good to excellent yields (Table 2). No impact of the functional groups of the anilines on the yield was observed. The reactions of 3 with aliphatic or benzylic amines under our optimized conditions proved to be unsuccessful and resulted in the formation of complex mixtures. Therefore, an additional ligand optimization was carried out for the reaction of 3 with benzylamine. It was found that DavePhos in the presence of Pd2dba3 improved the yield of product 5h drastically to 67% (Table 2). Consequently, our newly optimized conditions allowed the synthesis of products 5h–j in moderate to good yields (Table 2).
Table 2: Synthesis of 5a–ja.
| 5 | R | Yield (%)b |
| a | Ph | 63 |
| b | 4-MeC6H4 | 75 |
| c | 4-FC6H4 | 79 |
| d | 3-(CF3)C6H4 | 81 |
| e | 4-(MeO)C6H4 | 65 |
| f | 3,4-(MeO)2C6H3 | 51 |
| g | 4-t-BuC6H4 | 84 |
| h | Bn | 67c |
| i | n-C7H15 | 53c |
| j | Cyclohexyl | 57c |
aConditions: 1.5 equiv amine 4, 3.0 equiv NaOt-Bu, 5 mol % Pd2(dba)3, 10 mol % ligand, toluene, 110 ºC, 12 h; bIsolated yields; cDavePhos as ligand.
The structure of 5c was independently confirmed by X-ray crystal structure analysis. Figure 2 shows the planar benzofuroindol core structure with an orthogonally oriented aryl substituent located on the nitrogen atom.
![[1860-5397-15-276-2]](/bjoc/content/figures/1860-5397-15-276-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Ortep of 5c (propability of ellipsoids: 45%).
Figure 2: Ortep of 5c (propability of ellipsoids: 45%).
Nucleotide pyrophosphatase activity
Nucleotide pyrophosphatases belong to the family of ecto-nucleotidases [34,35]. They can hydrolyze nucleotides, dinucleotides, and nucleotide sugars, e.g., ATP, ADP, NAD+, ADP-ribose and diadenosine polyphosphates [36]. The isozymes of NPPs are involved in the pathophysiology of different diseases, such as calcification, cancer, and insulin resistance [34,35]. All derivatives of compounds 5 were tested for human recombinant NPPs, i.e., NPP1–3. In addition, we compared the obtained results with those of recently reported diindolofurans 6a–e (Figure 3) [31]. All compounds show significant inhibition of enzyme h-NPP-3 (Table 3) and most of them of the enzyme h-NPP-1.
![[1860-5397-15-276-3]](/bjoc/content/figures/1860-5397-15-276-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 3: Biological activity of 5 and 6.
|
h-NPP-1
IC50 (µM) ± SEMa |
h-NPP-3
IC50(µM) ± SEMa |
|
| 5a | – | 1.38 ± 0.03 |
| 5b | 2.84 ± 0.06 | 0.59 ± 0.02 |
| 5c | 1.29 ± 0.07 | 3.14 ± 0.09 |
| 5d | 3.57 ± 0.03 | 0.49 ± 0.04 |
| 5e | – | 0.26 ± 0.01 |
| 5h | 2.62 ± 0.03 | 0.27 ± 0.06 |
| 5i | 3.27 ± 0.08 | 2.55 ± 0.07 |
| 5j | 6.14 ± 0.09 | 2.39 ± 0.05 |
| 6a | 0.11 ± 0.06 | 0.61 ± 0.09 |
| 6b | – | 0.13 ± 0.06 |
| 6c | – | 0.28 ± 0.04 |
| 6d | 1.38 ± 0.09 | 0.18 ± 0.01 |
| 6e | 0.53 ± 0.09 | 0.21 ± 0.04 |
aThe IC50 is the concentration at which 50% of the enzyme activity is inhibited.
Compound 5a, containing a phenyl substituent, and compound 5e, containing a p-methoxyphenyl group, showed a selective inhibitory response towards nucleotide pyrophosphatase enzyme h-NPP-3. In case of 5e, an inhibitory value IC50 ± SEM = 0.26 ± 0.01 µM was observed which, thus, might be considered as potential inhibitor of h-NPP-3. Compound 5c with an inhibitory value for h-NPP-1 of IC50 ± SEM = 1.29 ± 0.07 µM was more active against NPP1 than against NPP3, while compounds 5b, 5d, and 5h were more active against NPP3 than NPP1. Compounds 5j and 5i, containing aliphatic groups, were found to be much less active against both of the two enzymes.
Compounds 6b and 6c with fluorinated functional groups (F-C6H4 and CF3C6H4) proved to be highly selective towards h-NPP-3 (IC50 ± SEM = 0.13 ± 0.06 and 0.28 ± 0.04, respectively). In fact, they had no interaction with enzyme h-NPP-1. Compound 6a, containing a tolyl group, was active against both h-NPP-1 and h-NPP-3, but was more selective to h-NPP-1 with an inhibitory value of IC50 ± SEM = 0.11 ± 0.06 µM. In contrast, compounds 6d with a methoxy group and 6e with a benzyl group were more active against NPP-3 than against NPP-1.
Compounds 5e and 6d with the methoxy functional group showed high inhibition of h-NPP-3. This suggests that the methoxy group could enhance the inhibition of h-NPP-3. Furo[3,2-b,4,5-b']diindoles 6 exhibited an even stronger activity than derivatives 5 which might be caused by their bigger heterocyclic moiety. All compounds 5 and 6 were active to inhibit enzymes h-NPP-3, which suggests that the furoindole core structure is the main pharmacophore for the inhibition against h-NPP, while changes of the substitution pattern allow for a modification of the selectivity and activity of these compounds to these enzymes.
