Abstract
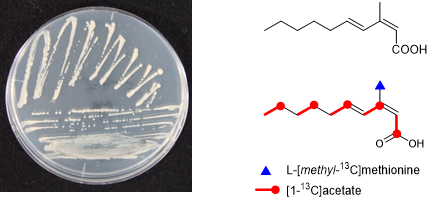
(2Z,4E)-3-Methyl-2,4-decadienoic acid (1) was identified as a major metabolite from a culture extract of a marine bacterium Microbulbifer which was collected from a stony coral Porites sp. NMR-based spectroscopic analysis revealed that 1 is an unsaturated fatty acid in which a methyl group is located in an uncommon position as a natural product. Feeding experiments of 13C-labeled precursors clarified that ʟ-methionine-derived methylation takes place at the carbon which is derived from the carbonyl carbon of acetate. Compound 1 showed weak growth inhibition against Saccharomyces cerevisiae.
Graphical Abstract
Introduction
Marine microbial symbionts are currently recognized as a reservoir of new bioactive compounds [1]. The most well-studied host animal is the sponge from which a vast array of natural products has been isolated and symbiotic bacteria are suggested to be responsible for the biosynthesis of such natural products [2]. Although it is well established that corals are associated with diverse microbes, coral-associated bacteria and their secondary metabolites have received lesser attention [3]. To date, a couple of new compounds were discovered from soft coral-associated bacteria such as pseudoalteromones from Pseudoalteromonas isolated from the cultured octocoral Lobophytum crassum [4,5] and macrolactin V from Bacillus amyloliquefaciens associated with a gorgonian coral Junceella juncea [6], but there is no report on the compounds from stony coral-associated bacteria except our recently published work [7].
Microbulbifer is a genus of Gram-negative bacteria belonging to the class Gammaproteobacteria [8]. Members of this genus are frequently isolated from halophilic environments including marine solar saltern [9], marine sediment [10], and marine invertebrates [11]. The Microbulbifer species are considered to be marine obligate bacteria as they require sodium salt for growth [8]. In our continuing investigation of natural products from marine-derived bacteria, (2Z,4E)-3-methyl-2,4-decadienoic acid (1) was obtained from the culture extract of Microbulbifer sp. C4-6 isolated from a stony coral Porites sp. (Figure 1). Compound 1 is known as a synthetic compound [12] but this is its first discovery as a natural product. In addition, 1 is biosynthetically unique: it has an uncommon methylation pattern in its carbon chain, derived from the C-methylation with ʟ-methionine at a carbon originated from a carbonyl carbon of acetate. In this paper, we report the isolation and structure determination of 1 and its biosynthetic origin proven by the feeding experiments of 13C-labeled precursors.
![[1860-5397-15-225-1]](/bjoc/content/figures/1860-5397-15-225-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of (2Z,4E)-3-methyl-2,4-decadienoic acid (1).
Figure 1: Structure of (2Z,4E)-3-methyl-2,4-decadienoic acid (1).
Results and Discussion
The bacterial strain C4-6 was isolated from a Scleractinian (stony) coral Porites sp. On the basis of 16S rRNA gene sequence, this strain was identified as Microbulbifer. Strain C4-6 was cultured in A11M seawater medium at 30 °C for five days, and the whole culture broth was extracted with 1-butanol. The extract was consecutively fractionated by normal- and reversed-phase column chromatographies, followed by HPLC purification on a C18 column to yield compound 1.
