Abstract
Butyrolactol A is an antifungal polyketide of Streptomyces bearing an uncommon tert-butyl starter unit and a polyol system in which eight hydroxy/acyloxy carbons are contiguously connected. Except for its congener butyrolactol B, there exist no structurally related natural products to date. In this study, inspired by our previous genomic analysis, incorporation of 13C- and 2H-labeled precursors into butyrolactol A was investigated. Based on the labeling pattern and sequencing analytical data, we confirmed that the tert-butyl group is derived from valine and its C-methylation with methionine and the polyol carbons are derived from a glycolysis intermediate, possibly hydroxymalonyl-ACP.

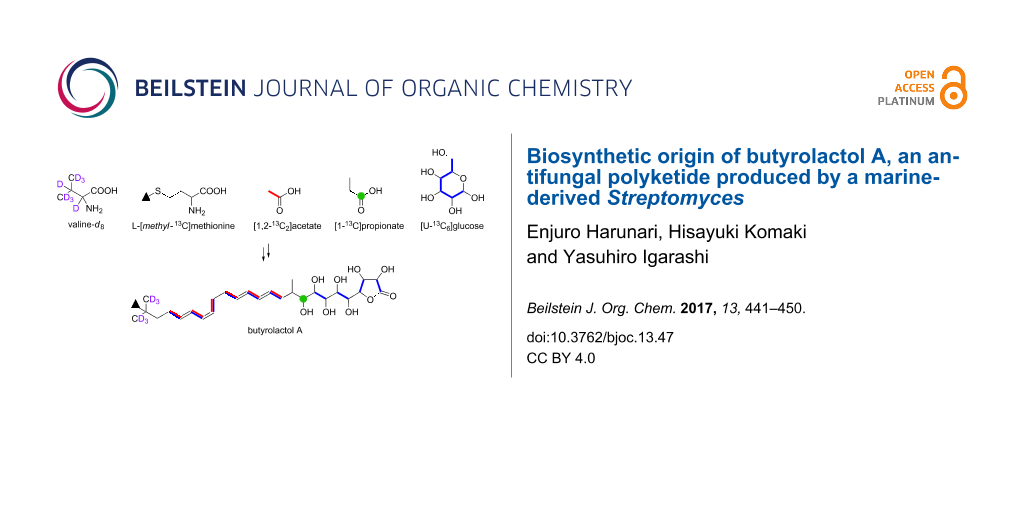
Graphical Abstract
Introduction
Actinomycetes produce structurally diverse secondary metabolites with pharmaceutically useful bioactivities. Importantly, members of the genus Streptomyces have been the main source of drug discovery programs due to their high capacity in secondary metabolism including polyketides, peptides, terpenoids, alkaloids, and amino acid/carbohydrate/nucleic acid derivatives [1,2]. One of the largest groups of bacterial secondary metabolites is polyketide from which a range of clinically used drugs have been developed. Polyketides still remain in the focus of drug development because of their structural complexity that can provide attractive templates for new pharmacophores [3]. While the frequency of discovering new skeletons from actinomycetes seems declining, biosynthetic analysis of structurally unique known compounds and the following bioengineering of biosynthetic genes are currently becoming an essential part of the creation of new drug-like structures [4-8].
Butyrolactol A (1) is an antifungal polyketide first isolated from Streptomyces rochei S785-16 [9] (Figure 1). The left half of 1 is the hydrophobic unconjugated tetraene system including one Z-olefin with a terminal tert-butyl group, whereas the hydrophilic polyol system bearing a γ-lactone terminus constitutes the right half of the molecule. To date, no structurally related natural products are known except for its demethyl congener butyrolactol B that was also isolated from the same strain and has an isopropyl group instead of the tert-butyl terminus [9]. Very recently, isolation of butyrolactols C and D was presented but the details are not available in public domains [10]. 1 has a broad antimicrobial activity against fungi ranging from Candida albicans to Trichophyton mentagrophytes with comparative activity to nystatin [9]. Despite the uniqueness of the structure and the antifungal potency, no further research has been reported for 1.
![[1860-5397-13-47-1]](/bjoc/content/figures/1860-5397-13-47-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The structure of butyrolactol A (1).
Figure 1: The structure of butyrolactol A (1).
There are two interesting aspects in the structure of butyrolactol A (1). First, among the polyketides, a tert-butyl group has been found exclusively in metabolites of marine cyanobacteria except for 1 [11-14] (Figure 2). Although no experimental evidence is available, pivaloyl-CoA (2,2-dimethylpropanoyl-CoA) is supposed to be a starter for its biosynthesis [15]. Additionally, trimethylation of malonyl-CoA is proposed for the synthesis of the tert-butyl starter in the biosynthesis of apratoxin A [16]. Another intriguing feature of this molecule is the highly oxygenated carbon chain in which eight hydroxy groups, one of which is used for lactone formation, are contiguously aligned. A 1,3-diol is a common structural element in aliphatic polyketides because the incorporation of malonate-precursors gives rise to the alternative alignment of the methylene and the oxygenated carbons. Meanwhile, a 1,2-diol in polyketides is known to be formed by hydroxylation of methylene carbons as seen in the biosynthesis of erythromycin or amphotericin B [17,18]. The contiguously hydroxylated carbon chain of 1 is quite unusual as a polyketide. Examples of similar but shorter polyol carbon chains are ossamycin [19], IB-96212 [20], and antifungalmycin [21], all of which are the secondary metabolites of actinomycetes (Figure 3).
![[1860-5397-13-47-2]](/bjoc/content/figures/1860-5397-13-47-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cyanobacterial polyketides bearing a tert-butyl group.
Figure 2: Cyanobacterial polyketides bearing a tert-butyl group.
![[1860-5397-13-47-3]](/bjoc/content/figures/1860-5397-13-47-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Actinomycete metabolites possessing a contiguous 1,2-diol system.
