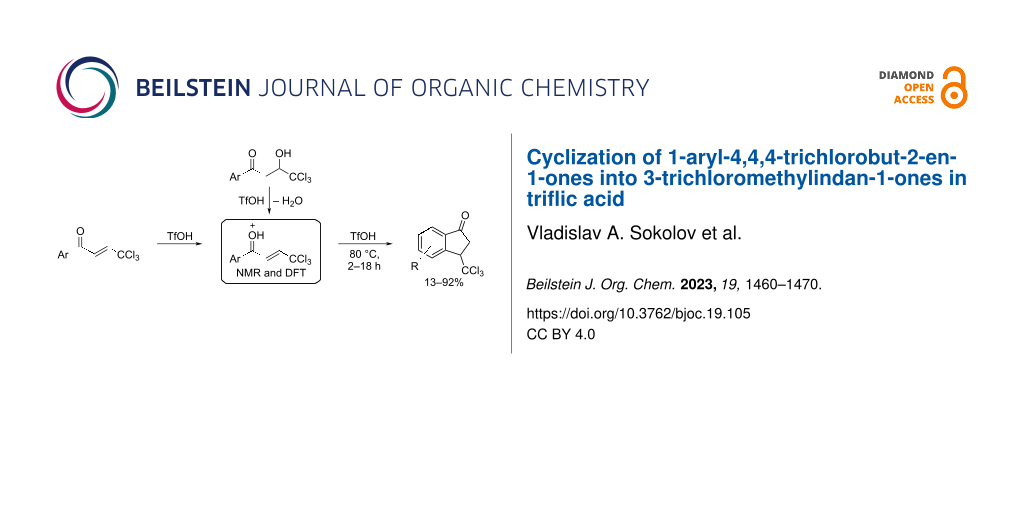
Abstract
Trichloromethyl-substituted enones (1-aryl-4,4,4-trichlorobut-2-en-1-ones, ArCOCH=CHCCl3, CCl3-enones) undergo intramolecular transformation into 3-trichloromethylindan-1-ones (CCl3-indanones) in Brønsted superacid CF3SO3H (triflic acid, TfOH) at 80 °C within 2–10 h in yields up to 92%. Protonation of the carbonyl oxygen of the starting CCl3-enones by TfOH affords the key reactive intermediates, the O-protonated forms ArC(=OH+)CH=CHCCl3, which are then cyclized into the target CCl3-indanones. These cations have been studied experimentally by means of NMR spectroscopy in TfOH and theoretically by DFT calculations. Under the same superacidic conditions in TfOH, CCl3-hydroxy ketones (1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones; ArCOCH2CH(OH)CCl3) undergo dehydration to the corresponding CCl3-enones, which are further cyclized into CCl3-indanones. The yields of CCl3-indanones starting from CCl3-hydroxy ketones are up to 86% in TfOH at 80 °C within 3–18 h.

Graphical Abstract
Introduction
Superelectrophilic activation of organic compounds under the action of strong Brønsted and Lewis acids is an effective method for the synthesis of various carbocycles and heterocycles, and polyfunctional compounds (see books [1,2] and reviews [3-10]). Protonation (or coordination) of basic centers (carbons of unsaturated bonds and heteroatoms) of organic molecules in Brønsted (or Lewis) acids gives rise to not only monocations, but also to highly reactive dicationic (and even higher charged) species. Thus, different conjugated enones afford O,C-diprotonated forms under superelectrophilic activation conditions. These dications can participate in electrophilic aromatic substitution reactions with arenes ([11] and references therein).
Recently, we have shown that the reaction of (E)-5,5,5-trichloropent-3-en-2-one [Cl3CCH=CHC(=O)Me] with arenes in Brønsted superacid TfOH (triflic acid, CF3SO3H) furnishes 3-methyl-1-trichloromethylindenes (Scheme 1a) [11]. Based on NMR analysis in TfOH and theoretical DFT calculations, it has been found that the reaction proceeds through an intermediate formation of the O-protonated form of the starting compound [Cl3CCH=CHC(=OH+)Me]. The presence of two strong electron-withdrawing substituents, the trichloromethyl group (CCl3) and a protonated carbonyl (C(OH+)Me), at the carbon–carbon double bond makes this O-protonated species electrophilic enough to react with arenes (Scheme 1a). The second protonation of the C=C bond is hampered due to a strong acceptor character of the substituents, contrary to other more donating enones.
![[1860-5397-19-105-i1]](/bjoc/content/inline/1860-5397-19-105-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Generation of O-protonated and O,C-diprotonated species from substituted conjugated enones under superelectrophilic activation and their subsequent transformations.
