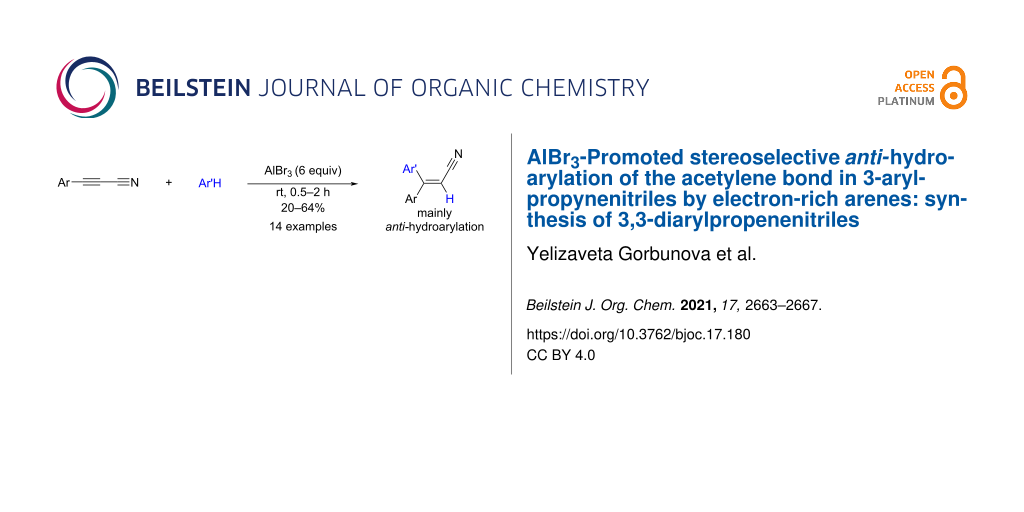
Abstract
Reactions of 3-arylpropynenitriles (ArC≡CCN) with electron-rich arenes (Ar′H, benzene and its polymethylated derivatives) under the action of aluminum bromide (AlBr3, 6 equiv) at room temperature for 0.5–2 h result in the stereoselective formation of 3,3-diarylpropenenitriles (Ar(Ar′)C=CHCN) in yields of 20–64%, as products of mainly anti-hydroarylation of the acetylene bond. The obtained 3,3-diarylpropenenitriles in triflic acid CF3SO3H (TfOH) at room temperature for 1 h are cyclized into 3-arylindenones in yields of 55–70%.

Graphical Abstract
Introduction
Conjugated acetylene nitriles (propynenitriles, R–C≡C–C≡N) are versatile building blocks in organic synthesis for the preparation of a plethora of functionalized compounds and heterocycles. The presence of conjugated acetylene and nitrile bonds in these compounds leads to an enhancement of reactivity of both functional groups. Thus, propynenitriles take part in electrophilic [1,2] and nucleophilic [3-6] addition reactions onto the acetylene bond leading to various substituted nitriles. Reactions onto both acetylene and nitrile groups are widely used for the construction of various heterocyclic systems [7-15].
Recently, we have shown that reactions of 3-arylpropenenitriles (cinnamonitriles, ArCH=CHCN) with arenes (Ar′H) under the superelectrophilic activation by the Brønsted superacid CF3SO3H (TfOH, triflic acid) or the strong Lewis acid AlBr3 result in the formation of 3,3-diarylpropanenitriles (Ar(Ar′)CHCH2CN) through the regioselective hydroarylation of the carbon–carbon double bond. In TfOH, the reactions proceed further to 3-arylindanones, as products of the intramolecular aromatic acylation of the 3,3-diarylpropanenitriles by the electrophilically activated nitrile group [16,17]. Based on this study and our work on electrophilic transformations of alkynes [18-20], we investigated reactions of 3-arylpropynenitriles under electrophilic activation conditions (see [21] for the chemistry of superelectrophilic species). The goal of this work was to study the reactions of 3-arylpropynenitriles with arenes under the action of the strong Lewis acids, aluminum halogenides AlX3 (X = Cl, Br) and the Brønsted superacid TfOH (CF3SO3H).
Results and Discussion
It was found that acetylene nitriles 1a–c reacted with arenes in the presence of excess AlBr3 (6 equiv) at room temperature for 0.5–2 h to afford E,Z-3,3-diarylpropenenitriles 2a–o as products of the regioselective hydroarylation of the acetylene bond (Scheme 1). These reactions proceeded only with an excess of AlBr3 (6 equiv). Thus, running the reaction of nitrile 1a with benzene under the action of less amount AlBr3 (1–4 equiv) resulted in an incomplete conversion of the starting compound 1a and a low yield of the target product 2a (<10%).