Figure 3: Actinomycete metabolites possessing a contiguous 1,2-diol system.
In our investigation on secondary metabolites of marine actinomycetes, butyrolactol A (1) was found to be produced by a Streptomyces strain collected from deep sea water of the Toyama Bay, Japan. In order to get insight into the construction of the above-mentioned unusual structures, we performed an in silico analysis of the biosynthetic genes of 1 through draft genome sequencing and proposed its biosynthetic pathway [22]. In this study, biosynthetic precursors of 1 were investigated for further genetic and enzymatic studies.
Results and Discussion
It was obvious from its structure that 1 was synthesized through the malonate pathway. First, [1,2-13C2]acetate was fed to the culture to ensure the alignment of malonate units. In the 13C NMR spectrum, split signals arising from 13C–13C couplings were observed for six pairs of carbons: C-11/C-12, C-13/C-14, C-15/C-16, C-17/C-18, C-19/C-20, and C-21/C-22 (Table 1, Figure S1 in Supporting Information File 1). In the 2D-INADEQUATE spectrum of 13C-labeled 1 obtained with a parameter set optimized for 1JCC 50 Hz, cross peaks derived from the intact 13C2 acetate units were detected for the carbon pairs mentioned above (Table 1, Figure 4a).
Table 1: Incorporation of 13C-labeled precursors into 1.
| Position | δC | [1,2-13C2]acetate | [U-13C6]glucose | [1-13C]propionate | L-[methyl-13C]methionine | ||||
|---|---|---|---|---|---|---|---|---|---|
|
1JCC
(Hz) |
2D-INADEQUATE |
1JCC
(Hz) |
2D-INADEQUATE | relative enrichmentsa | |||||
| 1 | 175.3 | 56 | 2 | 0.9 | 0.9 | ||||
| 2 | 74.7 | 56 | 1 | 1.0 | 1.0 | ||||
| 3 | 72.9 | 39 | 4 | 0.9 | 1.0 | ||||
| 4 | 80.0 | 39 | 3 | 1.0 | 1.1 | ||||
| 5 | 66.9 | 44 | 6 | 1.0 | 1.0 | ||||
| 6 | 68.9 | 44 | 5 | 0.9 | 0.9 | ||||
| 7 | 68.9 | 45 | 8 | 1.0 | 1.1 | ||||
| 8 | 69.7 | 45 | 7 | 1.0 | 1.1 | ||||
| 9 | 73.1 | 4.8 | 1.2 | ||||||
| 10 | 36.2 | 1.1 | 1.1 | ||||||
| 11 | 36.6 | 41 | 12 | 40 | 12 | 1.2 | 1.2 | ||
| 12 | 131.9 | 41 | 11 | 40 | 11 | 1.3 | 1.2 | ||
| 13 | 131.9 | 55 | 14 | 57 | 14 | 1.3 | 1.2 | ||
| 14 | 131.3 | 55 | 13 | 57 | 13 | 1.2 | 1.0 | ||
| 15 | 131.3 | 43 | 16 | 43 | 16 | 1.2 | 1.0 | ||
| 16 | 32.6 | 43 | 15 | 43 | 15 | 1.2 | 1.3 | ||
| 17 | 27.5 | 42 | 18 | 43 | 18 | 1.4 | 1.3 | ||
| 18 | 129.2 | 42 | 17 | 43 | 17 | 1.2 | 1.0 | ||
| 19 | 129.6 | 55 | 20 | 55 | 20 | 1.4 | 1.1 | ||
| 20 | 125.7 | 55 | 19 | 55 | 19 | 1.3 | 1.1 | ||
| 21 | 135.9 | 43 | 22 | 43 | 22 | 1.3 | 1.0 | ||
| 22 | 28.1 | 43 | 21 | 43 | 21 | 1.2 | 1.1 | ||
| 23 | 43.7 | 1.6 | 1.2 | ||||||
| 24 | 30.6 | 1.2 | 1.0 | ||||||
| 25 | 29.7 | 0.9 | 7.1 | ||||||
| 26 | 29.7 | 0.9 | 7.1 | ||||||
| 27 | 29.7 | 0.9 | 7.1 | ||||||
| 28 | 16.2 | 1.1 | 1.2 | ||||||
aThe 13C signal intensity of each peak in labeled 1 divided by that of the corresponding signal in unlabeled 1, respectively, normalized to give an enrichment ratio of 1 for the unenriched C-2 peak.
![[1860-5397-13-47-4]](/bjoc/content/figures/1860-5397-13-47-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Feeding experiments of 13C-labeled precursors into 1 detected by 2D-INADEQUATE NMR experiments. (a) [1,2-13C2]acetate; (b) [U-13C6]glucose.
Figure 4: Feeding experiments of 13C-labeled precursors into 1 detected by 2D-INADEQUATE NMR experiments. (a)...
According to the incorporation result of the doubly labeled acetate, malonyl-CoA is not the extender unit for the lactone (C-1 to C-4) and the pentaol (C-5 to C-9) moieties (Figure 3), suggesting that the contiguous polyol system is not formed by methylene hydroxylation. Another possible pathway for 1,2-diol formation is the incorporation of hydroxymalonyl-ACP from a glycolytic intermediate for chain elongation [23]. To investigate this possibility, we conducted a feeding experiment of [U-13C6]glucose which could label carbons derived from malonyl-CoA and hydroxymalonyl-ACP. In the 13C NMR spectrum, 13C–13C couplings were observed for C-1/C-2, C-3/C-4, C-5/C-6, C-7/C-8 in addition to the carbon pairs C-11/C-12, C-13/C-14, C-15/C-16, C-17/C-18, C-19/C-20, and C-21/C-22 (Table 1, Figure S3 in Supporting Information File 1). The 2D-INADEQUATE spectrum showed cross peaks for the above-mentioned two-carbon units derived from the glycolytic degradation of [U-13C6]glucose (Table 1, Figure 4b). Combined with the acetate-labeling result, this labeling pattern suggested that the carbons from C-1 to C-8 are derived from hydroxymalonyl-ACP. This conclusion is supported by the sequencing analysis of the gene cluster for butyrolactol biosynthesis (Figure 5, Table 2). Four genes coding homologues of enzymes involved in hydroxymalonyl-ACP formation in the zwittermicin biosynthesis (ZmaN, ZmaD, ZmaG, and ZmaE) (Figure 6) [24] are present in the downstream of the butyrolactol PKS cluster. Genes coding for O-methyltransferase homologues responsible for O-methylation of hydroxymalonyl-ACP were not found near the cluster. Hydroxymalonyl-ACP was first identified as an unusual polyketide extender for zittermicin from Bacillus cereus [25]. The occurrence of this uncommon extender unit is limited to some bacterial species Bacillus [25,26], Xenorhabdus [27], Paenibacillus [28], and Streptomyces [29,30].