Scheme 1: Generation of O-protonated and O,C-diprotonated species from substituted conjugated enones under su...
As a continuation of the research on the electrophilic activation of electron-poor alkenes bearing two electron-withdrawing substituents at the C=C bond, we initiated this study on transformations of 1-aryl-4,4,4-trichlorobut-2-en-1-ones under superelectrophilic activation conditions (Scheme 1b). The main goals of this work were the investigation of the protonation of CCl3-enones (1-aryl-4,4,4-trichlorobut-2-en-1-ones) by NMR spectroscopy and DFT calculations, and to study their intramolecular cyclization in triflic acid into the synthetically and medicinally relevant (see recent reviews [12-18]) indan-1-ones.
Results and Discussion
The synthesis of 1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones (CCl3-hydroxy ketones) 1a–o was carried out by condensation of acetophenones with chloral under reflux in acetic acid using the known literature procedure [19] (Scheme 2). Based on another literature approach [20], compounds 1p–v were obtained by acylation of electron-donating arenes with Wynberg lactone [21] (Scheme 3). Additionally, exact structures of compounds 1g,h,s,t,v were confirmed by X-ray analysis (see Supporting Information File 1).
![[1860-5397-19-105-i2]](/bjoc/content/inline/1860-5397-19-105-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones 1a–o by condensation of acetophenones with chloral in refluxing acetic acid.
Scheme 2: Synthesis of 1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones 1a–o by condensation of acetophenones wit...
![[1860-5397-19-105-i3]](/bjoc/content/inline/1860-5397-19-105-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones 1p–v by acylation of electron-donating arenes with Wynberg lactone.
Scheme 3: Synthesis of 1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones 1p–v by acylation of electron-donating ar...
The hydroxy ketones 1 were used as precursors for the synthesis of 1-aryl-4,4,4-trichlorobut-2-en-1-ones (CCl3-enones) 2 by dehydration of compounds 1 with p-toluenesulfonic acid monohydrate at reflux in toluene [19] (Scheme 4). By this route, mainly E-isomers of compounds 2 were formed except for compounds 2c,i,m which were obtained as mixtures of E,Z-isomers (see Experimental section). However, under the reaction conditions in the presence of TsOH, the hydroxy ketones 1k,p–s bearing strong electron-donating substituents in the aromatic ring gave oligomeric materials. Presumably, in these cases, after dehydration and formation of the corresponding enone 2, the latter underwent subsequent cationic oligomerization.
![[1860-5397-19-105-i4]](/bjoc/content/inline/1860-5397-19-105-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 1-aryl-4,4,4-trichlorobut-2-en-1-ones 2 by dehydration of hydroxy ketones 1.
Scheme 4: Synthesis of 1-aryl-4,4,4-trichlorobut-2-en-1-ones 2 by dehydration of hydroxy ketones 1.
Next we studied the intramolecular cyclization of compounds 1 and 2 in TfOH. It was found that enones 2 were transformed into the corresponding 3-trichloromethylindan-1-ones 3 upon heating in neat TfOH at 80 °C for 2–10 h (Scheme 5). Under the same reaction conditions, hydroxy ketones 1 were cyclized into indanones 3 as well (Scheme 6). The structure of compound 3a was confirmed by X-ray analysis (see Supporting Information File 1). Both, hydroxy ketone 1 and the corresponding enone 2, can be converted into the same indanone 3 in comparable yields; see pairs of reactions for 1a and 2a (indanone 3a), 1d and 2d (indanone 3d), 1i and 3i (indanone 3i), and 1n and 3n (indanone 3n) in Scheme 5, Scheme 6 and the Experimental section).
![[1860-5397-19-105-i5]](/bjoc/content/inline/1860-5397-19-105-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cyclization of 1-aryl-4,4,4-trichlorobut-2-en-1-ones 2 into 3-trichloromethylindan-1-ones 3 in TfOH.
Scheme 5: Cyclization of 1-aryl-4,4,4-trichlorobut-2-en-1-ones 2 into 3-trichloromethylindan-1-ones 3 in TfOH....
![[1860-5397-19-105-i6]](/bjoc/content/inline/1860-5397-19-105-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Cyclization of 1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones 1 into 3-trichloromethylindan-1-ones 3 in TfOH.
Scheme 6: Cyclization of 1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones 1 into 3-trichloromethylindan-1-ones 3 ...