The molecular formula of 1 was determined to be C11H18O2 with three degrees of unsaturation on the basis of its NMR and HR-ESI-TOFMS (m/z 181.1230 [M − H]−; calcd for C11H17O2, 181.1229) data. The UV spectrum of 1 in methanol exhibited an absorption maximum at 262 nm. The IR absorption bands at 1678 and 2800–3200 cm−1 suggested the presence of carboxyl group. The 1H and 13C NMR data of 1 (Table 1) showed the presence of 11 carbon signals, which were identified by the assistance of a DEPT135 spectrum as two methyls, four sp3 methylenes, three sp2 methines (δC 140.6, 127.7, 115.1), and two sp2 nonprotonated carbons including one carbonyl group (δC 171.3, 153.9). Besides, the 1H NMR spectrum showed characteristic resonances, especially for two methyl groups at δH 2.02 (3H, s) and 0.90 (3H, t) in the highfield region and for multiple methylene signals, suggesting the presence of an alkyl chain. COSY analysis established two separate spin systems, H4/H5/H6/H7 and H8/H9/H10. These partial structures were joined by the mutual HMBC correlations between H7 and H9. Furthermore, long-range correlations from H11 to C2, C3, and C4 and from H2 to C1, C3, and C4 established the carbon connectivity among these carbons to complete the structure of 1 (Figure 2). The double bond geometry was determined on the basis of NOESY analysis and 3JHH coupling constants. NOE was detected between H2 and H11 but not between H2 and H4, reflecting the Z-configuration for the C2–C3 double bond. Similarly, NOESY correlations for H4/H6 and H5/H11 reflected the E-configuration for the C4–C5 double bond, which was corroborated by 3JHH vicinal coupling constant (JH4,H5 = 15.8 Hz).
Table 1: 1H and 13C NMR data for compound 1 in CDCl3.
| position | δCa | δH mult (J in Hz)b | HMBCb,c |
|---|---|---|---|
| 1 | 171.3, C | ||
| 2 | 114.8, CH | 5.63, s | 1, 3, 4, 11 |
| 3 | 153.8, C | ||
| 4 | 127.5, CH | 7.55, d (15.8) | 2, 3, 6, 11 |
| 5 | 140.3, CH | 6.22, dt (15.8, 7.2) | 3, 6, 7 |
| 6 | 33.3, CH2 | 2.24, dt (7.2, 7.2) | 4, 5, 7, 8 |
| 7 | 28.7, CH2 | 1.46, m | 5, 8, 9 |
| 8 | 31.4, CH2 | 1.32d, m | 6, 7, 10 |
| 9 | 22.4, CH2 | 1.32d, m | |
| 10 | 13.9, CH3 | 0.90, t (7.1) | 8, 9 |
| 11 | 21.3, CH3 | 2.02, s | 2, 3, 4 |
aRecorded at 125 MHz (reference δC 77.0). bRecorded at 500 MHz (reference δH 7.26). cHMBC correlations are from proton(s) stated to the indicated carbon. dOverlapping signals.
![[1860-5397-15-225-2]](/bjoc/content/figures/1860-5397-15-225-2.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 2: COSY and key HMBC correlations for 1.
Figure 2: COSY and key HMBC correlations for 1.
In fungi and certain kinds of bacteria, methyl substituents in the fatty acid carbon chain or the polyketide chain are derived from the methyl group of S-adenosylmethionine (SAM) (Figure 3A) [13,14]. This methylation reaction usually occurs at the nucleophilic carbons originated from the methyl carbon of acetate (C2) since SAM acts as an electrophilic methyl donor. In most of the bacteria including actinomycetes, methyl branching in polyketide chain is derived from methylmalonyl CoA, thereby the methylation position must be also the α-position (C2) of the acetate unit (Figure 3B) [13]. In contrast, methylation at the carbons derived from the carbonyl carbon of the acetate (C1) is quite unusual in fatty acids and polyketides. This unusual methylation pattern is reported for the polyketides of eukaryotic algae dinoflagellates. Some of the methyl groups in dinoflagellate compounds are positioned at the carbons derived from the carbonyl carbon of acetate (C1). Precursor labeling studies indicated that the origin of the methyl carbon is not SAM but one carbon fragment derived from the cleavage of another acetate unit (Figure 3C) [15]. The only single example of C-methylation with SAM at a carbon derived from the carbonyl carbon of acetate (C1; Figure 3D) is reported for sphingolipid biosynthesis in the yeast, Pichia pastoris [16]. SAM-dependent C-methylation takes place at the alkenyl carbon C9 of glucosylceramide, yielding a cationic intermediate, from which deprotonation occurs at C9 to give an internal olefin (Scheme 1).