![[1860-5397-13-47-5]](/bjoc/content/figures/1860-5397-13-47-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Organization of the biosynthesis gene cluster for 1. Blue, transcriptional regulator; pink, PKS for polyketide backbone of 1; yellow, genes for biosynthesis of hydroxymalonyl-ACP; gray, transporter.
Figure 5: Organization of the biosynthesis gene cluster for 1. Blue, transcriptional regulator; pink, PKS for...
Table 2: Annotated putative ORFs in biosynthetic gene cluster and neighboring genes of 1.
| Orf10- | Accession no. |
Size
(aa) |
Proposed function | BLAST search | |
|---|---|---|---|---|---|
| Protein homolog, Origin, Accession number | %a | ||||
| 8 | WP_055469543 | 127 | HxlR family transcriptional regulator | HxlR family transcriptional regulator, Streptomyces sp. NRRL F-7442, KOX41174 | 99/100 |
| 10b | WP_030405160 | 71 | acetyl-CoA carboxylase biotin carboxyl carrier protein subunit | acetyl-CoA carboxylase, Streptomyces sp. NRRL F-7442, KOX41173 | 100/100 |
| 11b | WP_055469545 | 6,065 | PKS | FscE, Streptomyces cattleya, AEW99638 | 76/82 |
| 12b | WP_055469546 | 473 | propionyl-CoA carboxylase subunit beta | propionyl-CoA carboxylase subunit beta, Streptomyces sp. NRRL F-7442, KOX41172 | 99/99 |
| 13 | WP_055469547 | 676 | helix-turn-helix transcriptional regulator | regulator, Streptomyces sp. NRRL F-7442, KOX41171 | 99/99 |
| 14 | WP_055469666 | 2,075 | PKS | polyketide synthase type I, Streptomyces cattleya, AEW99622 | 71/80 |
| 15 | WP_055469548 | 3,365 | PKS | FscC, Streptomyces cattleya, AEW99623 | 71/79 |
| 16 | WP_055469549 | 3,462 | PKS | polyketide synthase (fragment), Streptomyces cattleya, KOX46585 | 99/99 |
| 17 | WP_055469550 | 3,135 | PKS | polyketide synthase, Streptomyces sp. NRRL F-7442, KOX46586 | 99/99 |
| 18 | WP_055469551 | 1,169 | PKS | short-chain dehydrogenase, Streptomyces sp. NRRL-7442, KOX46587 | 99/99 |
| 19 | WP_055469552 | 301 | 3-hydroxyacyl-CoA dehydratase | 3-hydroxybutyryl-CoA dehydrogenase, Streptomyces sp. NRRL F-7442, KOX46623 | 98/99 |
| 20 | WP_030403675 | 88 | ACP | Acyl carrier protein, Streptomyces sp. NRRL F-7442, KOX46588 | 100/100 |
| 21 | WP_055469553 | 381 | acyl-CoA dehydratase | acyl-CoA dehydrogenase, Streptomyces sp. NRRL F-7442, KOX46589 | 99/99 |
| 22 | WP_055469554 | 356 | glyceroyl-ACP biosynthesis protein | FkbH, Streptomyces sp. NRRL F-7442, KOX46590 | 99/99 |
| 23 | WP_030403672 | 261 | thioesterase | thioesterase, Streptomyces sp. NRRL F-7442, KOX46591 | 100/100 |
| 24 | WP_055469555 | 448 | MFS transporter | major facilitator superfamily permease, Streptomyces cattleya, AEW99632 | 78/84 |
| 25 | WP_055469556 | 408 | hypothetical protein | uncharacterized protein, Streptomyces sp. NRRL F-7442, KOX46624 | 99/100 |
| 26b | WP_055469557 | 253 | multidrug ABC transporter permease | multidrug ABC transporter permease, Streptomyces sp. NRRL F-7442, KOX46592 | 99/99 |
| 27b | not registered in GenBank | 373 | ABC transporter ATP-binding protein | ABC transporter related protein, Streptomyces cattleya, AEW99635 | 82/89 |
| 28b | WP_055469558 | 868 | hypothetical protein | beta-ketoacyl synthase, Streptomyces sp. NRRL F-7442, KOX46593 | 99/99 |
| 29b | WP_055469559 | 290 | ketopantoate reductase | ketopantoate reductase, Streptomyces sp. NRRL F-7442, KOX46594 | 98/99 |
| 30 | WP_055469560 | 209 | TetR family transcriptional regulator | TetR family transcriptional regulator, Streptomyces sp. NRRL F-7442, KOX46595 | 99/99 |
| 31 | WP_059296555 | 65 | chitinase | secreted chitinase, Streptomyces coelicolor, NP_733504 | 83/87 |
| 32b | WP_055469561 | 407 | FAD-dependent oxidoreductase | FAD-dependent oxidoreductase, Streptomyces sp. NRRL F-7442, KOX46596 | 98/99 |
| 33 | WP_030403663 | 168 | MarR family transcriptional regulator | MarR family transcriptional regulator, Streptomyces sp. NRRL F-7442, KOX46597.1 | 100/100 |
aIdentity/similarity; bencoded in complementary strand.