Docking studies of h-ENPP1 inhibitors
Molecular docking of the most potent compounds 5c and 6a (for ENPP1) and for 6e (exhibiting dual inhibition for both isozymes) were performed to identify binding interactions, as illustrated in Figure 4. The 3D binding interaction study of suramin revealed a number of binding interactions with amino acid residues. Due to its bulky structure, suramin showed linkage inside and over the surface of the enzyme pocket. The important residues involved in hydrogen bonding were His380, Asn277, Ser378, Ser381, Tyr382, Lys391, Ser387 and Ser386. In addition, the study also showed binding with the zinc ion and π-interactions, in particular, π–π T-shaped and amide–π shaped coupling with Ser377 and Tyr382. The molecular docking study of compound 5c exhibited π–π T-shaped interactions connecting the indole and furan rings with Tyr340. However, π–alkyl linkage was observed between the benzofuran moiety of compound 5c and amino acid Lys295. The fluorine atom of the 4-fluorophenyl group was involved in the binding with the zinc ion and Ser377. Hydrogen bonding was found between the oxygen of the benzofuran ring and the hydrogens of Lys291. When compound 6a was docked inside the active pocket of the homology model, it represented π–π stacked and π–alkyl attachment of the indole rings on both sides of the molecule with amino acids His380 and Lys295. However, the zinc–metal interaction was exhibited by the substituted phenyl moiety on the indole ring. The oxygen atom of the furan ring of dual inhibitor 6e showed a single hydrogen bond with Phe548. Asp423 was found to be involved in two π–cation bindings with the indole ring, whereas π–π stacked, π–π T-shaped and π–alkyl related bindings were noticed, connecting Ile235, Phe220, Ala546, Trp322 and Ile419. The fluorine atom was perceived interacting with Ile419.
![[1860-5397-15-276-4]](/bjoc/content/figures/1860-5397-15-276-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Illustration of binding poses of selected inhibitors for the ENPP1 homology model: (a): suramin, (b): 5c, (c): 6a, (d): 6e.
Figure 4: Illustration of binding poses of selected inhibitors for the ENPP1 homology model: (a): suramin, (b...
Docking studies of h-ENPP3 inhibitors
The binding of ENPP3 was studied for selective inhibitor 5e. A dual affinity was observed for 5h and 6e. Suramin was used as a positive control. The 3D illustrations of these docked compounds are shown in Figure 5. The binding was observed in a large area of the modelled ENPP3 protein, inside and over the border of the active pocket, due to the bulky structure of suramin. The amino acids Asn290, Ser292, Asn245, Glu275, Tyr289, Leu239, Ser326, Thr205, His329 and Glu322 were involved in the hydrogen binding with suramin. One π–alkyl interaction was found with Pro288 and one metal interaction with zinc. Amino acid Tyr289 was connected with the sulfur atom as shown in Figure 5a. The hydrogen atoms of the methoxy group, located on the phenyl ring of compound 5e, were engaged in carbon–hydrogen bonds with Asp225 and Glu400, as depicted in Figure 5b. However, phenyl rings of both indole and benzofuran scaffolds were inclined towards Leu239 and His329, respectively. When compound 5h was taken into account, it was noticed that both five and six-membered rings of the benzofuran moiety were linked via π–π stacked relation with His483. However, the aromatic ring of indole demonstrated π–alkyl bonding by Leu239. The additional π–anion interaction was observed with the phosphate group and benzofuran. The 3D docking of compound 6e revealed distinct bonding, due to the presence of a fluorine atom on the phenyl ring. The fluorine atom was attached to the side chains of Asn226 and Asp225. The aromatic ring of indole was coupled via π–alkyl line interactions with Leu239 (Figure 5d).
![[1860-5397-15-276-5]](/bjoc/content/figures/1860-5397-15-276-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: 3D poses of docked selected inhibitors inside homology model of ENPP3. (a): suramin, (b): 5e, (c): 5h, (d): 6e.
Figure 5: 3D poses of docked selected inhibitors inside homology model of ENPP3. (a): suramin, (b): 5e, (c): ...
The docking studies of selected inhibitors on isozymes ENPP1 and ENPP3 modelled proteins were in accordance with in vitro experimental studies.
Conclusion
In conclusion, we have reported a convenient strategy for the preparation of benzo[4,5]furo[3,2-b]indoles based on Suzuki–Miyaura coupling reactions followed by Pd-catalyzed double C,N-coupling. All products are potent inhibitors of nucleotide pyrophosphatases with sensitivities and selectivities several times greater than those of previously reported inhibitors of h-NPP-1 and h-NPP-3 [37]. Biological activities of these compounds are prone to changes of the substituents. The activities and selectivity of the compounds reported make them ideal targets for further applications in medicinal chemistry.
Experimental
General
All reagents were obtained from commercial sources and used without further purification. Reactions were conducted in oven-dried glassware under an inert atmosphere of argon. Concentrations (c) in the general procedures refer to the limiting reagent and are given in mmol/mL. Thin-layer chromatography (TLC) was carried out on Merck aluminium foil backed sheets precoated with 0.2 mm Kielselgel 60 F254. The spots were visualized by UV irradiation (λ = 254 nm). Column chromatography was performed on Fluka silica gel 60 (0.063–0.200 mm, 70–320 mesh) on a glass column. For the cation exchange column dowex 50WX8 H+ was used. Melting points (mp) were determined by the instrument Elektrothermal. 1H and 13C NMR spectra («Mercury-300 Varian» 300 MHz with Bruker AV 500 (75 MHz), Bruker AV 300 III (62.9 MHz), respectively) were recorded using TMS as an internal standard (0 ppm). NMR spectra are calibrated by solvent at 7.26 (CDCl3) and 77.23 (CDCl3) for 13C NMR spectra. 13C NMR spectra were measured proton decoupled. MS and HRMS spectra were recorded with a Finnigan MAT 95 XP spectrometer. IR spectra were recorded with a Nicolet 6700 FT-IR spectrometer.