![[1860-5397-15-225-3]](/bjoc/content/figures/1860-5397-15-225-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Methylation pattern that can occur in fatty acids and polyketides.
Figure 3: Methylation pattern that can occur in fatty acids and polyketides.
![[1860-5397-15-225-i1]](/bjoc/content/inline/1860-5397-15-225-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: C-Methylation of alkenyl carbon in sphingolipid biosynthesis.
Scheme 1: C-Methylation of alkenyl carbon in sphingolipid biosynthesis.
In the case of 1, methylation at the C3 carbon is inconsistent with the regular methylation pattern that occurs in fatty acids synthesized by the FAS (fatty acid synthase) or polyketides from the PKS (polyketide synthase) system of bacterial groups. Therefore, the biosynthetic origin of 1 was investigated by feeding experiments of 13C-labeled precursors (Table 2). Firstly, in order to verify the origin of the carbon backbone, [1-13C]acetate was fed to the culture which gave a high enrichment of the carbons at C1, C3, C5, C7, and C9 (Figure 4). A feeding experiment with ʟ-[methyl-13C]methionine was then carried out. The high level of enrichment was observed only for C11 in the 13C NMR spectrum, thereby confirming that the methyl group is derived from methionine via SAM. In addition to the fungal sphingolipids described above, methyl branches with SAM-origin are also found in some bacterial fatty acids like tuberculostearic acid, a major constituent of mycobacterial membrane phospholipids [17]. Methylation at the olefinic double bond of oleic acid part in phospholipids is catalyzed by SAM-dependent methyltransferase, followed by 1,2-hydride shift and deprotonation, and a subsequent reduction of the exo-methylene intermediate gives rise to a methyl group (Scheme 2) [18]. The presence of the exo-methylene intermediate was experimentally proved but the enzyme responsible for the double bond reduction has not been identified [18]. Based on the C-methylation pattern found in the biosynthesis of sphingolipid and tuberculostearic acid, we propose a possible mechanism of methylation for compound 1 although further chemical/biochemical investigation is required (Scheme 3). First, 2,4-decadienoic acid is formed and methylation takes place at C3 to give a putative cationic intermediate. The following proton loss from C3 is probable to give directly 1 in a similar fashion to the glucosylceramide biosynthesis. Branched chain fatty acids are commonly present in cell membranes, and mono-methylation at the iso or anteiso position is the most common [19]. Branching at other positions is less common but represents a significant proportion of branched-chain fatty acids in some organisms [19]. Tuberculostearic acid (10-methyloctadecanoic acid (18:1 me(10)), 10- methylhexadecanoic acid (16:1 me(10)), and 10-methylnonadecanoic acid (19:1 me(10)) are the major constituents of the cell wall of Mycobacterium phlei [20]. C9-Methylated glucosylceramides also possess a methyl-branching in the middle of the aliphatic carbon chain of the sphingosine part. C9-Methylated sphingolipids have not been found from plants and animals but are widely produced by many fungi and are thought to play an important role in the interaction between fungi and their host organisms. C9-Methylated sphingolipids are also found from some marine invertebrates such as sea anemone and starfish although their biological function is still unclear in these organisms. C-Methylation at C9 of fungal sphingosines is similarly catalyzed by SAM-dependent methyltransferases [16]. The methylation takes place at the alkenyl carbon (C9) which is originally derived from the carbonyl carbon of an acetate unit: C1 and C2 of sphingosines are derived from ʟ-serine and carbons from C3 to C18 are from palmitoyl-CoA. To the best of our knowledge, compound 1 is the first example of a simple fatty acid in which the carbonyl carbon of an acetate unit is methylated with SAM (Figure 3D).