There are some features of this cyclization. The electron-poor enones 2l,m are unreactive and do not give rise to the corresponding indanones due to the low nucleophilicity of the nitro-substituted aromatic ring. On the other hand, the electron-rich enones 2o,t,u, bearing electron-donating substituents in the aromatic rings, afford complex mixtures of oligomeric materials. The cyclization of hydroxy ketone 1k into indanone 3k is accompanied by demethylation of one of the methoxy groups (Scheme 6). The positions of hydroxy and methoxy groups in the aryl ring of compound 3k were determined by NOESY correlations between protons in this structure (see Supporting Information File 1). Surprisingly, enone 2j transformed into the phenoxy-substituted indanone 3j in a low yield of 13% (Scheme 5). The formation of the latter represents an interesting rearrangement with the intermolecular transfer of a phenyl group in the starting methoxy-substituted enone 2j under the rather harsh reaction conditions (TfOH at 80 °C).
To study the cyclization further, reactions of some hydroxy ketones 1 were run for shorter time (1–4.5 h) in TfOH at 80 °C, i.e., under conditions of incomplete conversion of the starting compounds. It was found that, apart from the target indanones 3, substantial amounts of the corresponding enones 2 were detected (Table 1). This means that, in the superacid TfOH, hydroxy ketones 1 may initially undergo dehydration to enones 2, which are subsequently cyclized into indanones 3. From a synthetic point of view, the use of hydroxy ketones 1 as starting compounds for the cyclization without additional preparation and isolation of the corresponding enones 2 is more economical as it reduces the number of steps in the synthesis.
Table 1: Transformations of hydroxy ketones 1a,c,f,i into the corresponding enones 2 and indanones 3 in TfOH at 80 °C for shorter reaction time under the conditions of incomplete conversion of the starting compounds.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-105-i8.svg?max-width=637&scale=1.0)
|
||||
| Entry | Starting hydroxy ketone 1 |
Reaction
time, h |
Ratio of compounds
hydroxy ketone 1/enone 2/indanone 3 |
Substituent, R in 1, 2, and 3 |
| 1 | 1a | 1 |
1a/2a/3a
1:2.6:5 |
H |
| 2 | 1c | 3 |
1c/2c/3c
5:1.8:1 |
Me |
| 3 | 1f | 3 |
1f/2f/3f
0:2:1 |
Br |
| 4 | 1i | 4.5 |
1i/2i/3i
0:1:5 |
CF3 |
aRatio of compounds 1, 2, and 3 was determined by NMR.
Other Brønsted and Lewis acids were also tested for this cyclization. Thus, enones 2a and 2e were not transformed into the corresponding indanones 3 in neat sulfuric acid (H2SO4) at room temperature for 3 days. That is in accord with literature data [19], where H2SO4 was used for dehydration of hydroxy ketones 1 into enones 2, and no cyclization into indanones was detected in this acid. However, in the current study, we carried out the reaction of electron-donating naphthyl-substituted enone 2n in H2SO4 at room temperature, which resulted in the quantitative formation of indanone 3n after 3 days. The reactions of enones 2a,e,n with an excess (5 equiv) of AlBr3 or AlCl3 in CH2Cl2 solution at room temperature for 3 days afforded complex mixtures of compounds.
Then, the protonation of compounds 1 and 2 in TfOH was investigated by means of NMR spectroscopy. According to the 1H, 13C, and 19F NMR data, hydroxy ketones 1 and enones 2 afford stable O-protonated forms A and B, respectively, in TfOH at room temperature (Table 2 and spectra in Supporting Information File 1).
Table 2: 1H, 13C, and 19F NMR data of compounds 1, and 2 in CDCl3 and their O-protonated forms A, and B, respectively, in TfOH.