General procedure for the preparation of 3-bromo-2-(2-bromophenyl)benzofuran (3): Analogously as previously described [31], 2,3-dibromobenzofuran (1), 2-bromophenylboronic acid (2), Pd(PPh3)4 (5 mol %) and K3PO4 were added to a Schlenk flask under Argon atmosphere. To the mixture 70 mL 1,4-dioxane and 10 mL distilled water were added. The reaction was heated at the desired temperature until the reaction was completed. The mixture was allowed to reach room temperature, diluted with water and extracted with dichloromethane. The organic layer was dried over Na2SO4, filtered and the solvent was evaporated in vacuo. The brown residue was purified by column chromatography (silica gel, heptane/ethyl acetate) to yield 3-bromo-2-(2-bromophenyl)benzofuran (3).
General procedure for double C–N coupling with amine derivatives, exemplified by the synthesis of benzo[4,5]furo[3,2-b]indoles 5a–j. Similarly as previously described [31], 1.1 equiv of amine 4 was added to a pressure tube charged with 3, Pd2(dba)3 (5 mol %), BINAP (10 mol %) and 3 equiv of NaOt-Bu under Argon. The mixture was dissolved in anhydrous toluene (10 mL). The tube was sealed with a Teflon valve and stirred at 110 °C for 12 h. After the reaction was completed, the mixture was allowed to reach room temperature, worked up with water and extracted with dichloromethane. The combined organic layers were dried over sodium sulfate and concentrated under vacuum. The crude material was purified by flash column chromatography on silica gel (heptane/EtOAc) to yield benzo[4,5]furo[3,2-b]indoles 5a–j.
Nucleotide pyrophosphatase inhibition assay
The inhibition potential of all the sulphonate derivatives against NPP1 and NPP3, was determined by considering an already reported colorimetric method with minor modifications [38-41]. The reaction buffer was comprised of 50 mM Tris-hydrochloric acid, 5 mM magnesium chloride (MgCl2) and 0.1 mM zinc chloride (ZnCl2) with final pH 9.5. For initial screening of compounds, first enzyme and substrate parameters (concentration, temperature, time) were optimized. The reaction assay was performed at 100 µL well volume containing assay buffer, 10 µL of 1 mM test compound prepared in 10% DMSO, enzymes NPP1 (35 ng), NPP3 (94 ng) and assay buffer. The reaction mixture was incubated at 37 °C, 10 minutes for NPP1 and NPP3. Absorbance was taken at wavelength of 405 nm as pre-read by using a microplate reader (BioTek FLx800, Instruments, Inc. USA). After pre-read the artificial substrate p-nitrophenyl 5'-thymidine monophosphate (pNP-TMP), 400 µM for NPP1 and 600 µM in case of NPP3, was added followed by a second incubation at 37 °C, 15 minutes for NPP1 and NPP3. Absorbance was taken as after read. All the experiments were performed in triplicate. Compounds exhibiting more than 50% inhibition against either of NPP1 and NPP3 were further subjected to serial dilutions for the determination of IC50 values, using the nonlinear curve fitting program PRISM 5.0 (Graph Pad, San Diego, California, USA).
Molecular docking studies
Homology modelling of human ENPP1 and ENPP3
Homology models of ENPP1 and ENPP3 were developed by our research group [34-36,42] because the X-ray crystallographic structures were not available in protein data bank (PDB). For the prediction of the 3D homology prototype, the reported structure of mouse ENPP1 with PDB ID 4B56, was used as standard pattern [43]. Molecular Operating Environment (2014. 0901) was used for this purpose. The modeled crystal structures manifested 80% for ENPP1 and 52% in case of ENPP3, amino acids sequence similarity with template, mouse ENPP1. The RMSD for ENPP1 was 0.613 Å and 1.349 Å for ENPP3, over 816 and 811 residues in comparison to the template, respectively. The Ramachandran plot represented promising stereochemical properties as above 98% amino acids were engaged in core and selected region of homology models [44].
Preparation of ligands and proteins
The structures of the most active compounds were drawn by utilizing the MOE builder tool [45]. After introducing hydrogen atoms and charges to the prepared structures, the MMFF94x force field was applied for energy minimization with root mean square deviation (RMSD) of 0.01 kcal/mol Å [46]. Modeled protein structures of ENPP1 and ENPP3 were protonated and the energy was minimized with the help of the AMBER99 force field and an RMSD gradient of 0.05 kcal/mol Å.
Docking analysis
Apparent binding interactions and binding energies of the selected compounds within the binding pocket of modeled proteins were found out by using MOE (2014.0901). MOE site finder was used to locate the binding site while default setting of the triangular matcher placement method in combination with GBVI/WSA ∆G, were used to find docking calculations and scoring functions. Out of 40 conformations for each compound, the conformations with lowest free binding energies and S score were selected for visualization by using Discovery studio visualizer DS [47,48].
X-ray structure determination
An X-ray quality crystal of 5d was selected in Fomblin YR-1800 perfluoroether (Alfa Aesar) at ambient temperature. The sample was cooled to 123(3) K during the measurement. The data was collected on a Bruker D8 QUEST diffractometer using Mo Kα radiation (λ = 0.71073 Å). The structure was solved by direct methods (SHELXTL) and refined by full-matrix least squares procedures (SHELXL-2014/7) [49,50]. Semi-empirical absorption corrections were applied (SADABS) [51]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included in the refinement at calculated positions using a riding model. Refinement details can be found in the cif file CCDC 1432173, which can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Supporting Information
| Supporting Information File 1: Additional experimental and analytical data, and NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 1.9 MB | Download |
Acknowledgements
Financial support by the State of Vietnam (scholarship for T. Q. H.) and by the State of Mecklenburg-Vorpommern (scholarship for H. H. D.) is gratefully acknowledged. Financial support by the DAAD (Programm Deutsch-Pakistanische Hochschulzusammenarbeit) is gratefully acknowledged. We are grateful to Prof. Dr. José Luis Millán for providing plasmids of APLs. J. I. is thankful to the Higher Education Commission of Pakistan for the financial support through Project No.Ph-V-MG-3/Peridot/R&D/HEC/2019 and 6927/NRPU/R&D/17. J. S. received support from the Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN-2016-05867).