Table 2: Incorporation of 13C-labeled precursors into 1.
| relative enrichmenta | |||
|---|---|---|---|
| position | δCb | [1-13C]acetate | ʟ-[methyl-13C]-methionine |
| 1 | 171.3 | 8.99 | 1.10 |
| 2 | 114.8 | 0.91 | 1.12 |
| 3 | 153.8 | 10.13 | 0.84 |
| 4 | 127.5 | 1.00 | 1.00 |
| 5 | 140.3 | 11.19 | 0.99 |
| 6 | 33.3 | 1.15 | 1.12 |
| 7 | 28.7 | 11.35 | 1.12 |
| 8 | 31.4 | 1.19 | 1.20 |
| 9 | 22.4 | 12.01 | 1.13 |
| 10 | 13.9 | 1.37 | 1.17 |
| 11 | 21.3 | 1.20 | 17.74 |
aThe 13C signal intensity of each peak in the labeled 1 divided by that of the corresponding signal in the unlabeled, normalized to the peak area of C4 to give an enrichment ratio for enriched peak. The numbers in bold type indicate 13C-enriched atoms from 13C-labeled precursors. bRecorded at 125 MHz (reference δC 77.0).
![[1860-5397-15-225-4]](/bjoc/content/figures/1860-5397-15-225-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Incorporation of 13C-labeled precursors into 1.
Figure 4: Incorporation of 13C-labeled precursors into 1.
![[1860-5397-15-225-i2]](/bjoc/content/inline/1860-5397-15-225-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Biosynthesis of tuberculostearic acid in Mycobacterium.
Scheme 2: Biosynthesis of tuberculostearic acid in Mycobacterium.
![[1860-5397-15-225-i3]](/bjoc/content/inline/1860-5397-15-225-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Possible methylation mechanism for compound 1.
Scheme 3: Possible methylation mechanism for compound 1.
Compound 1 was not active against Staphylococcus aureus FDA209P JC-1, Micrococcus luteus ATCC9341, Bacillus subtilis ATCC6633, Escherichia coli NIHJ JC-2, Ralstonia solanacearum SUPP1541, Rhizobium radiobacter NBRC14554, and Candida albicans NBRC0197 (MIC > 100 μg/mL) but weakly active against Saccharomyces cerevisiae S100 (MIC 100 μg/mL).
Conclusion
In summary, chemical investigation of metabolites in a marine-derived Microbulbifer sp. led to the discovery of a unique unsaturated fatty acid, (2Z,4E)-3-methyl-2,4-decadienoic acid (1) in which the carbon originated from the carbonyl carbon of an acetate unit is methylated, providing a quite rare case of C-methylation pattern in fatty acid/polyketide biosynthesis. Compound 1 and its (2E,4E)-isomer were reported previously as synthetic compounds but this is the first finding of 1 as a natural product [12,21]. Very recently, benzoate derivatives were reported from a marine-derived Microbulbifer [22]. Therefore, this is the second report on the small molecule from this underexplored taxon. According to the genome sequence database, biosynthetic genes for NRPS and siderophore are present in Microbulbifer species which will be pursued in our future investigation.
Experimental
General experimental procedures
The UV spectrum was recorded on a Shimadzu UV-1800 spectrophotometer. The IR spectrum was measured on a Perkin-Elmer Spectrum 100. NMR spectra were obtained on a Bruker AVANCE 500 spectrometer in CDCl3 using the signals of the residual solvent proton (δH 7.26) and carbon (δH 77.0) as internal standards. HR-ESI-TOFMS were recorded on a Bruker microTOF focus. Sodium [1-13C]acetate was purchased from Cambridge Isotope Laboratories, Inc., and ʟ-[methyl-13C]methionine from Sigma-Aldrich Co. LLC.
Microorganism
Strain C4-6 was isolated from a cultured stony coral (Porites sp.) obtained from an aquarium vendor in Nagasaki, Japan. The coral specimen was washed with 70% ethanol and then washed with sterile natural seawater. A fragment of the coral (ca. 1 g) was homogenized by mortar and pestle with an equal amount of sterile natural seawater (1 mL), and 10-fold serial dilution was carried out up to 10−5 and 0.1 mL of each dilution was spread onto marine agar 2216 (Difco). The plates were kept at 23 °C, and a single colony was repeatedly transferred onto the same agar medium to obtain the pure isolate of strain C4-6. The strain was identified as a member of the genus Microbulbifer on the basis of 99.0% similarity in the 16S rRNA gene sequence (1405 nucleotides; DDBJ accession number LC456787) to Microbulbifer variabilis Ni-2088T (accession number AB167354).
