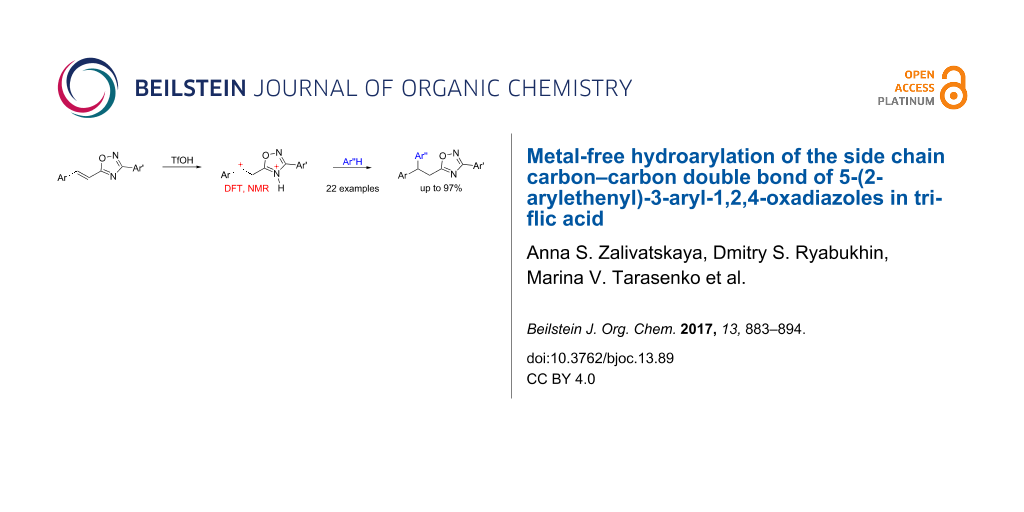
Abstract
The metal-free reaction of 5-(2-arylethenyl)-3-aryl-1,2,4-oxadiazoles with arenes in neat triflic acid (TfOH, CF3SO3H), both under thermal and microwave conditions, leads to 5-(2,2-diarylethyl)-3-aryl-1,2,4-oxadiazoles. The products are formed through the regioselective hydroarylation of the side chain carbon–carbon double bond of the starting oxadiazoles in yields up to 97%. According to NMR data and DFT calculations, N4,C-diprotonated forms of oxadiazoles are the electrophilic intermediates in this reaction.

Graphical Abstract
Introduction
Oxadiazoles are an important class of heterocyclic compounds and great attention has been paid to their synthesis and to the studies of their chemical, physical and biological properties (see numerous reviews [1-8]). The oxadiazole ring represents an essential part of the pharmacophore in many drugs. These compounds possess different kinds of biological activities, such as analgesic [9], anti-inflammatory [10], antimicrobial [11], antidiabetic [12], and anticancer [13] to name a few. Some representatives of 1,2,4-oxadiazole-based drugs are shown in Figure 1. Libexin and oxolamine are used as antitussive (cough) agents [14], butalamine is a coronary vasodilator and local anesthetic [15], and ataluren finds application for the treatment of fibrosis [16]. Often, oxadiazole derivatives act as inhibitors of bacterial phenylalanyl-tRNA-synthetase [17], phosphodiesterase 4B2 [18], y-secretase [19] and phenol-substituted 1,2,4-oxadiazoles exhibit powerful anti-oxidant properties [20]. Moreover, they have antihypertensive [21] and antituberculosis [22] activities. In medicinal chemistry the oxadiazole ring is considered as bioisosteric replacements for ester or amide groups [23]. Thus, the further development of syntheses of 1,2,4-oxadiazoles and the investigation of their properties are of ongoing interest in chemistry and medicine.
![[1860-5397-13-89-1]](/bjoc/content/figures/1860-5397-13-89-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on our previous works on reactions of cinnamides [24] and 5-styryl-2H-tetrazoles [25] with arenes under superelectrophilic activation with Brønsted or Lewis superacids, we turned our attention towards the hydroarylation of the C=C double bond in 5-styryl-substituted oxadiazoles, such as (E)-5-(2-arylethenyl)-3-aryl-1,2,4-oxadiazoles 1 (Scheme 1). The main goals of the current work were to investigate the reactions of oxadiazoles 1 with different arenes under the conditions of superelectrophilic activation and to study protonated forms of the oxadiazoles as reactive intermediates by means of NMR and DFT calculations.
![[1860-5397-13-89-i1]](/bjoc/content/inline/1860-5397-13-89-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The hydroarylation of 5-(2-arylethenyl)-3-aryl-1,2,4-oxadiazoles 1 under superelectrophilic activation leading to compounds 2.
Scheme 1: The hydroarylation of 5-(2-arylethenyl)-3-aryl-1,2,4-oxadiazoles 1 under superelectrophilic activat...
It should be noted, that the metal-catalyzed hydroarylation of C=C bonds is widely used in organic synthesis [26,27]. The most efficient catalysts for these purposes are complexes of the transition metals Pt [28], Au [29], Ru [30], Rh [31-33], Ni [34], Pd [35], and Pd/Ag [36]. However, a metal-free hydroarylation variant of C=C bonds under the action of Brønsted or Lewis superacids has been developed [37,38] and we were able to extend the scope of this reaction [24,25].
The expected reaction products, oxadiazoles 2 (Scheme 1) are structurally close to many biologically active compounds and drugs [39-49], which contain a chain of three carbon atoms, two aryl rings on one end of this chain, and further functional groups on the other end of it (Figure 2, compare also with libexin in Figure 1). One may suppose that compounds of type 2 having the same structural fragments, i.e., three carbon atoms (one of them part of the oxadiazole moiety), two aryl groups, and the four remaining atoms of the oxadiazole ring, as a functional group, may also show biological activity. Thus, the synthesis of these particular 5-(2,2-diarylethyl)-substituted oxadiazoles 2 may be interesting for medicinal chemistry.
![[1860-5397-13-89-2]](/bjoc/content/figures/1860-5397-13-89-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: General structure for various biologically active compounds containing three carbon atoms, two aryl rings and functional groups.
Figure 2: General structure for various biologically active compounds containing three carbon atoms, two aryl...
Results and Discussion
According to literature data [50,51] the protonation of a 1,2,4-oxadiazole ring takes place mainly at the N4 nitrogen. However, also the N2 nitrogen may be protonated depending on the substituents attached to the heterocyclic system [50]. To investigate this issue in more detail we undertook a theoretical study on the protonation of 5-(2-phenylethenyl)-3-phenyl-1,2,4-oxadiazole (1a) by quantum-chemical calculations. Table 1 contains data obtained by DFT calculations for the different possible mono-, di- and tricationic species A–F derived from the protonation of 1a. Charge distributions, contributions of the atomic orbital into LUMO, global electrophilicity indices ω [52,53], and the Gibbs free energies ΔG298 of the protonation reactions with the hydroxonium ion (H3O+) were calculated.