References
-
Adolphson, R. L.; Finkel, M. P.; Robichaud, L. J. Int. J. Immunopharmacol. 1987, 9, 41–49. doi:10.1016/0192-0561(87)90109-3
Return to citation in text: [1] -
Adolphson, R. L.; Finkel, M. P.; Robichaud, L. J. Int. J. Immunopharmacol. 1987, 9, 51–60. doi:10.1016/0192-0561(87)90110-x
Return to citation in text: [1] -
Mullican, M. D.; Sorenson, R. J.; Connor, D. T.; Thueson, D. O.; Kennedy, J. A.; Conroy, M. C. J. Med. Chem. 1991, 34, 2186–2194. doi:10.1021/jm00111a039
Return to citation in text: [1] -
Unangst, P. C.; Carethers, M. E.; Webster, K.; Janik, G. M.; Robichaud, L. J. J. Med. Chem. 1984, 27, 1629–1633. doi:10.1021/jm00378a017
Return to citation in text: [1] -
Chen, Y.-F.; Lin, Y.-C.; Chen, J.-P.; Chan, H.-C.; Hsu, M.-H.; Lin, H.-Y.; Kuo, S.-C.; Huang, L.-J. Bioorg. Med. Chem. Lett. 2015, 25, 3873–3877. doi:10.1016/j.bmcl.2015.07.058
Return to citation in text: [1] -
Zhuang, S.-H.; Lin, Y.-C.; Chou, L.-C.; Hsu, M.-H.; Lin, H.-Y.; Huang, C.-H.; Lien, J.-C.; Kuo, S.-C.; Huang, L.-J. Eur. J. Med. Chem. 2013, 66, 466–479. doi:10.1016/j.ejmech.2013.06.012
Return to citation in text: [1] -
Nakashima, Y.; Kawashima, Y.; Amanuma, F.; Sota, K.; Tanaka, A.; Kameyama, T. Chem. Pharm. Bull. 1984, 32, 4271–4280. doi:10.1248/cpb.32.4271
Return to citation in text: [1] -
Butera, J. A.; Antane, S. A.; Hirth, B.; Lennox, J. R.; Sheldon, J. H.; Norton, N. W.; Warga, D.; Argentieri, T. M. Bioorg. Med. Chem. Lett. 2001, 11, 2093–2097. doi:10.1016/s0960-894x(01)00385-7
Return to citation in text: [1] [2] [3] -
Lee, S.; Chae, M. R.; Lee, B.-C.; Kim, Y.-C.; Choi, J. S.; Lee, S. W.; Cheong, J. H.; Park, C.-S. Mol. Pharmacol. 2016, 90, 140–150. doi:10.1124/mol.115.102939
Return to citation in text: [1] [2] -
Gormemis, A. E.; Ha, T. S.; Im, I.; Jung, K.-Y.; Lee, J. Y.; Park, C.-S.; Kim, Y.-C. ChemBioChem 2005, 6, 1745–1748. doi:10.1002/cbic.200400448
Return to citation in text: [1] [2] [3] -
dela Peña, I. C.; Yoon, S. Y.; Kim, S. M.; Lee, G. S.; Park, C.-S.; Kim, Y. C.; Cheong, J. H. Arch. Pharmacal Res. 2009, 32, 413–420. doi:10.1007/s12272-009-1315-x
Return to citation in text: [1] [2] -
Sung, H. H.; Kang, S. J.; Chae, M. R.; Kim, H. K.; Park, J. K.; Kim, C. Y.; Lee, S. W. J. Sex. Med. 2017, 14, 59–68. doi:10.1016/j.jsxm.2016.11.316
Return to citation in text: [1] [2] -
Lee, S. W.; Park, J. K.; So, I. S.; Sung, H. H.; Kim, J. I.; Kim, Y. C.; Park, J. S. Composition for treating or preventing erectile dysfunction comprising ldd175 as active ingredient. U.S. Patent US2016/0235710 A1, Aug 18, 2016.
Return to citation in text: [1] [2] -
Sui, Z.; Zhang, X.; Li, X. Novel tetracyclic heteroatom containing derivatives useful as sex steriod hormone receptor modulators. WO Patent WO2006/034090, March 30, 2006.
Return to citation in text: [1] -
Takamatsu, K.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2014, 16, 2892–2895. doi:10.1021/ol501037j
Return to citation in text: [1] -
Carril, M.; San Martin, R.; Domínguez, E.; Tellitu, I. Green Chem. 2007, 9, 219–220. doi:10.1039/b614218d
Return to citation in text: [1] -
Schroeder, D. C.; Corcoran, P. O.; Holden, C. A.; Mulligan, M. C. J. Org. Chem. 1962, 27, 586–591. doi:10.1021/jo01049a057
Return to citation in text: [1] -
Saito, K.; Chikkade, P. K.; Kanai, M.; Kuninobu, Y. Chem. – Eur. J. 2015, 21, 8365–8368. doi:10.1002/chem.201501116
Return to citation in text: [1] -
Zhao, H.; Koenig, S. G.; Dankwardt, J. W.; Singh, S. P. Org. Process Res. Dev. 2014, 18, 198–204. doi:10.1021/op4001737
Return to citation in text: [1] -
Milkiewicz, K. L.; Parks, D. J.; Lu, T. Tetrahedron Lett. 2003, 44, 4257–4260. doi:10.1016/s0040-4039(03)00895-5
Return to citation in text: [1] -
Pudlo, M.; Csányi, D.; Moreau, F.; Hajós, G.; Riedl, Z.; Sapi, J. Tetrahedron 2007, 63, 10320–10329. doi:10.1016/j.tet.2007.07.068
Return to citation in text: [1] -
Nozaki, K.; Takahashi, K.; Nakano, K.; Hiyama, T.; Tang, H.-Z.; Fujiki, M.; Yamaguchi, S.; Tamao, K. Angew. Chem., Int. Ed. 2003, 42, 2051–2053. doi:10.1002/anie.200250648
Return to citation in text: [1] -
Kuwahara, A.; Nakano, K.; Nozaki, K. J. Org. Chem. 2005, 70, 413–419. doi:10.1021/jo048472+
Return to citation in text: [1] -
Nakano, K.; Hidehira, Y.; Takahashi, K.; Hiyama, T.; Nozaki, K. Angew. Chem., Int. Ed. 2005, 44, 7136–7138. doi:10.1002/anie.200502855