Fermentation
The producing strain C4-6 was maintained on marine agar 2216 (Difco). A single colony of strain C4-6 was inoculated into a 500 mL K-1 flask containing 100 mL of marine broth 2216 (Difco) as a seed culture. The seed culture was incubated at 30 °C on a rotary shaker at 200 rpm for 2 days. Three mL of seed culture was inoculated into twenty-five 500 mL K-1 flasks each containing 100 mL of A11M production medium, which consists of glucose 0.2%, soluble starch 2.5%, yeast extract 0.5%, polypeptone (Wako Pure Chemical Industries, Ltd.) 0.5%, NZ-amine (Wako Pure Chemical Industries, Ltd.) 0.5%, CaCO3 0.3%, and Diaion HP-20 (Mitsubishi Chemical Co.) 1% in natural seawater (collected from Toyama Bay, Japan). The pH of the medium was adjusted to 7.0 before sterilization. The inoculated flasks were incubated at 30 °C on a rotary shaker at 200 rpm for 5 days.
Extraction and isolation
After fermentation, 100 mL of 1-butanol was added to each production culture flask, and the flasks were allowed to shake for 1 h on a rotary shaker at 200 rpm. The mixture was centrifuged at 6000 rpm for 10 min in order to separate organic layer and aqueous layer. The organic layer was concentrated in vacuo to afford 3.2 g of extract from 2.5 L of production culture. The extract was fractionated by silica gel column chromatography with a step gradient of CHCl3/MeOH (1:0, 20:1, 10:1, 4:1, 2:1, 1:1, and 0:1 v/v). Fraction 3 (10:1) was concentrated to give a brown oil (646 mg), which was then fractionated by reversed-phase ODS column chromatography with a gradient of MeCN/0.1% HCO2H (2:8, 3:7, 4:6, 5:5, 6:4, 7:3, and 8:2 v/v). Fraction 6 (7:3) was concentrated and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to give a semi-pure material (21 mg). Final purification was achieved by preparative HPLC (Cosmosil Cholester 5 µm, 10 × 250 mm, 4 mL/min, UV detection at 254 nm) with an isocratic elution of MeCN/0.1% HCO2H solution (50:50) to yield (2Z,4E)-3-methyl-2,4-decadienoic acid (1, 8.0 mg, tR 20.5 min).
(2Z,4E)-3-Methyl-2,4-decadienoic acid (1): colorless amorphous solid; UV (MeOH) λmax (log ε) 262 (4.16) nm; IR (ATR) νmax 2952, 2923, 2852, 2585, 1678, 1662 cm−1; 1H and 13C NMR data, see Table 1; HR-ESI-TOFMS m/z 181.1230 [M − H]− (calcd for C11H17O2, 181.1229).
Feeding experiment
Feeding experiments were carried out using 13C-labeled precursors, sodium [1-13C]acetate and ʟ-[methyl-13C]methionine. The fermentation, extraction and purification of labeled compounds were performed in the same manner as describe for the unlabeled compound. After 24 h of the inoculation into the production medium from the seed culture, supplementation of 13C-labeled precursors was commenced and carried out for four times at 24 h interval. After further 24 h incubation, the whole culture broth was extracted with 1-butanol.
1) Sodium [1-13C]acetate: 13C-Labeled 1 (1.6 mg) was obtained from 1 L of culture supplemented with sodium [1-13C]acetate (total 800 mg; 20 mg × 10 flasks × 4 days). The 13C NMR spectrum showed enriched signals at δ 170.2, 153.8, 140.3, 28.7, and 22.4 ppm.