Table 1: Selected electronic characteristics for cations A–F calculated by DFT from protonation of oxadiazole 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-89-i2.svg?max-width=637&scale=1.0)
|
|||||||
| Species | EHOMO, eV | ELUMO, eV | ωa, eV | q (Cβ)b, e | k (Cβ)LUMOc, % | q(N2)b, e | q(N4)b, e |
| A | −7.10 | −3.49 | 3.87 | −0.04 | 17.2 | −0.19 | −0.52 |
| B | −7.95 | −4.98 | 7.02 | 0.20 | 27.0 | −0.17 | −0.49 |
| C | −7.18 | −3.43 | 3.75 | −0.02 | 26.2 | −0.15 | 0.52 |
| D | −7.83 | −5.06 | 7.48 | 0.18 | 37.3 | −0.12 | −0.49 |
| E | −7.72 | −4.32 | 5.32 | 0.03 | 21.4 | −0.16 | −0.5 |
| F | −8.46 | −5.24 | 7.30 | 0.16 | 34.7 | −0.15 | −0.48 |
aGlobal electrophilicity index ω = (EHOMO + ELUMO)2/8 (ELUMO − EHOMO). bNatural charges. cContribution of atomic orbital into the molecular orbital.
Negative values for ΔG298 of the first protonation step show that this reaction is energetically favorable at both N2 (formation of species A) and N4 (formation of species C). A further protonation may occur at the second nitrogen atom of the oxadiazole system (formation of dication E) or at the carbon Cα of the side chain C=C double bond (formation of dications B or D). Finally, the third protonation giving rise to trication F is rather high in energy (ΔG298 121 kJ/mol for E→F) and therefore highly unlikely. Also the outlined route leading to species B is energetically unfavorable (ΔG298 = −40.3 + 66.7 = 26.4 kJ/mol) and the formation of this dication is not expected. In contrast, the generation of dication D should be possible (ΔG298 = −59.2 + 52.4 = −6.8 kJ/mol). Despite more negative values of ΔG298 calculated for the formation of the N2,N4-diprotonated form E from both A (ΔG298 = −40.3 + 9.7 = −30.6 kJ/mol for 1a→A→E) and C (ΔG298 = −59.2 + 28.7 = −30.5 kJ/mol for 1a→C→E), it was found by calculations, that species E had an imaginary frequency revealing that it may be a transition state rather than an intermediate species.
Thus, from a thermodynamic point of view, the first protonation of 5-styryl-substituted 1,2,4-oxadiazoles takes place at the N4 nitrogen atom leading to cation C (ΔG298 of this reaction has the lowest value of −59.2 kJ/mol) and the most probable dicationic species, obtained through protonation of species C, should be N4,C-diprotonated form D.
The calculated electronic characteristics of species A–F revealed that the dication D has the highest electrophilicity index ω (7.48 eV) among the other cationic species, even including trication F (Table 1). Therefore, dication D is expected to be an extremely reactive electrophile. Moreover, it has a large portion of the positive charge (0.18 e) located at the Cβ carbon atom and a high contribution to the LUMO (37.3%), thus making this carbon atom a reactive electrophilic center through charge and orbital control. One may not exclude that N2- and N4-monoprotonated forms A and C, respectively, also may behave as electrophiles. However, the calculated negative charges on the Cβ carbon atoms may prevent a chemical transformation on them.
Summarizing the calculation results, one may conclude that the most probable reactive electrophilic species, derived through the protonation of 5-styryl-substituted 1,2,4-oxadiazoles, is the N4,C-diprotonated species D from both the thermodynamic and electronic point of view.
Next, we carried out an NMR study of the protonation of oxadiazoles 1a and 1m in the superacid FSO3H at low temperature (−80 °C). The spectra are shown in Supporting Information File 1. Upon dissolving compounds 1a and 1m in fluorosulfonic acid, FSO3H in an NMR tube at −80 °C, the formation of N4-protonated species Ca and Cm, respectively, was detected (Figure 3). The proton bounded to nitrogen N4 resonates at δ ≈ 12.5–13 ppm at this temperature whereas at higher temperature (above −40 °C) this signal disappeared due to fast proton exchange with the superacidic medium (see Supporting Information File 1). The addition of the proton at the N4 position, rather than at N2, was proved by the NOESY correlation between this proton and the vinyl proton Hα (Figure 3). In the case of a protonation at N2, there should not be any correlation between the proton attached to N2 and Hα. The assignment of all signals in the 1H and 13C NMR spectra of cations Ca and Cm was undertaken on the basis of 1H–13C HSQC and 1H–15N HSQC spectra (see spectra in Supporting Information File 1). According to the NMR study and preparative experiments, the increasing reaction temperature in trifluoromethanesulfonic acid (TfOH, to 60 °C for 1a, and to room temperature for 1m) led to the formation of oligomeric material. Most likely, at higher temperature the second protonation at the C=C bond takes place giving rise to the corresponding unstable highly reactive dications D, which were not detected by NMR.
![[1860-5397-13-89-3]](/bjoc/content/figures/1860-5397-13-89-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected 1H, 13C, 15N NMR data for cations Ca and Cm generated by protonation of oxadiazoles 1a and 1m at the N4 nitrogen (FSO3H, −80 °C for Ca, and −60 °C for Cm, with CH2Cl2 as internal standard).
Figure 3: Selected 1H, 13C, 15N NMR data for cations Ca and Cm generated by protonation of oxadiazoles 1a and ...
It should be mentioned, that there have been reports [37,54-57] on NMR observations of O,C-diprotonated forms (dications) of conjugated enones in superacidic medium, which are structurally close to species D.
Thus, the NMR data reveal that the protonation of 5-styryl-substituted 1,2,4-oxadiazoles in superacids results in the formation of their relatively stable N4-protonated forms. However, these species do not react with aromatic π-nucleophiles (vide infra). Most probably, those reactive intermediates, generated under the protonation of substrates 1, are N4,C-diprotonated species D.
The experimental results from the hydroarylation reactions of the side chain C=C double bond of oxadiazoles 1a–n with various arenes under the action of different acidic reagents leading to oxadiazoles 2a–za are shown in Table 2 (see also X-ray structures of 2a and 2m in Figure 4). First, it should be emphasized that no reaction is observed under the conditions of generation of monoprotonated species C (see Table 1, and Figure 3). Thus, in FSO3H at low temperature (−80 to −60 °C) compounds 1e (Table 2, entries 14 and 15) and 1m (Table 2, entry 29) do not react with benzene. At higher temperature, fluorosulfonation of the aromatic ring is observed, especially with donating arenes (see example of this reaction in our work [24]). On the other hand, compounds 1e and 1m readily react with benzene in TfOH at room temperature (see Table 2, entries 16 and 28). Presumably, at higher temperatures the second protonation takes place at the C=C double bond giving rise to reactive dications D. The acidity of H2SO4, which is lower than that of FSO3H and TfOH, is not sufficient to promote this reaction. Thus, the protonation of the C=C bond of 1a in H2SO4 does not take place even at elevated temperature (75 °C, see Table 2, entry 1). Also, Lewis acids such as AlCl3 and AlBr3 are not effective in this transformation (Table 2, entries 2 and 3). The best results were obtained in neat TfOH.