![[1860-5397-13-47-6]](/bjoc/content/figures/1860-5397-13-47-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Biosynthetic pathway of hydroxymalonyl-ACP. Adapted from [24].
Figure 6: Biosynthetic pathway of hydroxymalonyl-ACP. Adapted from [24].
Methylmalonyl-CoA was readily predicted as the origin of the methyl-branched three-carbon fragment (C-9/C-10/C-28). Actually, intense enhancement of the C-9 carbon signal was observed by feeding of [1-13C]propionate (Table 1). The remaining carbons not labeled by malonate-type precursors were the terminal tert-butyl carbons (C-23 to C-27). The origin of the tert-butyl group in polyketide biosynthesis is still unknown, however, the tert-butyl functionality of bottromycin and polytheonamide was shown to be produced by radical C-methylation of the isopropyl group of valine [31,32]. By analogy, the tert-butyl portion of 1 was most likely supplied through the C-methylation of valine. To examine this possibility, feeding experiments of L-[methyl-13C]methionine and L-valine-d8 were carried out. As expected, the tert-butylmethyl carbons (C-25, C-26, C-27) were labeled as a result of L-[methyl-13C]methionine incorporation (Table 1). In addition, the 2H (deuterium) NMR spectrum showed deuterium signals for the methyl group (H-25, H-26, H-27) of L-valine-d8-labeled 1 (Figure 7a). The mass spectrum of the L-valine-d8-labeled 1 displayed the molecular ion with a mass increment of 6 Da (Figure 7b), corresponding to the incorporation of six deuterium atoms into the terminal methyl groups. Based on these results from precursor-feeding experiments, we concluded that the tert-butyl group and the adjacent methylene carbon (C-23) are derived from valine and the S-methyl carbon of methionine. It is controversial whether pivaloyl CoA is loaded onto the ACP as a starter or isobutyl-CoA is used as a starter and C-methylation takes place afterwards. The signature sequence region of the acyltransferase domain of the PKS starter loading module for butyrolactol biosynthesis (FAGHS) shares some amino acid residues with the known loading module of isobutyl CoA (bafilomycin: LAAHS [33], α-lipomycin: LAAHS [34], tautomycin: LAAHS [35]). Meanwhile, it is known that the substrate recognition is not strict for the loading module of avermectin (VPAHS) [36] and myxalamide (VAVHS) [37] which accept both isobutyl-CoA and 2-methylbutyl-CoA. In addition, genes coding for C-methyltransferase are not present near the butyrolactol PKS genes. Further enzymatic studies are necessary to establish the order of the starter loading/C-methylation events.
![[1860-5397-13-47-7]](/bjoc/content/figures/1860-5397-13-47-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Incorporation of L-valine-d8 into 1. (a) 1H NMR spectrum of natural 1 and 2H NMR spectrum of L-valine-d8-labeled 1. (b) ESIMS spectra of natural 1 and L-valine-d8-labeled 1.
Figure 7: Incorporation of L-valine-d8 into 1. (a) 1H NMR spectrum of natural 1 and 2H NMR spectrum of L-vali...
Conclusion
In summary, we elucidated the biosynthetic origin of butyrolactol A (1) on the basis of the feeding experiments of isotope-labeled precursors in combination with the bioinformatics analysis of its biosynthetic genes. The overall result of labeling experiments is summarized in Figure 8. The tert-butyl group was shown to be derived from the C-methylated isopropyl group of valine. This is the first study that experimentally identified the precursor of a tert-butyl group in a polyketide backbone. The unusual contiguous polyol system comprising eight hydroxylated carbons was proved to be arising from the chain extension using hydroxymalonyl-ACP by labeling experiments of [1,2-13C2]acetate and [U-13C6]glucose. This conclusion is consistent with our previous bioinformatic prediction that suggested the presence of genes necessary for the supply of hydroxymalonyl-ACP adjacent to the PKS gene cluster of the butyrolactol biosynthesis. The results obtained in this study provide useful information for further biosynthetic studies and genome mining of structurally unique/novel secondary metabolites.
![[1860-5397-13-47-8]](/bjoc/content/figures/1860-5397-13-47-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Incorporation of 13C- and 2H-labeled precursors into 1.
Figure 8: Incorporation of 13C- and 2H-labeled precursors into 1.
Experimental
General experimental procedures
Sodium [1,2-13C2]acetate and L-valine-d8 were purchased from Cambridge Isotope Laboratories, Inc. [U-13C6]Glucose, sodium [1-13C]propionate, and L-[methyl-13C]methionine were purchased from Sigma-Aldrich Co. LLC. 1H and 13C NMR spectra were obtained on a Bruker AVANCE 500 spectrometer in DMSO-d6 using the signal of the residual solvent signals (δH 2.50, δC 40.0) as an internal standard. The 2H NMR spectrum was obtained on a Bruker AVANCE 500 spectrometer in DMSO. Chemical shifts were referenced to the solvent signal (δH(D) 2.50). ESITOFMS were recorded on a Bruker microTOF focus.
Microorganism
Streptomyces sp. strain TP-A0882 was isolated from a deep seawater collected in the Toyama Bay, Japan. The strain was identified as a member of the genus Streptomyces on the basis of 99.9% 16S rRNA gene sequence identity (1533 nucleotides; NCBI GneBank number BBOK01000029.1) with Streptomyces diastaticus subsp. ardesiacus NRRL B-1773T (accession number DQ026631).