Return to citation in text: [1] -
Kitawaki, T.; Hayashi, Y.; Chida, N. Heterocycles 2005, 65, 1561–1567. doi:10.3987/com-05-10418
Return to citation in text: [1] -
Kitawaki, T.; Hayashi, Y.; Ueno, A.; Chida, N. Tetrahedron 2006, 62, 6792–6801. doi:10.1016/j.tet.2006.04.087
Return to citation in text: [1] -
Ueno, A.; Kitawaki, T.; Chida, N. Org. Lett. 2008, 10, 1999–2002. doi:10.1021/ol800602v
Return to citation in text: [1] -
Hung, T. Q.; Dang, T. T.; Villinger, A.; Sung, T. V.; Langer, P. Org. Biomol. Chem. 2012, 10, 9041–9044. doi:10.1039/c2ob26489g
Return to citation in text: [1] -
Hung, T. Q.; Hoang, D. H.; Thang, N. N.; Dang, T. T.; Ayub, K.; Villinger, A.; Friedrich, A.; Lochbrunner, S.; Flechsig, G.-U.; Langer, P. Org. Biomol. Chem. 2014, 12, 6151–6166. doi:10.1039/c4ob00841c
Return to citation in text: [1] -
Hung, T. Q.; Hancker, S.; Villinger, A.; Lochbrunner, S.; Dang, T. T.; Friedrich, A.; Breitsprecher, W.; Langer, P. Org. Biomol. Chem. 2015, 13, 583–591. doi:10.1039/c4ob01723d
Return to citation in text: [1] -
Hoang, H. D.; Janke, J.; Amirjanyan, A.; Ghochikyan, T.; Flader, A.; Villinger, A.; Ehlers, P.; Lochbrunner, S.; Surkus, A.-E.; Langer, P. Org. Biomol. Chem. 2018, 16, 6543–6551. doi:10.1039/c8ob01737a
Return to citation in text: [1] [2] [3] [4] -
Truong, M. A.; Nakano, K. J. Org. Chem. 2015, 80, 11566–11572. doi:10.1021/acs.joc.5b02086
Return to citation in text: [1] -
Benincori, T.; Brenna, E.; Sannicolò, F.; Trimarco, L.; Antognazza, P.; Cesarotti, E.; Demartin, F.; Pilati, T. J. Org. Chem. 1996, 61, 6244–6251. doi:10.1021/jo960211f
Return to citation in text: [1] -
Bollen, M.; Gijsbers, R.; Ceulemans, H.; Stalmans, W.; Stefan, C. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 393–432. doi:10.1080/10409230091169249
Return to citation in text: [1] [2] [3] -
Iqbal, J.; Lévesque, S. A.; Sévigny, J.; Müller, C. E. Electrophoresis 2008, 29, 3685–3693. doi:10.1002/elps.200800013
Return to citation in text: [1] [2] [3] -
Ausekle, E.; Ejaz, S. A.; Khan, S. U.; Ehlers, P.; Villinger, A.; Lecka, J.; Sévigny, J.; Iqbal, J.; Langer, P. Org. Biomol. Chem. 2016, 14, 11402–11414. doi:10.1039/c6ob02236g
Return to citation in text: [1] [2] -
Khan, I.; Ibrar, A.; Ejaz, S. A.; Khan, S. U.; Shah, S. J. A.; Hameed, S.; Simpson, J.; Lecka, J.; Sévigny, J.; Iqbal, J. RSC Adv. 2015, 5, 90806–90818. doi:10.1039/c5ra14836g
Return to citation in text: [1] -
Vollmayer, P.; Clair, T.; Goding, J. W.; Sano, K.; Servos, J.; Zimmermann, H. Eur. J. Biochem. 2003, 270, 2971–2978. doi:10.1046/j.1432-1033.2003.03674.x
Return to citation in text: [1] -
Kukulski, F.; Lévesque, S. A.; Lavoie, É. G.; Lecka, J.; Bigonnesse, F.; Knowles, A. F.; Robson, S. C.; Kirley, T. L.; Sévigny, J. Purinergic Signalling 2005, 1, 193–204. doi:10.1007/s11302-005-6217-x
Return to citation in text: [1] -
Jin-Hua, P.; Goding, J. W.; Nakamura, H.; Sano, K. Genomics 1997, 45, 412–415. doi:10.1006/geno.1997.4949
Return to citation in text: [1] -
Akhtar, T.; Hameed, S.; Al-Masoudi, N.; Loddo, R.; Colla, P. Acta Pharm. 2008, 58, 135–149. doi:10.2478/v10007-008-0007-2
Return to citation in text: [1] -
Albers, H. M. H. G.; van Meeteren, L. A.; Egan, D. A.; van Tilburg, E. W.; Moolenaar, W. H.; Ovaa, H. J. Med. Chem. 2010, 53, 4958–4967. doi:10.1021/jm1005012
Return to citation in text: [1] -
Jafari, B.; Yelibayeva, N.; Ospanov, M.; Ejaz, S. A.; Afzal, S.; Khan, S. U.; Abilov, Z. A.; Turmukhanova, M. Z.; Kalugin, S. N.; Safarov, S.; Lecka, J.; Sévigny, J.; Rahman, Q.; Ehlers, P.; Iqbal, J.; Langer, P. RSC Adv. 2016, 6, 107556–107571. doi:10.1039/c6ra22750c
Return to citation in text: [1] -
Jansen, S.; Perrakis, A.; Ulens, C.; Winkler, C.; Andries, M.; Joosten, R. P.; Van Acker, M.; Luyten, F. P.; Moolenaar, W. H.; Bollen, M. Structure 2012, 20, 1948–1959. doi:10.1016/j.str.2012.09.001
Return to citation in text: [1] -
Laskowski, R. A.; MacArthur, M. W.; Moss, D. S.; Thornton, J. M. J. Appl. Crystallogr. 1993, 26, 283–291. doi:10.1107/s0021889892009944
Return to citation in text: [1] -
MOE (Molecular Operating Environment) Version 2014.0901. Chemical Computing Group, (CCG). http://www.chemcomp.com/MOEMolecular_Operating_Environment.htm.