Table 2: Hydroarylation of oxadiazoles 1a–n with arenes under superelectrophilic activation leading to compounds 2a–za.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-89-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Starting materials | Reaction conditions | Reaction products 2, yield (%)a | |
| Oxadiazole 1 | Arene, Ar”H | |||
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-89-i4.svg?max-width=637&scale=1.0)
1a |
benzene | H2SO4, 75 °C, 24 h | 1ab |
| 2 | 1a | benzene | AlCl3, CH2Cl2, rt, 24 h | 1ab |
| 3 | 1a | benzene | AlBr3, 60 °C, 3 h |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-89-i5.svg?max-width=637&scale=1.0)
2a (60%) |
| 4 | 1a | benzene | TfOH, 60 °C, 2 h | 2a (77%) |
| 5 | 1a | benzene | TfOH, rt, 18 h | 2a (80%) |
| 6 | 1a | chlorobenzene | TfOH, rt, 18 h |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-89-i6.svg?max-width=637&scale=1.0)
2b (84%) ![[Graphic 6]](/bjoc/content/inline/1860-5397-13-89-i7.svg?max-width=637&scale=1.0)
2c (12%) |
| 7 | 1a |
1,2-dichloro-
benzene |
TfOH, rt, 24 h |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-89-i8.svg?max-width=637&scale=1.0)
2d (60%) ![[Graphic 8]](/bjoc/content/inline/1860-5397-13-89-i9.svg?max-width=637&scale=1.0)
2e (5%) |
| 8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-89-i10.svg?max-width=637&scale=1.0)
1b |
benzene | TfOH, 60 °C, 2 h |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-89-i11.svg?max-width=637&scale=1.0)
2f (89%) |
| 9 | 1b | benzene | TfOH-SbF5 (20 mol %), rt, 0.5 h | 2f (64%) |
| 10 | 1b |
tert-butyl-
benzene |
TfOH, 60 °C, 2 h | 2f (96%) |
| 11 | 1b | anisole | TfOH, 60 °C, 2 h |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-89-i12.svg?max-width=637&scale=1.0)
2g (39%) |
| 12 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-89-i13.svg?max-width=637&scale=1.0)
1c |
benzene | TfOH, rt,12 h |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-89-i14.svg?max-width=637&scale=1.0)
2h (80%) |
| 13 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-89-i15.svg?max-width=637&scale=1.0)
1d |
benzene | TfOH, rt, 2 h |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-89-i16.svg?max-width=637&scale=1.0)
2i (60%) ![[Graphic 16]](/bjoc/content/inline/1860-5397-13-89-i17.svg?max-width=637&scale=1.0)
2j (31%) |
| 14 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-89-i18.svg?max-width=637&scale=1.0)
1e |
benzene | FSO3H, −80 °C, 2 h | 1eb |
| 15 | 1e | benzene | FSO3H, −60 °C, 3 h | 1eb |
| 16 | 1e | benzene | TfOH, rt, 1 h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-89-i19.svg?max-width=637&scale=1.0)
2k (90%) |
| 17 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-89-i20.svg?max-width=637&scale=1.0)
1f |
benzene | TfOH, 60 °C, 3 h |
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-89-i21.svg?max-width=637&scale=1.0)
2l (93%) |
| 18 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-13-89-i22.svg?max-width=637&scale=1.0)
1g |
benzene | TfOH, rt, 52 h |
![[Graphic 22]](/bjoc/content/inline/1860-5397-13-89-i23.svg?max-width=637&scale=1.0)
2m (91%) |
| 19 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-13-89-i24.svg?max-width=637&scale=1.0)
1h |
benzene | TfOH, rt, 24 h |
![[Graphic 24]](/bjoc/content/inline/1860-5397-13-89-i25.svg?max-width=637&scale=1.0)
2n (90%) |
| 20 | 1h | anisole | TfOH, rt, 24 h |
![[Graphic 25]](/bjoc/content/inline/1860-5397-13-89-i26.svg?max-width=637&scale=1.0)
2o (50%) ![[Graphic 26]](/bjoc/content/inline/1860-5397-13-89-i27.svg?max-width=637&scale=1.0)
2p (25%) |
| 21 | 1h | chlorobenzene | TfOH, rt, 24 h |
![[Graphic 27]](/bjoc/content/inline/1860-5397-13-89-i28.svg?max-width=637&scale=1.0)
2q (50%) ![[Graphic 28]](/bjoc/content/inline/1860-5397-13-89-i29.svg?max-width=637&scale=1.0)
2r (8%) |
| 22 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-13-89-i30.svg?max-width=637&scale=1.0)
1i |
benzene | TfOH, rt, 20 h |
![[Graphic 30]](/bjoc/content/inline/1860-5397-13-89-i31.svg?max-width=637&scale=1.0)
2s (94%) |
| 23 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-13-89-i32.svg?max-width=637&scale=1.0)
1j |
benzene | TfOH, rt, 24 h |
![[Graphic 32]](/bjoc/content/inline/1860-5397-13-89-i33.svg?max-width=637&scale=1.0)
2t (97%) |
| 24 | 1j | toluene | TfOH, rt, 24 h |
![[Graphic 33]](/bjoc/content/inline/1860-5397-13-89-i34.svg?max-width=637&scale=1.0)
2u (60%) ![[Graphic 34]](/bjoc/content/inline/1860-5397-13-89-i35.svg?max-width=637&scale=1.0)
2v (5%) |
| 25 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-13-89-i36.svg?max-width=637&scale=1.0)
1k |
benzene | TfOH, rt, 12 h |
![[Graphic 36]](/bjoc/content/inline/1860-5397-13-89-i37.svg?max-width=637&scale=1.0)
2w (95%) |
| 26 | 1k | toluene | TfOH, rt, 12 h |
![[Graphic 37]](/bjoc/content/inline/1860-5397-13-89-i38.svg?max-width=637&scale=1.0)
2x (67%) |
| 27 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-13-89-i39.svg?max-width=637&scale=1.0)
1l |
benzene | TfOH, rt, 2 h |
![[Graphic 39]](/bjoc/content/inline/1860-5397-13-89-i40.svg?max-width=637&scale=1.0)
2y (92%) |
| 28 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-13-89-i41.svg?max-width=637&scale=1.0)
1m |
benzene | TfOH, rt, 2 h |
![[Graphic 41]](/bjoc/content/inline/1860-5397-13-89-i42.svg?max-width=637&scale=1.0)
2z (50%) |
| 29 | 1m | benzene | FSO3H, −80 °C, 2 h | 1mb |
| 30 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-13-89-i43.svg?max-width=637&scale=1.0)
1n |
benzene | TfOH, rt, 1 h |
![[Graphic 43]](/bjoc/content/inline/1860-5397-13-89-i44.svg?max-width=637&scale=1.0)
2za (53%) |
aIsolated yields. bQuantitative recovery of unreacted starting oxadiazole.