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-105-i9.svg?max-width=637&scale=1.0)
|
|||||||
| compounds 1, 2 and cations A, B, respectively | solvent | 1H NMR, δ, ppm, J, Hz | 13C NMR, δ, ppm | ||||
| H2 | H3 | C1 | C2 | C3 | C4 | ||
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-105-i10.svg?max-width=637&scale=1.0)
1a |
CDCl3 |
3.68 dd,
J = 17.4, 2.0; 3.52 dd, J = 17.4, 8.9 AB system |
4.91 ddd,
J = 8.9, 4.4, 2.0 |
197.1 | 40.7 | 79.0 | 102.5 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-19-105-i11.svg?max-width=637&scale=1.0)
Aa |
TfOHa |
4.54 dd,
J = 18.7, 4.1; 4.15 dd, J = 18.7, 7.6 AB system |
5.21 dd,
J = 7.6, 4.1 |
218.2 | 34.2 | 81.0 | 98.9 |
| Δδb |
0.86
0.63 |
0.30 | 21.1 | −6.5 | 2.0 | −3.6 | |
![[Graphic 5]](/bjoc/content/inline/1860-5397-19-105-i12.svg?max-width=637&scale=1.0)
1c |
CDCl3 |
3.66 dd,
J = 17.3, 2.0; 3.48 dd, J = 17.3, 9.0 AB system |
4.87 dd,
J = 8.9 |
196.8 | 40.5 | 79.1 | 102.5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-19-105-i13.svg?max-width=637&scale=1.0)
Ac |
TfOHa |
4.46 dd,
J = 18.3, 3.9; 4.07 dd, J = 18.3, 7.8 AB system |
5.15 dd,
J = 7.8, 3.9 |
214.5 | 33.9 | 80.9 | 99.0 |
| Δδb |
0.80
0.59 |
0.28 | 17.7 | −6.6 | 1.8 | −3.5 | |
![[Graphic 7]](/bjoc/content/inline/1860-5397-19-105-i14.svg?max-width=637&scale=1.0)
1d |
CDCl3 |
3.62 dd,
J = 17.3, 2.2; 3.50 dd, J = 17.3, 8.8 AB system |
4.89 dd,
J = 8.8, 2.2 |
195.4 | 40.7 | 79.0 | 102.6 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-19-105-i15.svg?max-width=637&scale=1.0)
Ad |
TfOHa |
4.49 dd,
J = 18.6, 4.0; 4.11 dd, J = 18.6, 7.6 AB system |
5.20 dd,
J = 7.6, 4.0 |
215.3 | 34.1 | 80.9 | 98.8 |
|
19F NMR, δ, ppm
−103.6 → −77.9 Δδa = 25.7 |
Δδb |
0.81
0.61 |
0.31 | 19.9 | −6.6 | 1.9 | −3.8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-19-105-i16.svg?max-width=637&scale=1.0)
(E)-2a |
CDCl3 |
7.27 d,
J = 17.6 |
7.42 d,
J = 17.6 |
189.0 | 124.3 | 145.6 | 93.1 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-19-105-i17.svg?max-width=637&scale=1.0)
Ba |
TfOHa |
7.86 d,
J = 12.0 |
7.90 d,
J = 12.0 |
204.9 | 120.7 | 159.0 | 90.0 |
| Δδb | 0.61 | 0.46 | 15.9 | −3.6 | 13.4 | −3.1 | |
![[Graphic 11]](/bjoc/content/inline/1860-5397-19-105-i18.svg?max-width=637&scale=1.0)
(E)-2c |
CDCl3 |
7.28 d,
J = 14.6 |
7.43 d,
J = 14.6 |
188.3 | 124.2 | 145.2 | 93.1 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-19-105-i19.svg?max-width=637&scale=1.0)
Bc |
TfOHa |
7.82 d,
J = 12.0 |
7.84 d,
J = 12.0 |
201.0 | 120.4 | 163.3 | 90.2 |
| Δδb | 0.54 | 0.41 | 12.7 | −3.8 | 8.1 | −2.9 | |
![[Graphic 13]](/bjoc/content/inline/1860-5397-19-105-i20.svg?max-width=637&scale=1.0)
(E)-2d |
CDCl3 |
7.26 d,
J = 14.5 |
7.38 d,
J = 14.5 |
187.3 | 123.9 | 146.8 | 93.0 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-19-105-i21.svg?max-width=637&scale=1.0)
Bd |
TfOH |
7.80 d,
J = 16.0 |
7.84 d,
J = 16.0 |
202.0 | 120.4 | 158.1 | 90.2 |
|
19F NMR, δ, ppm
−103.2 → −77.9 Δδa = 25.3 |
Δδa | 0.54 | 0.46 | 14.7 | −3.5 | 11.3 | −2.8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-19-105-i22.svg?max-width=637&scale=1.0)
2m |
CDCl3 |
7.35 d,
J = 14.6 |
7.42 d,
J = 14.6 |
187.5 | 124.1 | 146.9 | 92.4 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-19-105-i23.svg?max-width=637&scale=1.0)
Bm |
TfOHa |
7.85 d,
J = 14.9 |
7.96 d,
J = 14.9 |
208.8 | 121.6 | 162.9 | 90.2 |
| Δδb | 0.50 | 0.54 | 21.3 | −2.5 | 6.0 | −2.2 | |
аCH2Cl2 was used as internal standard; b∆δ = δacid − δCDCl3.