![[1860-5397-17-180-i1]](/bjoc/content/inline/1860-5397-17-180-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: AlBr3-promoted hydroarylation of the acetylene bond of 3-arylpropynenitriles 1a–c by arenes with the formation of 3,3-diarylpropenenitriles E-,Z-2a–o.
Scheme 1: AlBr3-promoted hydroarylation of the acetylene bond of 3-arylpropynenitriles 1a–c by arenes with th...
The yields of the compounds 2 were moderate (20–64%) that may be caused by possible transformations of the nitrile group in the electrophilic medium leading to oligomeric material [22]. The reaction proceeded rather regioselectively giving mainly Z-isomers of nitriles 2, as products of an anti-addition of hydrogen and the aryl group to the carbon–carbon triple bond. Only in three cases, mixtures of E,Z-isomers (2b,l,n) in a ratio of ≈1:1 were obtained. The E,Z-configuration of compounds 2a–o was determined by NOESY correlations between the vinyl proton and the aromatic protons or methyl groups in neighboring aryl substituents (see Supporting Information File 1). However, the configuration of nitrile 2o was unclear. Benzene, o-, m-, p-xylenes, and 1,2,4-trimethylbenzene (mesitylene) were included in the hydroarylation of nitriles 1. Reactions of nitriles 1a–c with o-xylene led to the formation of regioisomers derived from the electrophilic substitution at different positions of this arene. Thus, nitrile 1a gave two types of regioisomers 2n and 2o. After reactions of nitriles 1b,c with o-xylene, compounds 2l and 2m correspondingly were obtained; other regioisomers were not isolated in amounts that were high enough for their identification.
This transformation was also tested with another strong Lewis acid, aluminum chloride (AlCl3), for the reaction of nitrile 1a with benzene. However, in this case, mainly oligomeric material was obtained and the yield of the target reaction product 2a was lower (20%) than in the same reaction with AlBr3. This result revealed the less effectiveness of AlCl3 for the hydroarylation of nitriles 1.
One may propose the following reaction mechanism (Scheme 2). The coordination of AlBr3 to both the nitrile and acetylene bonds of the starting compound 1 furnishes the highly electrophilic species A bearing a positive charge on the acetylenic carbon atom C3. The subsequent reaction of species A with the arene molecule via electrophilic aromatic substitution results in the formation of species B. The most probably, this stage proceeds stereoselectively due to spatial factors, with the incoming arene Ar′H attacking the acetylene bond in an anti-position to the bulky AlBr3, that determines the final predominant formation of the mainly anti-hydroarylation products of the starting nitriles 1. At the last step of the reaction, a proton substitutes AlBr3, and final hydrolysis of the reaction mixture gives rise to nitriles 2.
![[1860-5397-17-180-i2]](/bjoc/content/inline/1860-5397-17-180-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible mechanism for reaction of acetylene nitriles 1 with arenes leading to nitriles 2.
Scheme 2: Plausible mechanism for reaction of acetylene nitriles 1 with arenes leading to nitriles 2.
It should be noted that this AlBr3-promoted hydroarylation of acetylene nitriles 1 (Scheme 1) is a novel transition-metal (Pd, Pt, Rh, etc.)-free stereoselective way for the synthesis of compounds 2. These compounds can be alternatively obtained by a Pd-catalyzed Heck reaction of 3-arylpropenenitriles with iodoarenes [23] or by a Cu-catalyzed hydroarylation of 3-arylpropynenitriles with arylboronic acid [24,25]. There is one example of use of dicyanoacetylene in a similar AlCl3-catalyzed hydroarylation of an acetylene bond in the synthesis of cyclophanes [26].
Additionally, the cyclization of two selected nitriles 2c,g into 3-arylindanones 3a,b correspondingly was carried out in triflic acid TfOH (CF3SO3H) at room temperature for 1 h (Scheme 3). The intramolecular aromatic substitution by the electrophilically activated nitrile group took place in the more electron-rich methylated aryl ring. A similar cyclization of 3,3-diarylpropanenitriles into 3-arylindanones in TfOH was described by us previously [17]. It should be specially emphasized that the synthesis of indenones is an important goal in organic chemistry since this structural motif is associated with interesting chemical and biological properties [27-32].