![[1860-5397-13-89-4]](/bjoc/content/figures/1860-5397-13-89-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structures of compounds 2a (left) (CCDC 1526767) and 2m (right) (CCDC 1526105); ellipsoid contours of probability levels are 50%.
Figure 4: X-ray crystal structures of compounds 2a (left) (CCDC 1526767) and 2m (right) (CCDC 1526105); ellip...
The substituents present in the aromatic ring of the styryl group of oxadiazoles 1 play a crucial role for the protonation and reactivity of these compounds. Thus, styryl-substituted oxadiazoles 1b,c and substrates 1f–k, bearing electron-accepting halogen substituents, need higher reaction temperatures up to 60 °C (Table 2, entries 4, 8, 10, 11, and 17) or longer reaction times of 24–52 h (Table 2, entries 18–24) at rt in TfOH. On the other hand, electron-donating groups attached to the styryl moiety of oxadiazoles 1d,e,l–n facilitate the protonation of the C=C double bond, resulting in a reduced reaction time to 1–2 h at room temperature (Table 2, entries 13, 16, 27, 28, and 30). Increasing the acidity of the reaction medium promotes the protonation of deactivated oxadiazoles. Thus, compound 1b in the system TfOH-SbF5 (20 mol %) reacted with benzene within 0.5 h at room temperature (Table 2, entry 9), but in less acidic neat TfOH the reaction took 2 h at 60 °C (Table 2, entry 8).
Different arenes may be involved in this reaction. The corresponding hydroarylation products were obtained by reaction with benzene, chlorobenzene, 1,2-dichlorobenzene, toluene, and anisole. Electron-rich polymethylated aromatics, such as isomeric xylenes, mesitylene, pseudocumene, or durene gave mixtures of oligomeric products. Probably, these products are formed through multiple electrophilic substitution reactions of these arenes by the reactive dication species D. When oxadiazole 1b reacted with tert-butylbenzene (Table 2, entry 10), product 2f lacking the tert-butyl group was isolated. In this case, an ipso-substitution of the tert-butyl group by a proton under the superacidic conditions took place. Reactions with some arenes gave regioisomeric products, for instance, anisole (2o + 2p, Table 2, entry 20), 1,2-dichlorobenzene (2d + 2e, Table 2, entry 7), chlorobenzene (2b + 2c, Table 2, entry 6 and 2q + 2r, entry 21), and toluene (2u + 2v, Table 2, entry 24). The exact structures of these regioisomers were determined on the basis of multiplet signals of the aromatic protons in the 1H NMR spectra. The observation of regioisomeric products points out the high reactivity of the intermediate dicationic species D. The formation hydroxy-substituted oxadiazoles 2g (Table 2, entry 11), 2o and 2p (Table 2, entry 20), 2w (Table 2, entry 25), and 2x (Table 2, entry 26) may be explained by demethylation of the corresponding methoxy group under action of TfOH at elevated temperature (60 °C) or for prolonged reaction times (12 or 24 h) at room temperature. See reviews [58,59] on the dealkylation of ethers by various Brønsted and Lewis acids.
Additionally, the reactions were carried out under microwave (MW) irradiation (Table 3) analogously to our recent study on the hydroarylation of styryl tetrazoles [25]. Indeed, under MW activation the reactions in TfOH proceeded within 5–20 min at 120 °C (Table 3, entries 4–9) with formation of oxadiazoles 2 in high yields (compare the yields under thermal and MW heating in Table 3). The MW-activated process without any acid (Table 3, entry 1) or in a weaker acid (H2SO4, Table 3, entry 2) did not proceed at all. Apart from that, the conversion of 1a in the presence of AlCl3 was rather low (Table 3, entry 3). It should be emphasized that the oxadiazole ring is stable under the superacidic conditions and no destruction was noticed.
Table 3: Hydroarylation of oxadiazoles 1 with arenes under microwave (MW) activation in TFOH at 120 °C.
![[Graphic 44]](/bjoc/content/inline/1860-5397-13-89-i45.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Starting materials | Reaction conditions | Reaction products, yield (%), MW irradiationa | Conditions, yield (%), conventional heating | |
| Oxadiazole 1 | Arene, Ar”H | ||||
| 1 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-13-89-i46.svg?max-width=637&scale=1.0)
1a |
benzene | 15 min (without any acid) | 1ab | |
| 2 | 1a | benzene |
H2SO4,
15 min |
1ab | H2SO4, 75 °C, 24 h: 1ab,c |
| 3 | 1a | benzene | AlCl3, CH2Cl2, 30 min |
1a (80%)
![[Graphic 46]](/bjoc/content/inline/1860-5397-13-89-i47.svg?max-width=637&scale=1.0)
2a (8%) |
AlCl3, CH2Cl2, rt, 24 h: 1ab,c |
| 4 | 1a | benzene |
TfOH,
10 min |
2a (92%) | TfOH, 60 °C, 2 h: 2a (77%)c |
| 5 | 1a |
chloro-
benzene |
TfOH,
20 min |
![[Graphic 47]](/bjoc/content/inline/1860-5397-13-89-i48.svg?max-width=637&scale=1.0)
2b (70%) ![[Graphic 48]](/bjoc/content/inline/1860-5397-13-89-i49.svg?max-width=637&scale=1.0)
2c (24%) |
TfOH, rt, 18 h: 2b (84%) + 2c (12%)c |
| 6 | 1a |
1,2-dichloro-
benzene |
TfOH,
20 min |
![[Graphic 49]](/bjoc/content/inline/1860-5397-13-89-i50.svg?max-width=637&scale=1.0)
2d (50 %) ![[Graphic 50]](/bjoc/content/inline/1860-5397-13-89-i51.svg?max-width=637&scale=1.0)
2e (20%) |
TfOH, rt, 18 h: 2d (60%) + 2e (5%)c |
| 7 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-13-89-i52.svg?max-width=637&scale=1.0)
1g |
benzene |
TfOH,
10 min |
![[Graphic 52]](/bjoc/content/inline/1860-5397-13-89-i53.svg?max-width=637&scale=1.0)
2m (94%) |
TfOH, rt, 52 h: 2m (91%)c |
| 8 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-13-89-i54.svg?max-width=637&scale=1.0)
1j |
benzene |
TfOH,
5 min |
![[Graphic 54]](/bjoc/content/inline/1860-5397-13-89-i55.svg?max-width=637&scale=1.0)
2t (76%) |
TfOH, rt, 24 h: 2t (97%)c
TfOH, 120 °C, 5 min: 2t (95%)d |
aIsolated yields. bQuantitative recovery of unreacted starting oxadiazole. cData from Table 2. dReaction was carried out in glass high pressure tube.
Conclusion
We have developed an efficient method for the hydroarylation of the C=C double bond of 5-(2-arylethenyl)-3-aryl-1,2,4-oxadiazoles based on their TfOH-promoted reaction with arenes under thermal or microwave activation to form 5-(2,2-diarylethyl)-3-aryl-1,2,4-oxadiazoles in high yields. The reactive electrophilic intermediates of this hydroarylation process are N4,C-diprotonated forms of the starting oxadiazoles.