In species A both oxygens should be protonated by the superacid. The differences in chemical shifts (∆δ = δacid − δCDCl3) for the corresponding atoms H2 and H3 in cations Aa,c,d and their neutral precursors 1a,c,d were 0.59–0.86 and 0.28–0.31 ppm (Table 2). These downfield shifts of the signals point to an inductively induced positive charge on these hydrogens due to the protonation of the oxygens of the carbonyl and hydroxy groups. According to the 13C NMR spectra, the largest downfield shift was observed for the carbonyl carbon С1, with ∆δ = 17.7–21.1 ppm, showing a substantial degree of protonation of the carbonyl group in TfOH.
The tendencies are the same for the protonation of enones 2a,c,d,m leading to cations Ba,c,d,m (Table 2). Thus, in the 1H NMR spectra, downfield shifts of vinyl protons H2 and H3 upon protonation were 0.50–0.61 and 0.41–0.54 ppm, respectively. In the 13C NMR spectra, ∆δ values for carbons С1 and С3 were 12.7–21.3 and 6.0–13.4 ppm, respectively. The NMR data revealed that the positive charge in the O-protonated forms B is substantially delocalized from the carbonyl group to vinyl carbon C3.
For fluorophenyl-substituted compounds and cations 1d and Ad, 2d and Bd, also a large downfield shift of the corresponding fluorine signals is observed in the 19F NMR spectra (∆δ = 25.3–25.7 ppm), which shows a delocalization of the positive charge from the carbonyl group to the aromatic ring.
It should be mentioned that cation Bm was generated by two ways in TfOH: either directly by protonation of enone 2m or from hydroxy ketone 1m. The latter was found to undergo fast dehydration into enone 2m in TfOH at room temperature.
Then, we carried out DFT calculations of cations Aa–Da derived from protonation of compounds 1a and 2a. Thermodynamics of their formation, as Gibbs energies ΔG298 of the corresponding reactions, energies of HOMO/LUMO, electrophilicity indices ω [22,23], charge distribution, and contribution of atomic orbital into LUMO in species Aa–Da were estimated (Table 3).
Table 3: Selected calculated (DFT) electronic characteristics of the protonated forms Aa, Ba, Ca, and Da generated from hydroxy ketone 1a and enone 2a, and values of Gibbs energies of reactions (ΔG, kJ/mol).
![[Graphic 17]](/bjoc/content/inline/1860-5397-19-105-i24.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Species | EHOMO, eV | ELUMO, eV | ω,a eV | q(C1),b e | q(C3),b e | k(C1)LUMO,с % | k(C3)LUMO,с % |
| 1 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-19-105-i25.svg?max-width=637&scale=1.0)
1a |
−7.34 | −2.09 | 2.1 | 0.60 | 0.08 | 15.6 | 18.2 |
| 2 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-19-105-i26.svg?max-width=637&scale=1.0)
2a |
−7.37 | −2.86 | 2.9 | 0.54 | −0.18 | 10.5 | 12.7 |
| 3 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-19-105-i27.svg?max-width=637&scale=1.0)
Ca |
−7.93 | −3.67 | 3.9 | 0.66 | 0.07 | 25.0 | 3.1 |
| 4 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-19-105-i28.svg?max-width=637&scale=1.0)
Aa |
−8.05 | −3.97 | 4.4 | 0.66 | 0.07 | 28.9 | 12.7 |
| 5 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-19-105-i29.svg?max-width=637&scale=1.0)
Ba |
−7.93 | −4.12 | 4.8 | 0.60 | -0.13 | 14.9 | 12.1 |
| 6 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-19-105-i30.svg?max-width=637&scale=1.0)
Ea |
−8.10 | −5.50 | 8.9 | 0.65 | −0.28 | 3.3 | 14.4 |
aGlobal electrophilicity index ω = (EHOMO + ELUMO)2/8(ELUMO−EHOMO); bnatural charges; ccontribution of atomic orbital to the molecular orbital.
The formation of O-monoprotonated species Ba and Ca from enone 2a and hydroxy ketone 1a is favorable, with negative ΔG298 values for protonation of −52 and −39 kJ/mol, respectively (see overview scheme of Table 3). The second protonation of the C=C bond of species Ba is very unfavorable. Moreover, the corresponding O,C-diprotonated form Da was found to be extremely unstable and spontaneously rearranges into species Ea via a shift of a chlorine atom. Therefore, the DFT calculations and NMR data for enones 2 in TfOH (Table 2) indicate that the formation of dications D does not take place. Species B should be the key reactive intermediates that undergo cyclization into indanones 3 with a negative Gibbs energy of −7 kJ/mol for the reaction Ba→3a.