Return to citation in text: [1] -
Labute, P. Protonate 3D: Assignment of Macromolecular Protonation State and Geometry, Chemical Computing Group, 2007. http://www.chemcomp.com/journal/proton.htm.
Return to citation in text: [1] -
Discovery Studio Modeling Environment, Release 4.0; Accelrys Software Inc.: San Diego, CA, 2013.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930
Return to citation in text: [1] -
SADABS, Version 2; University of Göttingen: Göttingen, Germany, 2004.
Return to citation in text: [1] -
SHELXS-2013; University of Göttingen: Göttingen,Germany, 2013.
Return to citation in text: [1]
| 49. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930 |
| 50. | SADABS, Version 2; University of Göttingen: Göttingen, Germany, 2004. |
| 1. | Adolphson, R. L.; Finkel, M. P.; Robichaud, L. J. Int. J. Immunopharmacol. 1987, 9, 41–49. doi:10.1016/0192-0561(87)90109-3 |
| 2. | Adolphson, R. L.; Finkel, M. P.; Robichaud, L. J. Int. J. Immunopharmacol. 1987, 9, 51–60. doi:10.1016/0192-0561(87)90110-x |
| 3. | Mullican, M. D.; Sorenson, R. J.; Connor, D. T.; Thueson, D. O.; Kennedy, J. A.; Conroy, M. C. J. Med. Chem. 1991, 34, 2186–2194. doi:10.1021/jm00111a039 |
| 4. | Unangst, P. C.; Carethers, M. E.; Webster, K.; Janik, G. M.; Robichaud, L. J. J. Med. Chem. 1984, 27, 1629–1633. doi:10.1021/jm00378a017 |
| 8. | Butera, J. A.; Antane, S. A.; Hirth, B.; Lennox, J. R.; Sheldon, J. H.; Norton, N. W.; Warga, D.; Argentieri, T. M. Bioorg. Med. Chem. Lett. 2001, 11, 2093–2097. doi:10.1016/s0960-894x(01)00385-7 |
| 9. | Lee, S.; Chae, M. R.; Lee, B.-C.; Kim, Y.-C.; Choi, J. S.; Lee, S. W.; Cheong, J. H.; Park, C.-S. Mol. Pharmacol. 2016, 90, 140–150. doi:10.1124/mol.115.102939 |
| 11. | dela Peña, I. C.; Yoon, S. Y.; Kim, S. M.; Lee, G. S.; Park, C.-S.; Kim, Y. C.; Cheong, J. H. Arch. Pharmacal Res. 2009, 32, 413–420. doi:10.1007/s12272-009-1315-x |
| 36. | Ausekle, E.; Ejaz, S. A.; Khan, S. U.; Ehlers, P.; Villinger, A.; Lecka, J.; Sévigny, J.; Iqbal, J.; Langer, P. Org. Biomol. Chem. 2016, 14, 11402–11414. doi:10.1039/c6ob02236g |
| 8. | Butera, J. A.; Antane, S. A.; Hirth, B.; Lennox, J. R.; Sheldon, J. H.; Norton, N. W.; Warga, D.; Argentieri, T. M. Bioorg. Med. Chem. Lett. 2001, 11, 2093–2097. doi:10.1016/s0960-894x(01)00385-7 |
| 9. | Lee, S.; Chae, M. R.; Lee, B.-C.; Kim, Y.-C.; Choi, J. S.; Lee, S. W.; Cheong, J. H.; Park, C.-S. Mol. Pharmacol. 2016, 90, 140–150. doi:10.1124/mol.115.102939 |
| 10. | Gormemis, A. E.; Ha, T. S.; Im, I.; Jung, K.-Y.; Lee, J. Y.; Park, C.-S.; Kim, Y.-C. ChemBioChem 2005, 6, 1745–1748. doi:10.1002/cbic.200400448 |
| 11. | dela Peña, I. C.; Yoon, S. Y.; Kim, S. M.; Lee, G. S.; Park, C.-S.; Kim, Y. C.; Cheong, J. H. Arch. Pharmacal Res. 2009, 32, 413–420. doi:10.1007/s12272-009-1315-x |
| 12. | Sung, H. H.; Kang, S. J.; Chae, M. R.; Kim, H. K.; Park, J. K.; Kim, C. Y.; Lee, S. W. J. Sex. Med. 2017, 14, 59–68. doi:10.1016/j.jsxm.2016.11.316 |
| 13. | Lee, S. W.; Park, J. K.; So, I. S.; Sung, H. H.; Kim, J. I.; Kim, Y. C.; Park, J. S. Composition for treating or preventing erectile dysfunction comprising ldd175 as active ingredient. U.S. Patent US2016/0235710 A1, Aug 18, 2016. |
| 34. | Bollen, M.; Gijsbers, R.; Ceulemans, H.; Stalmans, W.; Stefan, C. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 393–432. doi:10.1080/10409230091169249 |
| 35. | Iqbal, J.; Lévesque, S. A.; Sévigny, J.; Müller, C. E. Electrophoresis 2008, 29, 3685–3693. doi:10.1002/elps.200800013 |
| 7. | Nakashima, Y.; Kawashima, Y.; Amanuma, F.; Sota, K.; Tanaka, A.; Kameyama, T. Chem. Pharm. Bull. 1984, 32, 4271–4280. doi:10.1248/cpb.32.4271 |
| 33. | Benincori, T.; Brenna, E.; Sannicolò, F.; Trimarco, L.; Antognazza, P.; Cesarotti, E.; Demartin, F.; Pilati, T. J. Org. Chem. 1996, 61, 6244–6251. doi:10.1021/jo960211f |