Supporting Information
| Supporting Information File 1: Experimental part, NMR spectra and DFT calculations. | ||
| Format: PDF | Size: 8.9 MB | Download |
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grants no. 15-03-02936-a). X-ray studies were performed at the Center for X-ray Diffraction Studies, and spectra of cations in FSO3H at low temperature were obtained at the Center for Magnetic Resonance of Saint Petersburg State University, Saint Petersburg, Russia.
References
-
Pace, A.; Buscemi, S.; Piccionello, A. P.; Pibiri, I. Adv. Heterocycl. Chem. 2015, 116, 85–136. doi:10.1016/bs.aihch.2015.05.001
Return to citation in text: [1] -
Bora, R. O.; Dar, B.; Pradhan, V.; Farooqui, M. Mini-Rev. Med. Chem. 2014, 14, 355–369. doi:10.2174/1389557514666140329200745
Return to citation in text: [1] -
Bokach, N. A.; Kuznetsov, M. L.; Kukushkin, V. Yu. Coord. Chem. Rev. 2011, 255, 2946–2967. doi:10.1016/j.ccr.2011.07.001
Return to citation in text: [1] -
Pace, A.; Pierro, P. Org. Biomol. Chem. 2009, 7, 4337–4348. doi:10.1039/B908937C
Return to citation in text: [1] -
Xiong, C. S.; Drewe, J.; Kasibhatla, S. Curr. Med. Chem. 2006, 13, 2627–2644. doi:10.2174/092986706778201521
Return to citation in text: [1] -
Hemming, K. J. Chem. Res., Synop. 2001, 209–216. doi:10.3184/030823401103169603
Return to citation in text: [1] -
Vivona, N.; Buscemi, S. Heterocycles 1995, 41, 2095–2116. doi:10.3987/REV-95-473
Return to citation in text: [1] -
Tabrizi, M. A.; Baraldi, P. G.; Borea, P. A.; Varani, K. Chem. Rev. 2016, 116, 519–560. doi:10.1021/acs.chemrev.5b00411
Return to citation in text: [1] -
Afiatpour, P.; Srivastava, R. M.; de Olivereira, M. L.; Barraeiro, E. J. Braz. J. Med. Biol. Res. 1994, 27, 1403–1406.
Return to citation in text: [1] -
Nicolaides, D. N.; Fylaktakidou, K. C.; Litinas, K. E.; Hadjipavlou-Litina, D. Eur. J. Med. Chem. 1998, 33, 715–724. doi:10.1016/S0223-5234(98)80030-5
Return to citation in text: [1] -
Ding, D.; Boudreau, M. A.; Leemans, E.; Spink, E.; Yamaguchi, T.; Testero, S. A.; O'Daniel, P. I.; Lastochkin, E.; Chang, M.; Mobashery, S. Bioorg. Med. Chem. Lett. 2015, 25, 4854–4857. doi:10.1016/j.bmcl.2015.06.044
Return to citation in text: [1] -
Jones, R. M.; Leonard, J. N.; Buzard, D. J.; Lehmann, J. Expert Opin. Ther. Pat. 2009, 19, 1339–1359. doi:10.1517/13543770903153878
Return to citation in text: [1] -
Kumar, D.; Patel, G.; Johnson, E. O.; Shah, K. Bioorg. Med. Chem. Lett. 2009, 19, 2739–2741. doi:10.1016/j.bmcl.2009.03.158
Return to citation in text: [1] -
Coupar, I. M.; Hedges, A.; Metcalfe, H. L.; Turner, P. J. Pharm. Pharmacol. 1969, 21, 474–475. doi:10.1111/j.2042-7158.1969.tb08294.x
Return to citation in text: [1] -
Palazzo, G.; Corsi, G. Arzneim. Forsch. 1962, 12, 545–549.
Return to citation in text: [1] -
Jones, A. M.; Helm, J. M. Drugs 2009, 69, 1903–1910. doi:10.2165/11318500-000000000-00000
Return to citation in text: [1] -
Montgomery, J.; Toogood, P. L.; Hutchings, K. M.; Liu, J.; Narasimham, L.; Braden, T.; Dermyer, M. R.; Kulynych, A. D.; Smith, Y. D.; Warmus, J. S.; Taylor, C. Bioorg. Med. Chem. Lett. 2009, 19, 665–669. doi:10.1016/j.bmcl.2008.12.054
Return to citation in text: [1] -
Kumar, D.; Patel, G.; Vijayakrishan, L.; Dastidar, S. G.; Ray, A. Chem. Biol. Drug Des. 2012, 79, 810–818. doi:10.1111/j.1747-0285.2011.01304.x
Return to citation in text: [1] -
Mahavri, G. M.; Fauq, A. H. Tetrahedron Lett. 2010, 51, 6542–6544. doi:10.1016/j.tetlet.2010.10.025
Return to citation in text: [1] -
Saigo, K.; Kono, M.; Tagaki, Y..; Takenoguchi, M.; Hiramatsu, Y.; Tada, H.; Hishita, T.; Misawa, M.; Imoto, S.; Imashuku, S. J. Clin. Med. Res. 2013, 5, 57–60. doi:10.4021/jocmr1180w
Return to citation in text: [1] -
Yang, X.; Liu, G.; Li, H.; Zhang, Y.; Song, D.; Li, C.; Wang, R.; Liu, B.; Liang, W.; Jing, Y.; Zhao, G. J. Med. Chem. 2010, 53, 1015–1022. doi:10.1021/jm9011565
Return to citation in text: [1] -
Macaev, F.; Ribkovskaia, Z.; Pogrebnoi, S.; Boldescu, V.; Rusu, G.; Shvets, N.; Dimoglo, A.; Geronikaki, A.; Reynolds, R. Bioorg. Med. Chem. 2011, 19, 6792–6807. doi:10.1016/j.bmc.2011.09.038
Return to citation in text: [1] -
Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A. T. J. Med. Chem. 2012, 55, 1817–1830. doi:10.1021/jm2013248
Return to citation in text: [1] -
Zakusilo, D. N.; Ryabukhin, D. S.; Boyarskaya, I. A.; Yuzikhin, O. S.; Vasilyev, A. V. Tetrahedron 2015, 71, 102–108. doi:10.1016/j.tet.2014.11.033
Return to citation in text: [1] [2] [3] -
Lisakova, A. D.; Ryabukhin, D. S.; Trifonov, R. E.; Ostrovskii, V. A.; Boyarskaya, I. A.; Vasilyev, A. V. Synthesis 2017, 49, 579–586. doi:10.1055/s-0036-1588884