![[1860-5397-17-180-i3]](/bjoc/content/inline/1860-5397-17-180-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyclization of nitriles 2c,g into indanones 3a,b in TfOH.
Scheme 3: Cyclization of nitriles 2c,g into indanones 3a,b in TfOH.
We also conducted reactions of nitriles 1a–c with arenes in TfOH at room temperature, which, however, led to complex mixtures of reaction products. Only in the cases of the reaction of nitriles 1a,b with benzene in TfOH, the hydrophenylation products, i.e., nitriles 2a,b, were obtained (Scheme 4; compare with synthesis of 2a,b in Scheme 1).
![[1860-5397-17-180-i4]](/bjoc/content/inline/1860-5397-17-180-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Hydrophenylation of nitriles 1a,b by benzene in TfOH leading to nitriles 2a,b.
Scheme 4: Hydrophenylation of nitriles 1a,b by benzene in TfOH leading to nitriles 2a,b.
In general, the comparison of reactions of 3-arylpropenenitriles (cinnamonitriles; ArCH=CHCN) (see our work [17]) and 3-arylpropynenitriles 1 (this work) under electrophilic activation reveals unambiguously that the electrophilic intermediates generated from acetylene nitriles 1 possess higher reactivity. Thus, the hydroarylation of 3-arylpropynenitriles 1 promoted by AlBr3 proceeds at room temperature (Scheme 1), whereas the same reaction of 3-arylpropenenitriles needs elevated temperature up to 80 °C [17]. Reactions of acetylene nitriles 1 with arenes in TfOH have complex character, contrary to 3-arylpropenenitriles, which react smoothly with arenes in TfOH at room temperature [17].
Conclusion
We have developed a novel transition-metal (Pd, Pt, Rh, etc.)-free procedure for the regio- and stereoselective hydroarylation of the carbon–carbon triple bond in 3-arylpropynenitriles by arenes under electrophilic activation by aluminum bromide AlBr3. The obtained 3,3-diarylpropenenitriles were cyclized into 3-arylindenones in triflic acid CF3SO3H (TfOH).
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization, and 1H and 13C NMR spectra of compounds. | ||
| Format: PDF | Size: 3.6 MB | Download |
References
-
Guan, Z.; Liu, Z.; Shi, W.; Chen, H. Tetrahedron Lett. 2017, 58, 3602–3606. doi:10.1016/j.tetlet.2017.07.104
Return to citation in text: [1] -
Perlmutter, P.; Chin Teo, C. Tetrahedron Lett. 1984, 25, 5951–5952. doi:10.1016/s0040-4039(01)81730-5
Return to citation in text: [1] -
Kishida, Y.; Terada, A. Chem. Pharm. Bull. 1968, 16, 1351–1359. doi:10.1248/cpb.16.1351
Return to citation in text: [1] -
Zhou, W.; Zhang, Y.; Li, P.; Wang, L. Org. Biomol. Chem. 2012, 10, 7184–7196. doi:10.1039/c2ob25969a
Return to citation in text: [1] -
Trofimov, B. A.; Andriyankova, L. V.; Nikitina, L. P.; Belyaeva, K. V.; Mal’kina, A. G.; Afonin, A. V.; Ushakov, I. A. Tetrahedron Lett. 2013, 54, 4693–4696. doi:10.1016/j.tetlet.2013.06.095
Return to citation in text: [1] -
Yadla, R.; Rehman, H.; Rao, J. M.; Mahesh, V. K. Tetrahedron 1989, 45, 7093–7098. doi:10.1016/s0040-4020(01)89177-8