According to the calculations, the subsequent complete protonation of the hydroxy group in species Ca leading to dication Aa is thermodynamically unfavorable (ΔG298 = 68 kJ/mol) as well (see overview scheme of Table 3). NMR experiments allowed us to observe the formation of O,O-diprotonated species in the superacid TfOH (Table 2). The dication Aa should not be transformed to species Da analogously to cation Ba. The cyclization of dication Aa into indanone 3a is very favorable (ΔG298 = −101.3 kJ/mol). However, the formation of both enones 2 and indanones 3 from hydroxy ketones 1 is observed in TfOH (Table 1). We assume that, at first, hydroxy ketones 1 undergo dehydration into enones 2, which then cyclize into the target indanones 3. Despite thermodynamic gain in energy, the cyclization of dications Aa in compounds 3 may have a high activation barrier.
The calculations showed that the largest part of the positive charge in the key reactive species Ba is localized on the carbonyl carbon C1 (0.60 e). This carbon atom gives 14.9% contribution to the LUMO. Contrary to that, the carbon C3 bears no positive charge (−0.13 e) and contributes 12.1% to the LUMO. The intramolecular cyclization of cation Ba takes place between the atom C3 and the ortho-carbons of the phenyl ring. Thus, electrophilic properties of atom C3 should be mainly explained by orbital factors, rather than charge ones.
Summarizing the data obtained during the synthesis of indanones 3 (Scheme 5, Scheme 6 and Table 1), NMR, and DFT studies on intermediate cations (Table 2 and Table 3), one may propose plausible reaction mechanisms for the cyclization of compounds 1 and 2 into indanones 3 in TfOH (Scheme 7). Protonation of the carbonyl oxygen of enone 2 gives rise to cation B which is followed by cyclization into indanone 3 through mesomeric form B'. The hydroxy ketone 1 is protonated at the oxygen atoms leading to cation A, followed by dehydration resulting in cation B, which is cyclized into indanone 3. The transformation of species B into indanone 3 can be also considered as a variant of Nazarov cyclization. It should be noted that such 3-trichloromethylindanones 3 have been obtained for the first time in this study, which structurally resemble the known 3-dichloromethylindanones [24] and 3-trifluoromethylindanones [25-27].
![[1860-5397-19-105-i7]](/bjoc/content/inline/1860-5397-19-105-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Plausible mechanisms for the cyclization of compounds 1 and 2 into indanones 3 in TfOH.
Scheme 7: Plausible mechanisms for the cyclization of compounds 1 and 2 into indanones 3 in TfOH.
Conclusion
A method for the synthesis of CCl3-indanones (3-trichloromethylindan-1-ones), as a novel class of indanones, has been developed and involves an intramolecular cyclization of both, CCl3-enones (1-aryl-4,4,4-trichlorobut-2-en-1-ones) and CCl3-hydroxy ketones (1-aryl-4,4,4-trichloro-3-hydroxybutan-1-ones) in Brønsted superacid TfOH at an elevated temperature of 80 °C within 2–18 h. In both cases, the reaction proceeds through an intermediate formation of the O-protonated carbonyl form of the CCl3-enones, that are finally cyclized into the target CCl3-indanones.
Supporting Information
| Supporting Information File 1: Experimental, characterization data and copies of spectra. | ||
| Format: PDF | Size: 8.2 MB | Download |
References
-
Olah, G. A.; Prakash, G. K. S.; Molnár, Á.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2008. doi:10.1002/9780470421604
Return to citation in text: [1] -