Return to citation in text: [1] [2] [3] -
Andreatta, J. R.; McKeown, B. A.; Gunnoe, T. B. J. Organomet. Chem. 2011, 696, 305–315. doi:10.1016/j.jorganchem.2010.09.030
Return to citation in text: [1] -
Foley, N. A.; Lee, J. P.; Ke, Z.; Gunnoe, T. B.; Cundari, T. R. Acc. Chem. Res. 2009, 42, 585–597. doi:10.1021/ar800183j
Return to citation in text: [1] -
Bowring, M. A.; Bergman, R. G.; Tilley, T. D. Organometallics 2011, 30, 1295–1298. doi:10.1021/om2000458
Return to citation in text: [1] -
Tarselli, M. A.; Liu, A.; Gagné, M. R. Tetrahedron 2009, 65, 1785–1789. doi:10.1016/j.tet.2008.10.110
Return to citation in text: [1] -
Cheng, H.; Dong, W.; Dannenberg, C. A.; Dong, S.; Guo, Q.; Bolm, C. ACS Catal. 2015, 5, 2770–2773. doi:10.1021/acscatal.5b00258
Return to citation in text: [1] -
Sun, Z.-M.; Zhang, J.; Manan, R. S.; Zhao, P. J. Am. Chem. Soc. 2010, 132, 6935–6937. doi:10.1021/ja102575d
Return to citation in text: [1] -
Zhao, D.; Nimphius, C.; Lindale, M.; Glorius, F. Org. Lett. 2013, 15, 4504–4507. doi:10.1021/ol402053n
Return to citation in text: [1] -
Xue, F.; Wang, D.; Li, X.; Wan, B. Org. Biomol. Chem. 2013, 11, 7893–7898. doi:10.1039/C3OB41342J
Return to citation in text: [1] -
Bair, J. S.; Schramm, Y.; Sergeev, A. G.; Clot, E.; Eisenstein, O.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 13098–13101. doi:10.1021/ja505579f
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Persiani, D. Org. Lett. 2008, 10, 1597–1600. doi:10.1021/ol800266e
Return to citation in text: [1] -
Karshtedt, D.; Bell, A. T.; Tilley, T. D. Organometallics 2004, 23, 4169–4171. doi:10.1021/om0495325
Return to citation in text: [1] -
Koltunov, K. Yu.; Walspurger, S.; Sommer, J. Eur. J. Org. Chem. 2004, 4039–4047. doi:10.1002/ejoc.200400313
Return to citation in text: [1] [2] -
King, F. D.; Caddick, S. Org. Biomol. Chem. 2012, 10, 3244–3252. doi:10.1039/c2ob00012a
Return to citation in text: [1] -
Simons, F. E. R.; Frith, E. M.; Simons, K. J. J. Allergy Clin. Immunol. 1982, 70, 458–464. doi:10.1016/0091-6749(82)90009-4
Return to citation in text: [1] -
Piper, H. M.; Hutler, J. F.; Spieckerman, P. G. Arzneim. Forsch. 1985, 35, 1495–1501.
Return to citation in text: [1] -
Certa, H.; Blashke, G. Arch. Pharm. 1990, 323, 744–747.
Return to citation in text: [1] -
Chen, Z.; Miller, W. S.; Shan, S.; Valenzano, K. J. Bioorg. Med. Chem. Lett. 2003, 13, 3247–3252. doi:10.1016/S0960-894X(03)00665-6
Return to citation in text: [1] -
Goehring, R. R.; Whitehead, J. F. W.; Brown, K.; Islam, K.; Wen, X.; Zhou, X.; Chen, Z.; Valenzano, K. J.; Miller, W. S.; Shan, S.; Kyle, D. J. Bioorg. Med. Chem. Lett. 2004, 14, 5045–5050. doi:10.1016/j.bmcl.2004.08.001
Return to citation in text: [1] -
Burrows, J. N.; Cumming, J. G.; Fillery, S. M.; Hamlin, G. A.; Hudson, J. A.; Jackson, R. J.; McLaughlin, S.; Shaw, J. S. Bioorg. Med. Chem. Lett. 2005, 15, 25–28. doi:10.1016/j.bmcl.2004.10.044
Return to citation in text: [1] -
Marcusson, J. O.; Norinder, U.; Högberg, T.; Ross, S. B. Eur. J. Pharmacol. 1992, 215, 191–198. doi:10.1016/0014-2999(92)90028-3
Return to citation in text: [1] -
Vatter, H.; Seifert, V. Cardiovasc. Drug Rev. 2006, 24, 63–76. doi:10.1111/j.1527-3466.2006.00063.x
Return to citation in text: [1] -
Hou, X.; Ge, Z.; Wang, T.; Guo, W.; Cui, J.; Cheng, T.; Lai, C.; Li, R. Bioorg. Med. Chem. Lett. 2006, 16, 4214–4219. doi:10.1016/j.bmcl.2006.05.085
Return to citation in text: [1] -
Wang, J.; Song, Y.; Zhang, J.; Li, X.; Ling, X.; Li, R.; Cui, J. Chromatographia 2010, 72, 459–464. doi:10.1365/s10337-010-1697-4
Return to citation in text: [1] -
Hou, X.; Ge, Z.; Wang, T.; Guo, W.; Wu, J.; Cui, J.; Lai, C.; Li, R. Arch. Pharm. 2011, 344, 320–332. doi:10.1002/ardp.201000259
Return to citation in text: [1] -
Trifonov, R. E.; Volovodenko, A. P.; Vergizov, S. N.; Shirinbekov, N. I.; Gindin, V. A.; Koren, A. O.; Ostrovskii, V. A. Helv. Chim. Acta 2005, 88, 1790–1797. doi:10.1002/hlca.200590140
Return to citation in text: [1] [2] -
Hartley, R. F.; Huang, Y.; Cassidy, M.; Razler, T. M.; Qian, F.; Hussain, M. A. J. Pharm. Sci. 2012, 101, 3124–3133. doi:10.1002/jps.23050
Return to citation in text: [1] -
Parr, R. G.; von Szentpály, L.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x
Return to citation in text: [1] -
Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p
Return to citation in text: [1] -
Koltunov, K. Y.; Shakirov, M. M.; Repinskaya, I. B.; Koptug, V. A. Zh. Org. Khim. 1991, 27, 2622–2623.
Return to citation in text: [1] -
Koltunov, K. Y.; Repinskaya, I. B. Zh. Org. Khim. 1993, 30, 90–93.