Fermentation
Strain TP-A0882 growing on a plate culture was inoculated into a 500 mL K-1 flask containing 100 mL of the V-22 seed medium consisting of soluble starch 1.0%, glucose 0.5%, NZ-case (Wako Pure Chemical Industries, Ltd.) 0.3%, yeast extract (Kyokuto Pharmaceutical Industrial Co., Ltd.), 0.2%, Tryptone (Difco Laboratories) 0.5%, K2HPO4 0.1%, MgSO4·7H2O 0.05%, and CaCO3 0.3% (pH 7.0). The flask was placed on a rotary shaker (200 rpm) at 30 °C for 4 days. Then, the seed culture (3 mL) was transferred into 500 mL K-1 flasks each containing 100 mL of the A-3M production medium consisting of soluble starch 2.0%, glycerol 2.0%, glucose 0.5%, Pharmamedia (Traders Protein) 1.5%, yeast extract 0.3%, and Diaion HP-20 resin (Mitsubishi Chemical Corporation) 1%. The pH of the medium was adjusted to 7.0 before sterilization. The inoculated flasks were placed on a rotary shaker (200 rpm) at 30 °C for 6 days.
Extraction and isolation
After incubation, 100 mL of 1-butanol was added to each flask, and the flasks were allowed to shake for an hour. The mixture was centrifuged at 6,000 rpm for 10 min and the organic layer was collected from the aqueous layer. The solvent was removed by evaporation to give 1.6 g of a crude extract from 1 L of culture. This crude extract was fractionated using silica gel column chromatography with a step gradient of CHCl3–MeOH (1:0, 20:1, 10:1, 4:1, 2:1, 1:1, and 0:1 v/v). Fraction 4 (4:1) containing 1 was concentrated to give 16.2 mg of dark yellow gum. The final purification was achieved by preparative HPLC (Cosmosil 5C18-AR-II, 10 × 250 mm, 4 mL/min) using a gradient of MeCN/0.1% HCO2H (MeCN concentration: 50–100% for 0–30 min) at 4 mL/min, yielding 1 (2.7 mg) with a retention time of 26.7 min.
Incorporation of 13C- and 2H-labeled precursors
Feeding experiments were performed for sodium [1,2-13C2]acetate, [U-13C6]glucose, sodium [1-13C]propionate, L-[methyl-13C]methionine, and L-valine-d8. Inoculation, cultivation, and purification were performed in the same manner as described above. Addition of 13C- and 2H-labeled precursors was initiated at 48 h after inoculation and periodically carried out every 24 h for four times. After further incubation for 24 h, the cultures were extracted with 1-butanol.
- Sodium [1,2-13C2]acetate: After feeding of sodium [1,2-13C2]acetate (total 800 mg; 20 mg × 10 flasks × 4 days), 3.6 mg of 13C-labeled 1 was obtained from 1 L of culture.
- [U-13C6]Glucose: After feeding of [U-13C6]glucose (total 800 mg; 20 mg × 10 flasks × 4 days), 2.5 mg of 13C-labeled 1 was obtained from 1 L of culture.
- Sodium [1-13C]propionate: After feeding of sodium [1-13C]propionate (total 800 mg; 20 mg × 10 flasks × 4 days), 2.1 mg of 13C-labeled 1 was obtained from 1 L of culture.
- L-[Methyl-13C]methionine: After feeding of L-[methyl-13C]methionine (total 80 mg; 2.0 mg × 10 flasks × 4 days), 3.2 mg of 13C-labeled 1 was obtained from 1 L of culture.
- L-Valine-d8: After feeding of L-valine-d8 (total 80 mg; 2.0 mg × 10 flasks × 4 days), 2.1 mg of deuterated 1 was obtained from 1 L of culture.
Supporting Information
| Supporting Information File 1: NMR spectra of 13C- and 2H-labeled 1. | ||
| Format: PDF | Size: 904.8 KB | Download |
References
-
Bérdy, J. J. Antibiot. 2005, 58, 1–26. doi:10.1038/ja.2005.1
Return to citation in text: [1] -
Bérdy, J. J. Antibiot. 2012, 65, 385–395. doi:10.1038/ja.2012.27
Return to citation in text: [1] -
Hertweck, C. Angew. Chem., Int. Ed. 2009, 48, 4688–4716. doi:10.1002/anie.200806121
Return to citation in text: [1] -
Sánchez, C.; Méndez, C.; Salas, J. A. J. Ind. Microbiol. Biotechnol. 2006, 33, 560–568. doi:10.1007/s10295-006-0092-5
Return to citation in text: [1] -
Salas, J. A.; Méndez, C. Trends Microbiol. 2007, 15, 219–232. doi:10.1016/j.tim.2007.03.004
Return to citation in text: [1] -
Wilkinson, B.; Micklefield, J. Nat. Chem. Biol. 2007, 3, 379–386. doi:10.1038/nchembio.2007.7
Return to citation in text: [1] -
Kim, W.; Lee, D.; Hong, S. S.; Na, Z.; Shin, J. C.; Roh, S. H.; Wu, C.-Z.; Choi, O.; Lee, K.; Shen, Y.-M.; Paik, S.-G.; Lee, J. J.; Hong, Y.-S. ChemBioChem 2009, 10, 1243–1251. doi:10.1002/cbic.200800763
Return to citation in text: [1] -
Kong, D.; Lee, M.-J.; Lin, S.; Kim, E.-S. J. Ind. Microbiol. Biotechnol. 2013, 40, 529–543. doi:10.1007/s10295-013-1258-6
Return to citation in text: [1] -
Kotake, C.; Yamasaki, T.; Moriyama, T.; Shinoda, M.; Komiyama, N.; Furumai, T.; Konishi, M.; Oki, T. J. Antibiot. 1992, 45, 1442–1450. doi:10.7164/antibiotics.45.1442
Return to citation in text: [1] [2] [3] -
Ko, K.; Ge, H. M.; Shin, J.; Oh, D. C. Planta Med. 2016, 82 (Suppl. Suppl.1), S1–S381. doi:10.1055/s-0036-1596637
Return to citation in text: [1] -
Dembitsky, V. M. Lipids 2006, 41, 309–340. doi:10.1007/s11745-006-5103-9
Return to citation in text: [1] -
Shao, C.-L.; Linington, R. G.; Balunas, M. J.; Centeno, A.; Boudreau, P.; Zhang, C.; Engene, N.; Spadafora, C.; Mutka, T. S.; Kyle, D. E.; Gerwick, L.; Wang, C.-Y.; Gerwick, W. H. J. Org. Chem. 2015, 80, 7849–7855. doi:10.1021/acs.joc.5b01264
Return to citation in text: [1] -
Salvador-Reyes, L. A.; Sneed, J.; Paul, V. J.; Luesch, H. J. Nat. Prod. 2015, 78, 1957–1962. doi:10.1021/acs.jnatprod.5b00293
Return to citation in text: [1] -
Ogawa, H.; Iwasaki, A.; Sumimoto, S.; Kanamori, Y.; Ohno, O.; Iwatsuki, M.; Ishiyama, A.; Hokari, R.; Otoguro, K.; Omura, S.; Suenaga, K. J. Nat. Prod. 2016, 79, 1862–1866. doi:10.1021/acs.jnatprod.6b00171
Return to citation in text: [1] -
Mander, L.; Liu, H.-W. Comprehensive Natural Products II: Chemistry and Biology, 1st ed.; Elsevier B.V.: Kidlington, UK, 2010.