Return to citation in text: [1] -
Zhang, J.; Zhang, Q.; Ji, X.; Meng, L.-G. Synlett 2019, 30, 1095–1099. doi:10.1055/s-0037-1610708
Return to citation in text: [1] -
Liu, P.; Clark, R. J.; Zhu, L. J. Org. Chem. 2018, 83, 5092–5103. doi:10.1021/acs.joc.8b00424
Return to citation in text: [1] -
Rama Rao, V. V. V. N. S.; Lingaiah, B. P. V.; Yadla, R.; Shanthan Rao, P.; Ravikumar, K.; Swamy, G. Y. S. K.; Narsimulu, K. J. Heterocycl. Chem. 2006, 43, 673–679. doi:10.1002/jhet.5570430321
Return to citation in text: [1] -
Trofimov, B. A.; Mal’kina, A. G.; Nosyreva, V. V.; Shemyakina, O. A.; Albanov, A. I.; Afonin, A. V.; Kazheva, O. N.; Alexandrov, G. G.; Dyachenko, O. A. Tetrahedron Lett. 2012, 53, 927–930. doi:10.1016/j.tetlet.2011.12.035
Return to citation in text: [1] -
Barton, P. Tetrahedron Lett. 2018, 59, 815–817. doi:10.1016/j.tetlet.2018.01.042
Return to citation in text: [1] -
Chen, Y.-R.; Duan, W.-L. J. Am. Chem. Soc. 2013, 135, 16754–16757. doi:10.1021/ja407373g
Return to citation in text: [1] -
Dückert, H.; Khedkar, V.; Waldmann, H.; Kumar, K. Chem. – Eur. J. 2011, 17, 5130–5137. doi:10.1002/chem.201003572
Return to citation in text: [1] -
Shi, Z.; Zhang, C.; Li, S.; Pan, D.; Ding, S.; Cui, Y.; Jiao, N. Angew. Chem., Int. Ed. 2009, 48, 4572–4576. doi:10.1002/anie.200901484
Return to citation in text: [1] -
McCauley, J. A.; Theberge, C. R.; Liverton, N. J. Org. Lett. 2000, 2, 3389–3391. doi:10.1021/ol006499j
Return to citation in text: [1] -
Gorbunova, Y.; Zakusilo, D. N.; Vasilyev, A. V. Tetrahedron Lett. 2019, 60, 961–964. doi:10.1016/j.tetlet.2019.02.047
Return to citation in text: [1] -
Gorbunova, Y.; Zakusilo, D. N.; Boyarskaya, I. A.; Vasilyev, A. V. Tetrahedron 2020, 76, 131264. doi:10.1016/j.tet.2020.131264
Return to citation in text: [1] [2] [3] [4] [5] -
Vasilyev, A. V. Russ. Chem. Rev. 2013, 82, 187–204. doi:10.1070/rc2013v082n03abeh004345
Return to citation in text: [1] -
Kazakova, A. N.; Vasilyev, A. V. Russ. J. Org. Chem. 2017, 53, 485–509. doi:10.1134/s1070428017040017
Return to citation in text: [1] -
Vasilyev, A. V. Adv. Org. Synth. 2018, 8, 81–120. doi:10.2174/9781681085647118080005
Return to citation in text: [1] -
Olah, G. A.; Klumpp, D. A. Superlectrophiles and their chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2008.
Return to citation in text: [1] -
Salnikov, G. E.; Genaev, A. M.; Vasiliev, V. G.; Shubin, V. G. Org. Biomol. Chem. 2012, 10, 2282–2288. doi:10.1039/c2ob06841a
Return to citation in text: [1] -
Masllorens, J.; Moreno-Mañas, M.; Pla-Quintana, A.; Pleixats, R.; Roglans, A. Synthesis 2002, 1903–1911. doi:10.1055/s-2002-33918
Return to citation in text: [1] -
Yamamoto, Y.; Asatani, T.; Kirai, N. Adv. Synth. Catal. 2009, 351, 1243–1249. doi:10.1002/adsc.200900067
Return to citation in text: [1] -
Yoo, K.; Kim, H.; Yun, J. Chem. – Eur. J. 2009, 15, 11134–11138. doi:10.1002/chem.200901262