Olah, G. A.; Klumpp, D. A. Superelectrophiles and Their Chemistry; John Wiley & Sons: New York, NY, USA, 2008. doi:10.1002/9780470185124
Return to citation in text: [1] -
Naredla, R. R.; Klumpp, D. A. Chem. Rev. 2013, 113, 6905–6948. doi:10.1021/cr4001385
Return to citation in text: [1] -
Kazakova, A. N.; Vasilyev, A. V. Russ. J. Org. Chem. 2017, 53, 485–509. doi:10.1134/s1070428017040017
Return to citation in text: [1] -
Klumpp, D. A.; Kennedy, S. ARKIVOC 2018, No. ii, 215–232. doi:10.24820/ark.5550190.p010.270
Return to citation in text: [1] -
Vasilyev, A. V. Adv. Org. Synth. 2018, 8, 81–120. doi:10.2174/9781681085647118080005
Return to citation in text: [1] -
Zhao, W.; Sun, J. Chem. Rev. 2018, 118, 10349–10392. doi:10.1021/acs.chemrev.8b00279
Return to citation in text: [1] -
Greb, L. Chem. – Eur. J. 2018, 24, 17881–17896. doi:10.1002/chem.201802698
Return to citation in text: [1] -
Fernandes, A. J.; Panossian, A.; Michelet, B.; Martin-Mingot, A.; Leroux, F. R.; Thibaudeau, S. Beilstein J. Org. Chem. 2021, 17, 343–378. doi:10.3762/bjoc.17.32
Return to citation in text: [1] -
Gigant, N. Synlett 2013, 24, 399–400. doi:10.1055/s-0032-1318074
Return to citation in text: [1] -
Shershnev, I. A.; Boyarskaya, I. A.; Vasilyev, A. V. Molecules 2022, 27, 6675. doi:10.3390/molecules27196675
Return to citation in text: [1] [2] -
Lazinski, L. M.; Royal, G.; Robin, M.; Maresca, M.; Haudecoeur, R. J. Med. Chem. 2022, 65, 12594–12625. doi:10.1021/acs.jmedchem.2c01150
Return to citation in text: [1] -
Das, S. ChemistrySelect 2021, 6, 4761–4781. doi:10.1002/slct.202101460
Return to citation in text: [1] -
Das, S.; Dutta, A. New J. Chem. 2021, 45, 4545–4568. doi:10.1039/d0nj06318e
Return to citation in text: [1] -
Khoroshilova, O. V.; Vasilyev, A. V. Organics 2021, 2, 348–364. doi:10.3390/org2040019
Return to citation in text: [1] -
Turek, M.; Szczęsna, D.; Koprowski, M.; Bałczewski, P. Beilstein J. Org. Chem. 2017, 13, 451–494. doi:10.3762/bjoc.13.48
Return to citation in text: [1] -
Menezes, J. C. J. M. D. S. RSC Adv. 2017, 7, 9357–9372. doi:10.1039/c6ra28613e
Return to citation in text: [1] -
Patil, S. A.; Patil, R.; Patil, S. A. Eur. J. Med. Chem. 2017, 138, 182–198. doi:10.1016/j.ejmech.2017.06.032
Return to citation in text: [1] -
Guirado, A.; Martiz, B.; Andreu, R.; Bautista, D.; Gálvez, J. Tetrahedron 2007, 63, 1175–1182. doi:10.1016/j.tet.2006.11.054
Return to citation in text: [1] [2] [3] -
Fujisawa, T.; Ito, T.; Fujimoto, K.; Shimizu, M.; Wynberg, H.; Staring, E. G. J. Tetrahedron Lett. 1997, 38, 1593–1596. doi:10.1016/s0040-4039(97)00120-2
Return to citation in text: [1] -
Ganta, A.; Shamshina, J. L.; Cafiero, L. R.; Snowden, T. S. Tetrahedron 2012, 68, 5396–5405. doi:10.1016/j.tet.2012.04.107
Return to citation in text: [1] -
Parr, R. G.; Szentpály, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x
Return to citation in text: [1] -
Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p
Return to citation in text: [1] -
Yang, J.; Ponra, S.; Li, X.; Peters, B. B. C.; Massaro, L.; Zhou, T.; Andersson, P. G. Chem. Sci. 2022, 13, 8590–8596. doi:10.1039/d2sc02685f
Return to citation in text: [1] -
Kundu, K.; McCullagh, J. V.; Morehead, A. T., Jr. J. Am. Chem. Soc. 2005, 127, 16042–16043. doi:10.1021/ja0564416
Return to citation in text: [1] -
Chaudhary, B.; Kulkarni, N.; Sharma, S. Org. Chem. Front. 2020, 7, 1512–1519. doi:10.1039/d0qo00330a
Return to citation in text: [1] -
Prakash, S. G. K.; Paknia, F.; Narayanan, A.; Rasul, G.; Mathew, T.; Olah, G. A. J. Fluorine Chem. 2012, 143, 292–302. doi:10.1016/j.jfluchem.2012.07.008
Return to citation in text: [1]