Return to citation in text: [1] -
Grindberg, R. V.; Ishoey, T.; Brinza, D.; Esquenazi, E.; Coates, R. C.; Liu, W.-t.; Gerwick, L.; Dorrestein, P. C.; Pevzner, P.; Lasken, R.; Gerwick, W. H. PLoS One 2011, 6, No. e18565. doi:10.1371/journal.pone.0018565
Return to citation in text: [1] -
Staunton, J.; Wilkinson, B. Chem. Rev. 1997, 97, 2611–2630. doi:10.1021/cr9600316
Return to citation in text: [1] -
Caffrey, P.; Lynch, S.; Flood, E.; Finnan, S.; Oliynyk, M. Cell Chem. Biol. 2001, 8, 713–723. doi:10.1016/S1074-5521(01)00046-1
Return to citation in text: [1] -
Kirst, H. A.; Mynderse, J. S.; Martin, J. W.; Baker, P. J.; Paschal, J. W.; Steiner, J. L. R.; Lobkovsky, E.; Clardy, J. J. Antibiot. 1996, 49, 162–167. doi:10.7164/antibiotics.49.162
Return to citation in text: [1] -
Fernández-Chimeno, R. I.; Cañedo, L.; Espliego, F.; Grávalos, D.; De La Calle, F.; Fernández-Puentes, J. L.; Romero, F. J. Antibiot. 2000, 53, 474–478. doi:10.7164/antibiotics.53.474
Return to citation in text: [1] -
Wang, Y.-F.; Wei, S.-J.; Zhang, Z.-P.; Zhan, T.-H.; Tu, G.-Q. Nat. Prod. Bioprospect. 2012, 2, 41–45. doi:10.1007/s13659-011-0037-1
Return to citation in text: [1] -
Komaki, H.; Ichikawa, N.; Hosoyama, A.; Fujita, N.; Igarashi, Y. FEMS Microbiol. Lett. 2015, 362, fnv155. doi:10.1093/femsle/fnv155
Return to citation in text: [1] -
Chan, Y. A.; Boyne, M. T.; Podevels, A. M.; Klimowicz, A. K.; Handelsman, J.; Kelleher, N. L.; Thomas, M. G. Proc. Natl. Acad. Sci. U. S. A. 2006, 39, 14349–14354. doi:10.1073/pnas.0603748103
Return to citation in text: [1] -
Park, H.; Kevany, B. M.; Dyer, D. H.; Thomas, M. G.; Forest, K. T. PLoS One 2014, 9, No. e110965. doi:10.1371/journal.pone.0110965
Return to citation in text: [1] [2] -
Emmert, E. A. B.; Klimowicz, A. K.; Thomas, M. G.; Handelsman, J. Appl. Environ. Microbiol. 2004, 70, 104–113. doi:10.1128/AEM.70.1.104-113.2004
Return to citation in text: [1] [2] -
Li, Y.; Li, Z.; Yamanaka, K.; Xu, Y.; Zhang, W.; Vlamakis, H.; Kolter, R.; Moore, B. S.; Qian, P.-Y. Sci. Rep. 2015, 5, No. 9383. doi:10.1038/srep09383
Return to citation in text: [1] -
Park, D.; Ciezki, K.; van der Hoeven, R.; Singh, S.; Reimer, D.; Bode, H. B.; Forst, S. Mol. Microbiol. 2009, 73, 938–949. doi:10.1111/j.1365-2958.2009.06817.x
Return to citation in text: [1] -
Müller, S.; Garcia-Gonzalez, E.; Genersch, E.; Süssmuth, R. D. Nat. Prod. Rep. 2015, 32, 765–778. doi:10.1039/C4NP00158C
Return to citation in text: [1] -
Sakuda, S.; Ono, M.; Ikeda, H.; Inagaki, Y.; Nakayama, J.; Suzuki, A.; Isogai, A. Tetrahedron Lett. 1997, 38, 7399–7402. doi:10.1016/S0040-4039(97)01734-6
Return to citation in text: [1] -
Ono, M.; Sakuda, S.; Ikeda, H.; Furihata, K.; Nakayama, J.; Suzuki, A.; Isogai, A. J. Antibiot. 1998, 51, 1019–1028. doi:10.7164/antibiotics.51.1019
Return to citation in text: [1] -
Huo, L.; Rachid, S.; Stadler, M.; Wenzel, S. C.; Müller, R. Chem. Biol. 2012, 19, 1278–1287. doi:10.1016/j.chembiol.2012.08.013
Return to citation in text: [1] -
Parent, A.; Guillot, A.; Benjdia, A.; Chartier, G.; Leprince, J.; Berteau, O. J. Am. Chem. Soc. 2016, 138, 15515–15518. doi:10.1021/jacs.6b06697
Return to citation in text: [1] -
Hwang, J. Y.; Kim, H. S.; Kim, S. H.; Oh, H. R.; Nam, D. H. AMB Express 2013, 3, No. 24. doi:10.1186/2191-0855-3-24
Return to citation in text: [1] -
Bihlmaier, C.; Welle, E.; Hofmann, C.; Welzel, K.; Vente, A.; Breitling, E.; Müller, M.; Glaser, S.; Bechthold, A. Antimicrob. Agents Chemother. 2006, 50, 2113–2121. doi:10.1128/AAC.00007-06