Return to citation in text: [1] -
Matsuda-Sentou, W.; Shinmyozu, T. Eur. J. Org. Chem. 2000, 3195–3203. doi:10.1002/1099-0690(200009)2000:18<3195::aid-ejoc3195>3.0.co;2-1
Return to citation in text: [1] -
Nigam, R.; Babu, K. R.; Ghosh, T.; Kumari, B.; Khan, F. A.; Das, P.; Anindya, R. Chem. Biol. Drug Des. 2021, 97, 1170–1184. doi:10.1111/cbdd.13839
Return to citation in text: [1] -
Hu, B.; Cheng, X.; Hu, Y.; Liu, X.; Karaghiosoff, K.; Li, J. Angew. Chem., Int. Ed. 2021, 60, 15497–15502. doi:10.1002/anie.202103465
Return to citation in text: [1] -
Song, J.; Sun, H.; Sun, W.; Fan, Y.; Li, C.; Wang, H.; Xiao, K.; Qian, Y. Adv. Synth. Catal. 2019, 361, 5521–5527. doi:10.1002/adsc.201901309
Return to citation in text: [1] -
Zhang, Y.; Sun, K.; Lv, Q.; Chen, X.; Qu, L.; Yu, B. Chin. Chem. Lett. 2019, 30, 1361–1368. doi:10.1016/j.cclet.2019.03.034
Return to citation in text: [1] -
Ramesh, K.; Satyanarayana, G. Eur. J. Org. Chem. 2018, 4135–4146. doi:10.1002/ejoc.201800591
Return to citation in text: [1] -
Ryabukhin, D. S.; Fukin, G. K.; Vasilyev, A. V. Tetrahedron 2014, 70, 7865–7873. doi:10.1016/j.tet.2014.09.006
Return to citation in text: [1]
| 1. | Guan, Z.; Liu, Z.; Shi, W.; Chen, H. Tetrahedron Lett. 2017, 58, 3602–3606. doi:10.1016/j.tetlet.2017.07.104 |
| 2. | Perlmutter, P.; Chin Teo, C. Tetrahedron Lett. 1984, 25, 5951–5952. doi:10.1016/s0040-4039(01)81730-5 |
| 18. | Vasilyev, A. V. Russ. Chem. Rev. 2013, 82, 187–204. doi:10.1070/rc2013v082n03abeh004345 |
| 19. | Kazakova, A. N.; Vasilyev, A. V. Russ. J. Org. Chem. 2017, 53, 485–509. doi:10.1134/s1070428017040017 |
| 20. | Vasilyev, A. V. Adv. Org. Synth. 2018, 8, 81–120. doi:10.2174/9781681085647118080005 |
| 17. | Gorbunova, Y.; Zakusilo, D. N.; Boyarskaya, I. A.; Vasilyev, A. V. Tetrahedron 2020, 76, 131264. doi:10.1016/j.tet.2020.131264 |
| 16. | Gorbunova, Y.; Zakusilo, D. N.; Vasilyev, A. V. Tetrahedron Lett. 2019, 60, 961–964. doi:10.1016/j.tetlet.2019.02.047 |
| 17. | Gorbunova, Y.; Zakusilo, D. N.; Boyarskaya, I. A.; Vasilyev, A. V. Tetrahedron 2020, 76, 131264. doi:10.1016/j.tet.2020.131264 |
| 7. | Zhang, J.; Zhang, Q.; Ji, X.; Meng, L.-G. Synlett 2019, 30, 1095–1099. doi:10.1055/s-0037-1610708 |
| 8. | Liu, P.; Clark, R. J.; Zhu, L. J. Org. Chem. 2018, 83, 5092–5103. doi:10.1021/acs.joc.8b00424 |
| 9. | Rama Rao, V. V. V. N. S.; Lingaiah, B. P. V.; Yadla, R.; Shanthan Rao, P.; Ravikumar, K.; Swamy, G. Y. S. K.; Narsimulu, K. J. Heterocycl. Chem. 2006, 43, 673–679. doi:10.1002/jhet.5570430321 |
| 10. | Trofimov, B. A.; Mal’kina, A. G.; Nosyreva, V. V.; Shemyakina, O. A.; Albanov, A. I.; Afonin, A. V.; Kazheva, O. N.; Alexandrov, G. G.; Dyachenko, O. A. Tetrahedron Lett. 2012, 53, 927–930. doi:10.1016/j.tetlet.2011.12.035 |
| 11. | Barton, P. Tetrahedron Lett. 2018, 59, 815–817. doi:10.1016/j.tetlet.2018.01.042 |
| 12. | Chen, Y.-R.; Duan, W.-L. J. Am. Chem. Soc. 2013, 135, 16754–16757. doi:10.1021/ja407373g |