| 5. | Chen, Y.-F.; Lin, Y.-C.; Chen, J.-P.; Chan, H.-C.; Hsu, M.-H.; Lin, H.-Y.; Kuo, S.-C.; Huang, L.-J. Bioorg. Med. Chem. Lett. 2015, 25, 3873–3877. doi:10.1016/j.bmcl.2015.07.058 |
| 6. | Zhuang, S.-H.; Lin, Y.-C.; Chou, L.-C.; Hsu, M.-H.; Lin, H.-Y.; Huang, C.-H.; Lien, J.-C.; Kuo, S.-C.; Huang, L.-J. Eur. J. Med. Chem. 2013, 66, 466–479. doi:10.1016/j.ejmech.2013.06.012 |
| 34. | Bollen, M.; Gijsbers, R.; Ceulemans, H.; Stalmans, W.; Stefan, C. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 393–432. doi:10.1080/10409230091169249 |
| 35. | Iqbal, J.; Lévesque, S. A.; Sévigny, J.; Müller, C. E. Electrophoresis 2008, 29, 3685–3693. doi:10.1002/elps.200800013 |
| 8. | Butera, J. A.; Antane, S. A.; Hirth, B.; Lennox, J. R.; Sheldon, J. H.; Norton, N. W.; Warga, D.; Argentieri, T. M. Bioorg. Med. Chem. Lett. 2001, 11, 2093–2097. doi:10.1016/s0960-894x(01)00385-7 |
| 10. | Gormemis, A. E.; Ha, T. S.; Im, I.; Jung, K.-Y.; Lee, J. Y.; Park, C.-S.; Kim, Y.-C. ChemBioChem 2005, 6, 1745–1748. doi:10.1002/cbic.200400448 |
| 15. | Takamatsu, K.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2014, 16, 2892–2895. doi:10.1021/ol501037j |
| 16. | Carril, M.; San Martin, R.; Domínguez, E.; Tellitu, I. Green Chem. 2007, 9, 219–220. doi:10.1039/b614218d |
| 17. | Schroeder, D. C.; Corcoran, P. O.; Holden, C. A.; Mulligan, M. C. J. Org. Chem. 1962, 27, 586–591. doi:10.1021/jo01049a057 |
| 18. | Saito, K.; Chikkade, P. K.; Kanai, M.; Kuninobu, Y. Chem. – Eur. J. 2015, 21, 8365–8368. doi:10.1002/chem.201501116 |
| 19. | Zhao, H.; Koenig, S. G.; Dankwardt, J. W.; Singh, S. P. Org. Process Res. Dev. 2014, 18, 198–204. doi:10.1021/op4001737 |
| 20. | Milkiewicz, K. L.; Parks, D. J.; Lu, T. Tetrahedron Lett. 2003, 44, 4257–4260. doi:10.1016/s0040-4039(03)00895-5 |
| 21. | Pudlo, M.; Csányi, D.; Moreau, F.; Hajós, G.; Riedl, Z.; Sapi, J. Tetrahedron 2007, 63, 10320–10329. doi:10.1016/j.tet.2007.07.068 |
| 31. | Hoang, H. D.; Janke, J.; Amirjanyan, A.; Ghochikyan, T.; Flader, A.; Villinger, A.; Ehlers, P.; Lochbrunner, S.; Surkus, A.-E.; Langer, P. Org. Biomol. Chem. 2018, 16, 6543–6551. doi:10.1039/c8ob01737a |
| 14. | Sui, Z.; Zhang, X.; Li, X. Novel tetracyclic heteroatom containing derivatives useful as sex steriod hormone receptor modulators. WO Patent WO2006/034090, March 30, 2006. |
| 32. | Truong, M. A.; Nakano, K. J. Org. Chem. 2015, 80, 11566–11572. doi:10.1021/acs.joc.5b02086 |
| 12. | Sung, H. H.; Kang, S. J.; Chae, M. R.; Kim, H. K.; Park, J. K.; Kim, C. Y.; Lee, S. W. J. Sex. Med. 2017, 14, 59–68. doi:10.1016/j.jsxm.2016.11.316 |
| 13. | Lee, S. W.; Park, J. K.; So, I. S.; Sung, H. H.; Kim, J. I.; Kim, Y. C.; Park, J. S. Composition for treating or preventing erectile dysfunction comprising ldd175 as active ingredient. U.S. Patent US2016/0235710 A1, Aug 18, 2016. |
| 10. | Gormemis, A. E.; Ha, T. S.; Im, I.; Jung, K.-Y.; Lee, J. Y.; Park, C.-S.; Kim, Y.-C. ChemBioChem 2005, 6, 1745–1748. doi:10.1002/cbic.200400448 |
| 22. | Nozaki, K.; Takahashi, K.; Nakano, K.; Hiyama, T.; Tang, H.-Z.; Fujiki, M.; Yamaguchi, S.; Tamao, K. Angew. Chem., Int. Ed. 2003, 42, 2051–2053. doi:10.1002/anie.200250648 |
| 23. | Kuwahara, A.; Nakano, K.; Nozaki, K. J. Org. Chem. 2005, 70, 413–419. doi:10.1021/jo048472+ |
| 24. | Nakano, K.; Hidehira, Y.; Takahashi, K.; Hiyama, T.; Nozaki, K. Angew. Chem., Int. Ed. 2005, 44, 7136–7138. doi:10.1002/anie.200502855 |
| 25. | Kitawaki, T.; Hayashi, Y.; Chida, N. Heterocycles 2005, 65, 1561–1567. doi:10.3987/com-05-10418 |
| 26. | Kitawaki, T.; Hayashi, Y.; Ueno, A.; Chida, N. Tetrahedron 2006, 62, 6792–6801. doi:10.1016/j.tet.2006.04.087 |
| 27. | Ueno, A.; Kitawaki, T.; Chida, N. Org. Lett. 2008, 10, 1999–2002. doi:10.1021/ol800602v |