Return to citation in text: [1] -
Zhao, C.; Ju, J.; Christenson, S. D.; Smith, W. C.; Song, D.; Zhou, X.; Shen, B.; Deng, Z. J. Bacteriol. 2006, 188, 4142–4147. doi:10.1128/JB.00173-06
Return to citation in text: [1] -
Ikeda, H.; Nonomiya, T.; Usami, M.; Ohta, T.; Ōmura, S. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9509–9514. doi:10.1073/pnas.96.17.9509
Return to citation in text: [1] -
Silakowski, B.; Nordsiek, G.; Kunze, B.; Blöcker, H.; Müller, R. Cell Chem. Biol. 2001, 8, 59–69. doi:10.1016/S1074-5521(00)00056-9
Return to citation in text: [1]
| 36. | Ikeda, H.; Nonomiya, T.; Usami, M.; Ohta, T.; Ōmura, S. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9509–9514. doi:10.1073/pnas.96.17.9509 |
| 37. | Silakowski, B.; Nordsiek, G.; Kunze, B.; Blöcker, H.; Müller, R. Cell Chem. Biol. 2001, 8, 59–69. doi:10.1016/S1074-5521(00)00056-9 |
| 1. | Bérdy, J. J. Antibiot. 2005, 58, 1–26. doi:10.1038/ja.2005.1 |
| 2. | Bérdy, J. J. Antibiot. 2012, 65, 385–395. doi:10.1038/ja.2012.27 |
| 9. | Kotake, C.; Yamasaki, T.; Moriyama, T.; Shinoda, M.; Komiyama, N.; Furumai, T.; Konishi, M.; Oki, T. J. Antibiot. 1992, 45, 1442–1450. doi:10.7164/antibiotics.45.1442 |
| 22. | Komaki, H.; Ichikawa, N.; Hosoyama, A.; Fujita, N.; Igarashi, Y. FEMS Microbiol. Lett. 2015, 362, fnv155. doi:10.1093/femsle/fnv155 |
| 9. | Kotake, C.; Yamasaki, T.; Moriyama, T.; Shinoda, M.; Komiyama, N.; Furumai, T.; Konishi, M.; Oki, T. J. Antibiot. 1992, 45, 1442–1450. doi:10.7164/antibiotics.45.1442 |
| 23. | Chan, Y. A.; Boyne, M. T.; Podevels, A. M.; Klimowicz, A. K.; Handelsman, J.; Kelleher, N. L.; Thomas, M. G. Proc. Natl. Acad. Sci. U. S. A. 2006, 39, 14349–14354. doi:10.1073/pnas.0603748103 |
| 4. | Sánchez, C.; Méndez, C.; Salas, J. A. J. Ind. Microbiol. Biotechnol. 2006, 33, 560–568. doi:10.1007/s10295-006-0092-5 |
| 5. | Salas, J. A.; Méndez, C. Trends Microbiol. 2007, 15, 219–232. doi:10.1016/j.tim.2007.03.004 |
| 6. | Wilkinson, B.; Micklefield, J. Nat. Chem. Biol. 2007, 3, 379–386. doi:10.1038/nchembio.2007.7 |
| 7. | Kim, W.; Lee, D.; Hong, S. S.; Na, Z.; Shin, J. C.; Roh, S. H.; Wu, C.-Z.; Choi, O.; Lee, K.; Shen, Y.-M.; Paik, S.-G.; Lee, J. J.; Hong, Y.-S. ChemBioChem 2009, 10, 1243–1251. doi:10.1002/cbic.200800763 |
| 8. | Kong, D.; Lee, M.-J.; Lin, S.; Kim, E.-S. J. Ind. Microbiol. Biotechnol. 2013, 40, 529–543. doi:10.1007/s10295-013-1258-6 |
| 20. | Fernández-Chimeno, R. I.; Cañedo, L.; Espliego, F.; Grávalos, D.; De La Calle, F.; Fernández-Puentes, J. L.; Romero, F. J. Antibiot. 2000, 53, 474–478. doi:10.7164/antibiotics.53.474 |
| 3. | Hertweck, C. Angew. Chem., Int. Ed. 2009, 48, 4688–4716. doi:10.1002/anie.200806121 |
| 21. | Wang, Y.-F.; Wei, S.-J.; Zhang, Z.-P.; Zhan, T.-H.; Tu, G.-Q. Nat. Prod. Bioprospect. 2012, 2, 41–45. doi:10.1007/s13659-011-0037-1 |
| 15. | Mander, L.; Liu, H.-W. Comprehensive Natural Products II: Chemistry and Biology, 1st ed.; Elsevier B.V.: Kidlington, UK, 2010. |
| 17. | Staunton, J.; Wilkinson, B. Chem. Rev. 1997, 97, 2611–2630. doi:10.1021/cr9600316 |
| 18. | Caffrey, P.; Lynch, S.; Flood, E.; Finnan, S.; Oliynyk, M. Cell Chem. Biol. 2001, 8, 713–723. doi:10.1016/S1074-5521(01)00046-1 |
| 11. | Dembitsky, V. M. Lipids 2006, 41, 309–340. doi:10.1007/s11745-006-5103-9 |