Return to citation in text: [1] -
Walspurger, S.; Vasilyev, A. V.; Sommer, J.; Pale, P. Tetrahedron 2005, 61, 3559–3564. doi:10.1016/j.tet.2005.01.110
Return to citation in text: [1] -
Rendy, R.; Zhang, Y.; McElrea, A.; Gomez, A.; Klumpp, D. A. J. Org. Chem. 2004, 69, 2340–2347. doi:10.1021/jo030327t
Return to citation in text: [1] -
Weissman, S. A.; Zewge, D. Tetrahedron 2005, 61, 7833–7863. doi:10.1016/j.tet.2005.05.041
Return to citation in text: [1] -
Ranu, B. C.; Bhar, S. Org. Prep. Proced. Int. 1996, 28, 371–409. doi:10.1080/00304949609356549
Return to citation in text: [1]
| 24. | Zakusilo, D. N.; Ryabukhin, D. S.; Boyarskaya, I. A.; Yuzikhin, O. S.; Vasilyev, A. V. Tetrahedron 2015, 71, 102–108. doi:10.1016/j.tet.2014.11.033 |
| 25. | Lisakova, A. D.; Ryabukhin, D. S.; Trifonov, R. E.; Ostrovskii, V. A.; Boyarskaya, I. A.; Vasilyev, A. V. Synthesis 2017, 49, 579–586. doi:10.1055/s-0036-1588884 |
| 39. | Simons, F. E. R.; Frith, E. M.; Simons, K. J. J. Allergy Clin. Immunol. 1982, 70, 458–464. doi:10.1016/0091-6749(82)90009-4 |
| 40. | Piper, H. M.; Hutler, J. F.; Spieckerman, P. G. Arzneim. Forsch. 1985, 35, 1495–1501. |
| 41. | Certa, H.; Blashke, G. Arch. Pharm. 1990, 323, 744–747. |
| 42. | Chen, Z.; Miller, W. S.; Shan, S.; Valenzano, K. J. Bioorg. Med. Chem. Lett. 2003, 13, 3247–3252. doi:10.1016/S0960-894X(03)00665-6 |
| 43. | Goehring, R. R.; Whitehead, J. F. W.; Brown, K.; Islam, K.; Wen, X.; Zhou, X.; Chen, Z.; Valenzano, K. J.; Miller, W. S.; Shan, S.; Kyle, D. J. Bioorg. Med. Chem. Lett. 2004, 14, 5045–5050. doi:10.1016/j.bmcl.2004.08.001 |
| 44. | Burrows, J. N.; Cumming, J. G.; Fillery, S. M.; Hamlin, G. A.; Hudson, J. A.; Jackson, R. J.; McLaughlin, S.; Shaw, J. S. Bioorg. Med. Chem. Lett. 2005, 15, 25–28. doi:10.1016/j.bmcl.2004.10.044 |
| 45. | Marcusson, J. O.; Norinder, U.; Högberg, T.; Ross, S. B. Eur. J. Pharmacol. 1992, 215, 191–198. doi:10.1016/0014-2999(92)90028-3 |
| 46. | Vatter, H.; Seifert, V. Cardiovasc. Drug Rev. 2006, 24, 63–76. doi:10.1111/j.1527-3466.2006.00063.x |
| 47. | Hou, X.; Ge, Z.; Wang, T.; Guo, W.; Cui, J.; Cheng, T.; Lai, C.; Li, R. Bioorg. Med. Chem. Lett. 2006, 16, 4214–4219. doi:10.1016/j.bmcl.2006.05.085 |
| 48. | Wang, J.; Song, Y.; Zhang, J.; Li, X.; Ling, X.; Li, R.; Cui, J. Chromatographia 2010, 72, 459–464. doi:10.1365/s10337-010-1697-4 |
| 49. | Hou, X.; Ge, Z.; Wang, T.; Guo, W.; Wu, J.; Cui, J.; Lai, C.; Li, R. Arch. Pharm. 2011, 344, 320–332. doi:10.1002/ardp.201000259 |
| 50. | Trifonov, R. E.; Volovodenko, A. P.; Vergizov, S. N.; Shirinbekov, N. I.; Gindin, V. A.; Koren, A. O.; Ostrovskii, V. A. Helv. Chim. Acta 2005, 88, 1790–1797. doi:10.1002/hlca.200590140 |
| 51. | Hartley, R. F.; Huang, Y.; Cassidy, M.; Razler, T. M.; Qian, F.; Hussain, M. A. J. Pharm. Sci. 2012, 101, 3124–3133. doi:10.1002/jps.23050 |
| 1. | Pace, A.; Buscemi, S.; Piccionello, A. P.; Pibiri, I. Adv. Heterocycl. Chem. 2015, 116, 85–136. doi:10.1016/bs.aihch.2015.05.001 |
| 2. | Bora, R. O.; Dar, B.; Pradhan, V.; Farooqui, M. Mini-Rev. Med. Chem. 2014, 14, 355–369. doi:10.2174/1389557514666140329200745 |
| 3. | Bokach, N. A.; Kuznetsov, M. L.; Kukushkin, V. Yu. Coord. Chem. Rev. 2011, 255, 2946–2967. doi:10.1016/j.ccr.2011.07.001 |
| 4. | Pace, A.; Pierro, P. Org. Biomol. Chem. 2009, 7, 4337–4348. doi:10.1039/B908937C |
| 5. | Xiong, C. S.; Drewe, J.; Kasibhatla, S. Curr. Med. Chem. 2006, 13, 2627–2644. doi:10.2174/092986706778201521 |
| 6. | Hemming, K. J. Chem. Res., Synop. 2001, 209–216. doi:10.3184/030823401103169603 |
| 7. | Vivona, N.; Buscemi, S. Heterocycles 1995, 41, 2095–2116. doi:10.3987/REV-95-473 |
| 8. | Tabrizi, M. A.; Baraldi, P. G.; Borea, P. A.; Varani, K. Chem. Rev. 2016, 116, 519–560. doi:10.1021/acs.chemrev.5b00411 |
| 12. | Jones, R. M.; Leonard, J. N.; Buzard, D. J.; Lehmann, J. Expert Opin. Ther. Pat. 2009, 19, 1339–1359. doi:10.1517/13543770903153878 |
| 22. | Macaev, F.; Ribkovskaia, Z.; Pogrebnoi, S.; Boldescu, V.; Rusu, G.; Shvets, N.; Dimoglo, A.; Geronikaki, A.; Reynolds, R. Bioorg. Med. Chem. 2011, 19, 6792–6807. doi:10.1016/j.bmc.2011.09.038 |
| 11. | Ding, D.; Boudreau, M. A.; Leemans, E.; Spink, E.; Yamaguchi, T.; Testero, S. A.; O'Daniel, P. I.; Lastochkin, E.; Chang, M.; Mobashery, S. Bioorg. Med. Chem. Lett. 2015, 25, 4854–4857. doi:10.1016/j.bmcl.2015.06.044 |
| 23. | Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A. T. J. Med. Chem. 2012, 55, 1817–1830. doi:10.1021/jm2013248 |
| 10. | Nicolaides, D. N.; Fylaktakidou, K. C.; Litinas, K. E.; Hadjipavlou-Litina, D. Eur. J. Med. Chem. 1998, 33, 715–724. doi:10.1016/S0223-5234(98)80030-5 |
| 20. | Saigo, K.; Kono, M.; Tagaki, Y..; Takenoguchi, M.; Hiramatsu, Y.; Tada, H.; Hishita, T.; Misawa, M.; Imoto, S.; Imashuku, S. J. Clin. Med. Res. 2013, 5, 57–60. doi:10.4021/jocmr1180w |