| 1. | Olah, G. A.; Prakash, G. K. S.; Molnár, Á.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2008. doi:10.1002/9780470421604 |
| 2. | Olah, G. A.; Klumpp, D. A. Superelectrophiles and Their Chemistry; John Wiley & Sons: New York, NY, USA, 2008. doi:10.1002/9780470185124 |
| 12. | Lazinski, L. M.; Royal, G.; Robin, M.; Maresca, M.; Haudecoeur, R. J. Med. Chem. 2022, 65, 12594–12625. doi:10.1021/acs.jmedchem.2c01150 |
| 13. | Das, S. ChemistrySelect 2021, 6, 4761–4781. doi:10.1002/slct.202101460 |
| 14. | Das, S.; Dutta, A. New J. Chem. 2021, 45, 4545–4568. doi:10.1039/d0nj06318e |
| 15. | Khoroshilova, O. V.; Vasilyev, A. V. Organics 2021, 2, 348–364. doi:10.3390/org2040019 |
| 16. | Turek, M.; Szczęsna, D.; Koprowski, M.; Bałczewski, P. Beilstein J. Org. Chem. 2017, 13, 451–494. doi:10.3762/bjoc.13.48 |
| 17. | Menezes, J. C. J. M. D. S. RSC Adv. 2017, 7, 9357–9372. doi:10.1039/c6ra28613e |
| 18. | Patil, S. A.; Patil, R.; Patil, S. A. Eur. J. Med. Chem. 2017, 138, 182–198. doi:10.1016/j.ejmech.2017.06.032 |
| 11. | Shershnev, I. A.; Boyarskaya, I. A.; Vasilyev, A. V. Molecules 2022, 27, 6675. doi:10.3390/molecules27196675 |
| 11. | Shershnev, I. A.; Boyarskaya, I. A.; Vasilyev, A. V. Molecules 2022, 27, 6675. doi:10.3390/molecules27196675 |
| 25. | Kundu, K.; McCullagh, J. V.; Morehead, A. T., Jr. J. Am. Chem. Soc. 2005, 127, 16042–16043. doi:10.1021/ja0564416 |
| 26. | Chaudhary, B.; Kulkarni, N.; Sharma, S. Org. Chem. Front. 2020, 7, 1512–1519. doi:10.1039/d0qo00330a |
| 27. | Prakash, S. G. K.; Paknia, F.; Narayanan, A.; Rasul, G.; Mathew, T.; Olah, G. A. J. Fluorine Chem. 2012, 143, 292–302. doi:10.1016/j.jfluchem.2012.07.008 |
| 3. | Naredla, R. R.; Klumpp, D. A. Chem. Rev. 2013, 113, 6905–6948. doi:10.1021/cr4001385 |
| 4. | Kazakova, A. N.; Vasilyev, A. V. Russ. J. Org. Chem. 2017, 53, 485–509. doi:10.1134/s1070428017040017 |
| 5. | Klumpp, D. A.; Kennedy, S. ARKIVOC 2018, No. ii, 215–232. doi:10.24820/ark.5550190.p010.270 |
| 6. | Vasilyev, A. V. Adv. Org. Synth. 2018, 8, 81–120. doi:10.2174/9781681085647118080005 |
| 7. | Zhao, W.; Sun, J. Chem. Rev. 2018, 118, 10349–10392. doi:10.1021/acs.chemrev.8b00279 |
| 8. | Greb, L. Chem. – Eur. J. 2018, 24, 17881–17896. doi:10.1002/chem.201802698 |
| 9. | Fernandes, A. J.; Panossian, A.; Michelet, B.; Martin-Mingot, A.; Leroux, F. R.; Thibaudeau, S. Beilstein J. Org. Chem. 2021, 17, 343–378. doi:10.3762/bjoc.17.32 |
| 10. | Gigant, N. Synlett 2013, 24, 399–400. doi:10.1055/s-0032-1318074 |
| 19. | Guirado, A.; Martiz, B.; Andreu, R.; Bautista, D.; Gálvez, J. Tetrahedron 2007, 63, 1175–1182. doi:10.1016/j.tet.2006.11.054 |
| 22. | Parr, R. G.; Szentpály, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x |
| 23. | Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p |
| 21. | Ganta, A.; Shamshina, J. L.; Cafiero, L. R.; Snowden, T. S. Tetrahedron 2012, 68, 5396–5405. doi:10.1016/j.tet.2012.04.107 |
| 24. | Yang, J.; Ponra, S.; Li, X.; Peters, B. B. C.; Massaro, L.; Zhou, T.; Andersson, P. G. Chem. Sci. 2022, 13, 8590–8596. doi:10.1039/d2sc02685f |
| 20. | Fujisawa, T.; Ito, T.; Fujimoto, K.; Shimizu, M.; Wynberg, H.; Staring, E. G. J. Tetrahedron Lett. 1997, 38, 1593–1596. doi:10.1016/s0040-4039(97)00120-2 |
| 19. | Guirado, A.; Martiz, B.; Andreu, R.; Bautista, D.; Gálvez, J. Tetrahedron 2007, 63, 1175–1182. doi:10.1016/j.tet.2006.11.054 |
| 19. | Guirado, A.; Martiz, B.; Andreu, R.; Bautista, D.; Gálvez, J. Tetrahedron 2007, 63, 1175–1182. doi:10.1016/j.tet.2006.11.054 |
© 2023 Sokolov et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.