| 12. | Shao, C.-L.; Linington, R. G.; Balunas, M. J.; Centeno, A.; Boudreau, P.; Zhang, C.; Engene, N.; Spadafora, C.; Mutka, T. S.; Kyle, D. E.; Gerwick, L.; Wang, C.-Y.; Gerwick, W. H. J. Org. Chem. 2015, 80, 7849–7855. doi:10.1021/acs.joc.5b01264 |
| 13. | Salvador-Reyes, L. A.; Sneed, J.; Paul, V. J.; Luesch, H. J. Nat. Prod. 2015, 78, 1957–1962. doi:10.1021/acs.jnatprod.5b00293 |
| 14. | Ogawa, H.; Iwasaki, A.; Sumimoto, S.; Kanamori, Y.; Ohno, O.; Iwatsuki, M.; Ishiyama, A.; Hokari, R.; Otoguro, K.; Omura, S.; Suenaga, K. J. Nat. Prod. 2016, 79, 1862–1866. doi:10.1021/acs.jnatprod.6b00171 |
| 19. | Kirst, H. A.; Mynderse, J. S.; Martin, J. W.; Baker, P. J.; Paschal, J. W.; Steiner, J. L. R.; Lobkovsky, E.; Clardy, J. J. Antibiot. 1996, 49, 162–167. doi:10.7164/antibiotics.49.162 |
| 9. | Kotake, C.; Yamasaki, T.; Moriyama, T.; Shinoda, M.; Komiyama, N.; Furumai, T.; Konishi, M.; Oki, T. J. Antibiot. 1992, 45, 1442–1450. doi:10.7164/antibiotics.45.1442 |
| 10. | Ko, K.; Ge, H. M.; Shin, J.; Oh, D. C. Planta Med. 2016, 82 (Suppl. Suppl.1), S1–S381. doi:10.1055/s-0036-1596637 |
| 16. | Grindberg, R. V.; Ishoey, T.; Brinza, D.; Esquenazi, E.; Coates, R. C.; Liu, W.-t.; Gerwick, L.; Dorrestein, P. C.; Pevzner, P.; Lasken, R.; Gerwick, W. H. PLoS One 2011, 6, No. e18565. doi:10.1371/journal.pone.0018565 |
| 25. | Emmert, E. A. B.; Klimowicz, A. K.; Thomas, M. G.; Handelsman, J. Appl. Environ. Microbiol. 2004, 70, 104–113. doi:10.1128/AEM.70.1.104-113.2004 |
| 26. | Li, Y.; Li, Z.; Yamanaka, K.; Xu, Y.; Zhang, W.; Vlamakis, H.; Kolter, R.; Moore, B. S.; Qian, P.-Y. Sci. Rep. 2015, 5, No. 9383. doi:10.1038/srep09383 |
| 24. | Park, H.; Kevany, B. M.; Dyer, D. H.; Thomas, M. G.; Forest, K. T. PLoS One 2014, 9, No. e110965. doi:10.1371/journal.pone.0110965 |
| 25. | Emmert, E. A. B.; Klimowicz, A. K.; Thomas, M. G.; Handelsman, J. Appl. Environ. Microbiol. 2004, 70, 104–113. doi:10.1128/AEM.70.1.104-113.2004 |
| 34. | Bihlmaier, C.; Welle, E.; Hofmann, C.; Welzel, K.; Vente, A.; Breitling, E.; Müller, M.; Glaser, S.; Bechthold, A. Antimicrob. Agents Chemother. 2006, 50, 2113–2121. doi:10.1128/AAC.00007-06 |
| 35. | Zhao, C.; Ju, J.; Christenson, S. D.; Smith, W. C.; Song, D.; Zhou, X.; Shen, B.; Deng, Z. J. Bacteriol. 2006, 188, 4142–4147. doi:10.1128/JB.00173-06 |
| 31. | Huo, L.; Rachid, S.; Stadler, M.; Wenzel, S. C.; Müller, R. Chem. Biol. 2012, 19, 1278–1287. doi:10.1016/j.chembiol.2012.08.013 |
| 32. | Parent, A.; Guillot, A.; Benjdia, A.; Chartier, G.; Leprince, J.; Berteau, O. J. Am. Chem. Soc. 2016, 138, 15515–15518. doi:10.1021/jacs.6b06697 |
| 33. | Hwang, J. Y.; Kim, H. S.; Kim, S. H.; Oh, H. R.; Nam, D. H. AMB Express 2013, 3, No. 24. doi:10.1186/2191-0855-3-24 |
| 29. | Sakuda, S.; Ono, M.; Ikeda, H.; Inagaki, Y.; Nakayama, J.; Suzuki, A.; Isogai, A. Tetrahedron Lett. 1997, 38, 7399–7402. doi:10.1016/S0040-4039(97)01734-6 |
| 30. | Ono, M.; Sakuda, S.; Ikeda, H.; Furihata, K.; Nakayama, J.; Suzuki, A.; Isogai, A. J. Antibiot. 1998, 51, 1019–1028. doi:10.7164/antibiotics.51.1019 |
| 24. | Park, H.; Kevany, B. M.; Dyer, D. H.; Thomas, M. G.; Forest, K. T. PLoS One 2014, 9, No. e110965. doi:10.1371/journal.pone.0110965 |
| 27. | Park, D.; Ciezki, K.; van der Hoeven, R.; Singh, S.; Reimer, D.; Bode, H. B.; Forst, S. Mol. Microbiol. 2009, 73, 938–949. doi:10.1111/j.1365-2958.2009.06817.x |
| 28. | Müller, S.; Garcia-Gonzalez, E.; Genersch, E.; Süssmuth, R. D. Nat. Prod. Rep. 2015, 32, 765–778. doi:10.1039/C4NP00158C |
© 2017 Harunari et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)