2) ʟ-[Methyl-13C]methionine: 13C-Labeled 1 (0.9 mg) was obtained from 1 L of culture supplemented with ʟ-[methyl-13C]methionine (total 80 mg; 2.0 mg × 10 flasks × 4 days). The 13C NMR spectrum showed an enriched signal at δ 21.3 ppm
Antimicrobial assay
Antimicrobial activity was evaluated by the liquid microculture method using round-bottomed 96-well microtiter plates against six bacteria, Bacillus subtilis ATCC6633, Micrococcus luteus ATCC9341, Staphylococcus aureus FDA209P JC-1, Ralstonia solanacearum SUPP1541, Rhizobium radiobacter NBRC14554, Escherichia coli NIHJ JC-2, and two yeasts Candida albicans NBRC0197 and Saccharomyces cerevisiae S100 as indication strains. Tryptic soy broth (Difco) and Sabouraud dextrose broth (Difco) were used for bacteria and yeasts, respectively. Compound 1 and reference drugs, kanamycin sulfate for bacteria and amphotericin B for yeasts, were made in 2-fold dilution series along the longer side of the plates by sequential transfer of 100 μL aliquots between the adjacent wells, to which the same amount of medium was pre-dispensed. To each well was added a 100 μL suspension of the indication strains prepared at 0.5 McFarland (≈108 cfu/mL) from a culture at the logarithmic growth phase. The solvent vehicle added to the top rows was set at the 0.5% of the final culture volume to avoid the effect on the growth of microbes. The plates were incubated for 48 h at 37 °C for bacteria and at 32 °C for yeasts. The tests were done in triplicate and the MIC values were read from the lowest drug concentrations at which no growth was observed.
Supporting Information
| Supporting Information File 1: 1D and 2D NMR spectra of 1; 13C NMR spectra of 13C-labeled 1. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Blunt, J. W.; Copp, B. R.; Keyzers, R. A.; Munro, M. H. G.; Prinsep, M. R. Nat. Prod. Rep. 2016, 33, 382–431. doi:10.1039/c5np00156k
Return to citation in text: [1] -
Brinkmann, C. M.; Marker, A.; Kurtböke, D. İ. Diversity 2017, 9, No. 40. doi:10.3390/d9040040
Return to citation in text: [1] -
Ritchie, K. Mar. Ecol.: Prog. Ser. 2006, 322, 1–14. doi:10.3354/meps322001
Return to citation in text: [1] -
Chen, Y.-H.; Lu, M.-C.; Chang, Y.-C.; Hwang, T.-L.; Wang, W.-H.; Weng, C.-F.; Kuo, J.; Sung, P.-J. Tetrahedron Lett. 2012, 53, 1675–1677. doi:10.1016/j.tetlet.2012.01.104
Return to citation in text: [1] -
Chen, Y.-H.; Kuo, J.; Su, J.-H.; Hwang, T.-L.; Chen, Y.-H.; Lee, C.-H.; Weng, C.-F.; Sung, P.-J. Mar. Drugs 2012, 10, 1566–1571. doi:10.3390/md10071566
Return to citation in text: [1] -
Gao, C.-H.; Tian, X.-P.; Qi, S.-H.; Luo, X.-M.; Wang, P.; Zhang, S. J. Antibiot. 2010, 63, 191–193. doi:10.1038/ja.2010.7
Return to citation in text: [1] -
Sharma, A. R.; Zhou, T.; Harunari, E.; Oku, N.; Trianto, A.; Igarashi, Y. J. Antibiot. 2019, 72, 634–639. doi:10.1038/s41429-019-0192-x
Return to citation in text: [1] -
Gonzalez, J. M.; Mayer, F.; Moran, M. A.; Hodson, R. E.; Whitman, W. B. Int. J. Syst. Bacteriol. 1997, 47, 369–376. doi:10.1099/00207713-47-2-369
Return to citation in text: [1] [2] -
Yoon, J.-H.; Jung, S.-Y.; Kang, S.-J.; Oh, T.-K. Int. J. Syst. Evol. Microbiol. 2007, 57, 2365–2369. doi:10.1099/ijs.0.65184-0
Return to citation in text: [1] -
Zhang, D.-S.; Huo, Y.-Y.; Xu, X.-W.; Wu, Y.-H.; Wang, C.-S.; Xu, X.-F.; Wu, M. Int. J. Syst. Evol. Microbiol. 2012, 62, 505–510. doi:10.1099/ijs.0.027714-0
Return to citation in text: [1] -
Lee, J.-Y.; Kim, P. S.; Hyun, D.-W.; Kim, H. S.; Shin, N.-R.; Jung, M.-J.; Yun, J.-H.; Kim, M.-S.; Whon, T. W.; Bae, J.-W. Int. J. Syst. Evol. Microbiol. 2017, 67, 998–1004. doi:10.1099/ijsem.0.001731
Return to citation in text: [1] -
Rousset, S.; Thibonnet, J.; Abarbri, M.; Duchêne, A.; Parrain, J.-L. Synlett 2000, 260–262. doi:10.1055/s-2000-6485
Return to citation in text: [1] [2] -
Dewik, P. M. Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; Wiley and Sons: Inc.: Chichester, United Kingdom, 2009.