| 28. | Hung, T. Q.; Dang, T. T.; Villinger, A.; Sung, T. V.; Langer, P. Org. Biomol. Chem. 2012, 10, 9041–9044. doi:10.1039/c2ob26489g |
| 29. | Hung, T. Q.; Hoang, D. H.; Thang, N. N.; Dang, T. T.; Ayub, K.; Villinger, A.; Friedrich, A.; Lochbrunner, S.; Flechsig, G.-U.; Langer, P. Org. Biomol. Chem. 2014, 12, 6151–6166. doi:10.1039/c4ob00841c |
| 30. | Hung, T. Q.; Hancker, S.; Villinger, A.; Lochbrunner, S.; Dang, T. T.; Friedrich, A.; Breitsprecher, W.; Langer, P. Org. Biomol. Chem. 2015, 13, 583–591. doi:10.1039/c4ob01723d |
| 31. | Hoang, H. D.; Janke, J.; Amirjanyan, A.; Ghochikyan, T.; Flader, A.; Villinger, A.; Ehlers, P.; Lochbrunner, S.; Surkus, A.-E.; Langer, P. Org. Biomol. Chem. 2018, 16, 6543–6551. doi:10.1039/c8ob01737a |
| 31. | Hoang, H. D.; Janke, J.; Amirjanyan, A.; Ghochikyan, T.; Flader, A.; Villinger, A.; Ehlers, P.; Lochbrunner, S.; Surkus, A.-E.; Langer, P. Org. Biomol. Chem. 2018, 16, 6543–6551. doi:10.1039/c8ob01737a |
| 37. | Khan, I.; Ibrar, A.; Ejaz, S. A.; Khan, S. U.; Shah, S. J. A.; Hameed, S.; Simpson, J.; Lecka, J.; Sévigny, J.; Iqbal, J. RSC Adv. 2015, 5, 90806–90818. doi:10.1039/c5ra14836g |
| 46. | MOE (Molecular Operating Environment) Version 2014.0901. Chemical Computing Group, (CCG). http://www.chemcomp.com/MOEMolecular_Operating_Environment.htm. |
| 47. | Labute, P. Protonate 3D: Assignment of Macromolecular Protonation State and Geometry, Chemical Computing Group, 2007. http://www.chemcomp.com/journal/proton.htm. |
| 48. | Discovery Studio Modeling Environment, Release 4.0; Accelrys Software Inc.: San Diego, CA, 2013. |
| 44. | Jansen, S.; Perrakis, A.; Ulens, C.; Winkler, C.; Andries, M.; Joosten, R. P.; Van Acker, M.; Luyten, F. P.; Moolenaar, W. H.; Bollen, M. Structure 2012, 20, 1948–1959. doi:10.1016/j.str.2012.09.001 |
| 45. | Laskowski, R. A.; MacArthur, M. W.; Moss, D. S.; Thornton, J. M. J. Appl. Crystallogr. 1993, 26, 283–291. doi:10.1107/s0021889892009944 |
| 34. | Bollen, M.; Gijsbers, R.; Ceulemans, H.; Stalmans, W.; Stefan, C. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 393–432. doi:10.1080/10409230091169249 |
| 35. | Iqbal, J.; Lévesque, S. A.; Sévigny, J.; Müller, C. E. Electrophoresis 2008, 29, 3685–3693. doi:10.1002/elps.200800013 |
| 36. | Ausekle, E.; Ejaz, S. A.; Khan, S. U.; Ehlers, P.; Villinger, A.; Lecka, J.; Sévigny, J.; Iqbal, J.; Langer, P. Org. Biomol. Chem. 2016, 14, 11402–11414. doi:10.1039/c6ob02236g |
| 42. | Albers, H. M. H. G.; van Meeteren, L. A.; Egan, D. A.; van Tilburg, E. W.; Moolenaar, W. H.; Ovaa, H. J. Med. Chem. 2010, 53, 4958–4967. doi:10.1021/jm1005012 |
| 43. | Jafari, B.; Yelibayeva, N.; Ospanov, M.; Ejaz, S. A.; Afzal, S.; Khan, S. U.; Abilov, Z. A.; Turmukhanova, M. Z.; Kalugin, S. N.; Safarov, S.; Lecka, J.; Sévigny, J.; Rahman, Q.; Ehlers, P.; Iqbal, J.; Langer, P. RSC Adv. 2016, 6, 107556–107571. doi:10.1039/c6ra22750c |
| 31. | Hoang, H. D.; Janke, J.; Amirjanyan, A.; Ghochikyan, T.; Flader, A.; Villinger, A.; Ehlers, P.; Lochbrunner, S.; Surkus, A.-E.; Langer, P. Org. Biomol. Chem. 2018, 16, 6543–6551. doi:10.1039/c8ob01737a |
| 38. | Vollmayer, P.; Clair, T.; Goding, J. W.; Sano, K.; Servos, J.; Zimmermann, H. Eur. J. Biochem. 2003, 270, 2971–2978. doi:10.1046/j.1432-1033.2003.03674.x |
| 39. | Kukulski, F.; Lévesque, S. A.; Lavoie, É. G.; Lecka, J.; Bigonnesse, F.; Knowles, A. F.; Robson, S. C.; Kirley, T. L.; Sévigny, J. Purinergic Signalling 2005, 1, 193–204. doi:10.1007/s11302-005-6217-x |
| 40. | Jin-Hua, P.; Goding, J. W.; Nakamura, H.; Sano, K. Genomics 1997, 45, 412–415. doi:10.1006/geno.1997.4949 |
| 41. | Akhtar, T.; Hameed, S.; Al-Masoudi, N.; Loddo, R.; Colla, P. Acta Pharm. 2008, 58, 135–149. doi:10.2478/v10007-008-0007-2 |
© 2019 Do et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)