| 58. | Weissman, S. A.; Zewge, D. Tetrahedron 2005, 61, 7833–7863. doi:10.1016/j.tet.2005.05.041 |
| 59. | Ranu, B. C.; Bhar, S. Org. Prep. Proced. Int. 1996, 28, 371–409. doi:10.1080/00304949609356549 |
| 9. | Afiatpour, P.; Srivastava, R. M.; de Olivereira, M. L.; Barraeiro, E. J. Braz. J. Med. Biol. Res. 1994, 27, 1403–1406. |
| 21. | Yang, X.; Liu, G.; Li, H.; Zhang, Y.; Song, D.; Li, C.; Wang, R.; Liu, B.; Liang, W.; Jing, Y.; Zhao, G. J. Med. Chem. 2010, 53, 1015–1022. doi:10.1021/jm9011565 |
| 25. | Lisakova, A. D.; Ryabukhin, D. S.; Trifonov, R. E.; Ostrovskii, V. A.; Boyarskaya, I. A.; Vasilyev, A. V. Synthesis 2017, 49, 579–586. doi:10.1055/s-0036-1588884 |
| 16. | Jones, A. M.; Helm, J. M. Drugs 2009, 69, 1903–1910. doi:10.2165/11318500-000000000-00000 |
| 18. | Kumar, D.; Patel, G.; Vijayakrishan, L.; Dastidar, S. G.; Ray, A. Chem. Biol. Drug Des. 2012, 79, 810–818. doi:10.1111/j.1747-0285.2011.01304.x |
| 37. | Koltunov, K. Yu.; Walspurger, S.; Sommer, J. Eur. J. Org. Chem. 2004, 4039–4047. doi:10.1002/ejoc.200400313 |
| 54. | Koltunov, K. Y.; Shakirov, M. M.; Repinskaya, I. B.; Koptug, V. A. Zh. Org. Khim. 1991, 27, 2622–2623. |
| 55. | Koltunov, K. Y.; Repinskaya, I. B. Zh. Org. Khim. 1993, 30, 90–93. |
| 56. | Walspurger, S.; Vasilyev, A. V.; Sommer, J.; Pale, P. Tetrahedron 2005, 61, 3559–3564. doi:10.1016/j.tet.2005.01.110 |
| 57. | Rendy, R.; Zhang, Y.; McElrea, A.; Gomez, A.; Klumpp, D. A. J. Org. Chem. 2004, 69, 2340–2347. doi:10.1021/jo030327t |
| 19. | Mahavri, G. M.; Fauq, A. H. Tetrahedron Lett. 2010, 51, 6542–6544. doi:10.1016/j.tetlet.2010.10.025 |
| 24. | Zakusilo, D. N.; Ryabukhin, D. S.; Boyarskaya, I. A.; Yuzikhin, O. S.; Vasilyev, A. V. Tetrahedron 2015, 71, 102–108. doi:10.1016/j.tet.2014.11.033 |
| 14. | Coupar, I. M.; Hedges, A.; Metcalfe, H. L.; Turner, P. J. Pharm. Pharmacol. 1969, 21, 474–475. doi:10.1111/j.2042-7158.1969.tb08294.x |
| 50. | Trifonov, R. E.; Volovodenko, A. P.; Vergizov, S. N.; Shirinbekov, N. I.; Gindin, V. A.; Koren, A. O.; Ostrovskii, V. A. Helv. Chim. Acta 2005, 88, 1790–1797. doi:10.1002/hlca.200590140 |
| 13. | Kumar, D.; Patel, G.; Johnson, E. O.; Shah, K. Bioorg. Med. Chem. Lett. 2009, 19, 2739–2741. doi:10.1016/j.bmcl.2009.03.158 |
| 17. | Montgomery, J.; Toogood, P. L.; Hutchings, K. M.; Liu, J.; Narasimham, L.; Braden, T.; Dermyer, M. R.; Kulynych, A. D.; Smith, Y. D.; Warmus, J. S.; Taylor, C. Bioorg. Med. Chem. Lett. 2009, 19, 665–669. doi:10.1016/j.bmcl.2008.12.054 |
| 52. | Parr, R. G.; von Szentpály, L.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x |
| 53. | Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p |
| 26. | Andreatta, J. R.; McKeown, B. A.; Gunnoe, T. B. J. Organomet. Chem. 2011, 696, 305–315. doi:10.1016/j.jorganchem.2010.09.030 |
| 27. | Foley, N. A.; Lee, J. P.; Ke, Z.; Gunnoe, T. B.; Cundari, T. R. Acc. Chem. Res. 2009, 42, 585–597. doi:10.1021/ar800183j |
| 24. | Zakusilo, D. N.; Ryabukhin, D. S.; Boyarskaya, I. A.; Yuzikhin, O. S.; Vasilyev, A. V. Tetrahedron 2015, 71, 102–108. doi:10.1016/j.tet.2014.11.033 |
| 25. | Lisakova, A. D.; Ryabukhin, D. S.; Trifonov, R. E.; Ostrovskii, V. A.; Boyarskaya, I. A.; Vasilyev, A. V. Synthesis 2017, 49, 579–586. doi:10.1055/s-0036-1588884 |
| 36. | Karshtedt, D.; Bell, A. T.; Tilley, T. D. Organometallics 2004, 23, 4169–4171. doi:10.1021/om0495325 |
| 37. | Koltunov, K. Yu.; Walspurger, S.; Sommer, J. Eur. J. Org. Chem. 2004, 4039–4047. doi:10.1002/ejoc.200400313 |
| 38. | King, F. D.; Caddick, S. Org. Biomol. Chem. 2012, 10, 3244–3252. doi:10.1039/c2ob00012a |
| 34. | Bair, J. S.; Schramm, Y.; Sergeev, A. G.; Clot, E.; Eisenstein, O.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 13098–13101. doi:10.1021/ja505579f |
| 35. | Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Persiani, D. Org. Lett. 2008, 10, 1597–1600. doi:10.1021/ol800266e |
| 30. | Cheng, H.; Dong, W.; Dannenberg, C. A.; Dong, S.; Guo, Q.; Bolm, C. ACS Catal. 2015, 5, 2770–2773. doi:10.1021/acscatal.5b00258 |
| 31. | Sun, Z.-M.; Zhang, J.; Manan, R. S.; Zhao, P. J. Am. Chem. Soc. 2010, 132, 6935–6937. doi:10.1021/ja102575d |
| 32. | Zhao, D.; Nimphius, C.; Lindale, M.; Glorius, F. Org. Lett. 2013, 15, 4504–4507. doi:10.1021/ol402053n |
| 33. | Xue, F.; Wang, D.; Li, X.; Wan, B. Org. Biomol. Chem. 2013, 11, 7893–7898. doi:10.1039/C3OB41342J |
| 28. | Bowring, M. A.; Bergman, R. G.; Tilley, T. D. Organometallics 2011, 30, 1295–1298. doi:10.1021/om2000458 |
| 29. | Tarselli, M. A.; Liu, A.; Gagné, M. R. Tetrahedron 2009, 65, 1785–1789. doi:10.1016/j.tet.2008.10.110 |
© 2017 Zalivatskaya et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)