Return to citation in text: [1] [2] -
Harunari, E.; Komaki, H.; Igarashi, Y. J. Antibiot. 2016, 69, 403–405. doi:10.1038/ja.2015.118
Return to citation in text: [1] -
Anttila, M.; Strangman, W.; York, R.; Tomas, C.; Wright, J. L. C. J. Nat. Prod. 2016, 79, 484–489. doi:10.1021/acs.jnatprod.5b00869
Return to citation in text: [1] -
Ternes, P.; Sperling, P.; Albrecht, S.; Franke, S.; Cregg, J. M.; Warnecke, D.; Heinz, E. J. Biol. Chem. 2006, 281, 5582–5592. doi:10.1074/jbc.m512864200
Return to citation in text: [1] [2] -
Meena, L. S.; Chopra, P.; Vishwakarma, R. A.; Singh, Y. Biol. Chem. 2013, 394, 871–877. doi:10.1515/hsz-2013-0126
Return to citation in text: [1] -
Buist, P. H. Nat. Prod. Rep. 2007, 24, 1110–1127. doi:10.1039/b508584p
Return to citation in text: [1] [2] -
Poger, D.; Caron, B.; Mark, A. E. J. Phys. Chem. B 2014, 118, 13838–13848. doi:10.1021/jp503910r
Return to citation in text: [1] [2] -
Campbell, I. M.; Naworal, J. J. Lipid Res. 1969, 10, 593–598.
Return to citation in text: [1] -
Kukovinets, O. S.; Lozhkina, E. A.; Odinokov, V. N.; Tolstikov, G. A. Bashk. Khim. Zh. 1995, 2, 35–36.
Return to citation in text: [1] -
Jayanetti, D. R.; Braun, D. R.; Barns, K. J.; Rajski, S. R.; Bugni, T. S. J. Nat. Prod. 2019, 82, 1930–1934. doi:10.1021/acs.jnatprod.9b00312
Return to citation in text: [1]
| 1. | Blunt, J. W.; Copp, B. R.; Keyzers, R. A.; Munro, M. H. G.; Prinsep, M. R. Nat. Prod. Rep. 2016, 33, 382–431. doi:10.1039/c5np00156k |
| 6. | Gao, C.-H.; Tian, X.-P.; Qi, S.-H.; Luo, X.-M.; Wang, P.; Zhang, S. J. Antibiot. 2010, 63, 191–193. doi:10.1038/ja.2010.7 |
| 15. | Anttila, M.; Strangman, W.; York, R.; Tomas, C.; Wright, J. L. C. J. Nat. Prod. 2016, 79, 484–489. doi:10.1021/acs.jnatprod.5b00869 |
| 4. | Chen, Y.-H.; Lu, M.-C.; Chang, Y.-C.; Hwang, T.-L.; Wang, W.-H.; Weng, C.-F.; Kuo, J.; Sung, P.-J. Tetrahedron Lett. 2012, 53, 1675–1677. doi:10.1016/j.tetlet.2012.01.104 |
| 5. | Chen, Y.-H.; Kuo, J.; Su, J.-H.; Hwang, T.-L.; Chen, Y.-H.; Lee, C.-H.; Weng, C.-F.; Sung, P.-J. Mar. Drugs 2012, 10, 1566–1571. doi:10.3390/md10071566 |
| 16. | Ternes, P.; Sperling, P.; Albrecht, S.; Franke, S.; Cregg, J. M.; Warnecke, D.; Heinz, E. J. Biol. Chem. 2006, 281, 5582–5592. doi:10.1074/jbc.m512864200 |
| 13. | Dewik, P. M. Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; Wiley and Sons: Inc.: Chichester, United Kingdom, 2009. |
| 14. | Harunari, E.; Komaki, H.; Igarashi, Y. J. Antibiot. 2016, 69, 403–405. doi:10.1038/ja.2015.118 |
| 2. | Brinkmann, C. M.; Marker, A.; Kurtböke, D. İ. Diversity 2017, 9, No. 40. doi:10.3390/d9040040 |
| 13. | Dewik, P. M. Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; Wiley and Sons: Inc.: Chichester, United Kingdom, 2009. |
| 10. | Zhang, D.-S.; Huo, Y.-Y.; Xu, X.-W.; Wu, Y.-H.; Wang, C.-S.; Xu, X.-F.; Wu, M. Int. J. Syst. Evol. Microbiol. 2012, 62, 505–510. doi:10.1099/ijs.0.027714-0 |
| 8. | Gonzalez, J. M.; Mayer, F.; Moran, M. A.; Hodson, R. E.; Whitman, W. B. Int. J. Syst. Bacteriol. 1997, 47, 369–376. doi:10.1099/00207713-47-2-369 |
| 9. | Yoon, J.-H.; Jung, S.-Y.; Kang, S.-J.; Oh, T.-K. Int. J. Syst. Evol. Microbiol. 2007, 57, 2365–2369. doi:10.1099/ijs.0.65184-0 |
| 12. | Rousset, S.; Thibonnet, J.; Abarbri, M.; Duchêne, A.; Parrain, J.-L. Synlett 2000, 260–262. doi:10.1055/s-2000-6485 |
| 8. | Gonzalez, J. M.; Mayer, F.; Moran, M. A.; Hodson, R. E.; Whitman, W. B. Int. J. Syst. Bacteriol. 1997, 47, 369–376. doi:10.1099/00207713-47-2-369 |
| 7. | Sharma, A. R.; Zhou, T.; Harunari, E.; Oku, N.; Trianto, A.; Igarashi, Y. J. Antibiot. 2019, 72, 634–639. doi:10.1038/s41429-019-0192-x |
| 11. | Lee, J.-Y.; Kim, P. S.; Hyun, D.-W.; Kim, H. S.; Shin, N.-R.; Jung, M.-J.; Yun, J.-H.; Kim, M.-S.; Whon, T. W.; Bae, J.-W. Int. J. Syst. Evol. Microbiol. 2017, 67, 998–1004. doi:10.1099/ijsem.0.001731 |
| 17. | Meena, L. S.; Chopra, P.; Vishwakarma, R. A.; Singh, Y. Biol. Chem. 2013, 394, 871–877. doi:10.1515/hsz-2013-0126 |
| 12. | Rousset, S.; Thibonnet, J.; Abarbri, M.; Duchêne, A.; Parrain, J.-L. Synlett 2000, 260–262. doi:10.1055/s-2000-6485 |
| 21. | Kukovinets, O. S.; Lozhkina, E. A.; Odinokov, V. N.; Tolstikov, G. A. Bashk. Khim. Zh. 1995, 2, 35–36. |
| 22. | Jayanetti, D. R.; Braun, D. R.; Barns, K. J.; Rajski, S. R.; Bugni, T. S. J. Nat. Prod. 2019, 82, 1930–1934. doi:10.1021/acs.jnatprod.9b00312 |
| 16. | Ternes, P.; Sperling, P.; Albrecht, S.; Franke, S.; Cregg, J. M.; Warnecke, D.; Heinz, E. J. Biol. Chem. 2006, 281, 5582–5592. doi:10.1074/jbc.m512864200 |
| 19. | Poger, D.; Caron, B.; Mark, A. E. J. Phys. Chem. B 2014, 118, 13838–13848. doi:10.1021/jp503910r |
| 19. | Poger, D.; Caron, B.; Mark, A. E. J. Phys. Chem. B 2014, 118, 13838–13848. doi:10.1021/jp503910r |
© 2019 Sharma et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)