| 13. | Dückert, H.; Khedkar, V.; Waldmann, H.; Kumar, K. Chem. – Eur. J. 2011, 17, 5130–5137. doi:10.1002/chem.201003572 |
| 14. | Shi, Z.; Zhang, C.; Li, S.; Pan, D.; Ding, S.; Cui, Y.; Jiao, N. Angew. Chem., Int. Ed. 2009, 48, 4572–4576. doi:10.1002/anie.200901484 |
| 15. | McCauley, J. A.; Theberge, C. R.; Liverton, N. J. Org. Lett. 2000, 2, 3389–3391. doi:10.1021/ol006499j |
| 17. | Gorbunova, Y.; Zakusilo, D. N.; Boyarskaya, I. A.; Vasilyev, A. V. Tetrahedron 2020, 76, 131264. doi:10.1016/j.tet.2020.131264 |
| 3. | Kishida, Y.; Terada, A. Chem. Pharm. Bull. 1968, 16, 1351–1359. doi:10.1248/cpb.16.1351 |
| 4. | Zhou, W.; Zhang, Y.; Li, P.; Wang, L. Org. Biomol. Chem. 2012, 10, 7184–7196. doi:10.1039/c2ob25969a |
| 5. | Trofimov, B. A.; Andriyankova, L. V.; Nikitina, L. P.; Belyaeva, K. V.; Mal’kina, A. G.; Afonin, A. V.; Ushakov, I. A. Tetrahedron Lett. 2013, 54, 4693–4696. doi:10.1016/j.tetlet.2013.06.095 |
| 6. | Yadla, R.; Rehman, H.; Rao, J. M.; Mahesh, V. K. Tetrahedron 1989, 45, 7093–7098. doi:10.1016/s0040-4020(01)89177-8 |
| 17. | Gorbunova, Y.; Zakusilo, D. N.; Boyarskaya, I. A.; Vasilyev, A. V. Tetrahedron 2020, 76, 131264. doi:10.1016/j.tet.2020.131264 |
| 24. | Yamamoto, Y.; Asatani, T.; Kirai, N. Adv. Synth. Catal. 2009, 351, 1243–1249. doi:10.1002/adsc.200900067 |
| 25. | Yoo, K.; Kim, H.; Yun, J. Chem. – Eur. J. 2009, 15, 11134–11138. doi:10.1002/chem.200901262 |
| 17. | Gorbunova, Y.; Zakusilo, D. N.; Boyarskaya, I. A.; Vasilyev, A. V. Tetrahedron 2020, 76, 131264. doi:10.1016/j.tet.2020.131264 |
| 23. | Masllorens, J.; Moreno-Mañas, M.; Pla-Quintana, A.; Pleixats, R.; Roglans, A. Synthesis 2002, 1903–1911. doi:10.1055/s-2002-33918 |
| 27. | Nigam, R.; Babu, K. R.; Ghosh, T.; Kumari, B.; Khan, F. A.; Das, P.; Anindya, R. Chem. Biol. Drug Des. 2021, 97, 1170–1184. doi:10.1111/cbdd.13839 |
| 28. | Hu, B.; Cheng, X.; Hu, Y.; Liu, X.; Karaghiosoff, K.; Li, J. Angew. Chem., Int. Ed. 2021, 60, 15497–15502. doi:10.1002/anie.202103465 |
| 29. | Song, J.; Sun, H.; Sun, W.; Fan, Y.; Li, C.; Wang, H.; Xiao, K.; Qian, Y. Adv. Synth. Catal. 2019, 361, 5521–5527. doi:10.1002/adsc.201901309 |
| 30. | Zhang, Y.; Sun, K.; Lv, Q.; Chen, X.; Qu, L.; Yu, B. Chin. Chem. Lett. 2019, 30, 1361–1368. doi:10.1016/j.cclet.2019.03.034 |
| 31. | Ramesh, K.; Satyanarayana, G. Eur. J. Org. Chem. 2018, 4135–4146. doi:10.1002/ejoc.201800591 |
| 32. | Ryabukhin, D. S.; Fukin, G. K.; Vasilyev, A. V. Tetrahedron 2014, 70, 7865–7873. doi:10.1016/j.tet.2014.09.006 |
| 22. | Salnikov, G. E.; Genaev, A. M.; Vasiliev, V. G.; Shubin, V. G. Org. Biomol. Chem. 2012, 10, 2282–2288. doi:10.1039/c2ob06841a |
| 21. | Olah, G. A.; Klumpp, D. A. Superlectrophiles and their chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2008. |
| 26. | Matsuda-Sentou, W.; Shinmyozu, T. Eur. J. Org. Chem. 2000, 3195–3203. doi:10.1002/1099-0690(200009)2000:18<3195::aid-ejoc3195>3.0.co;2-1 |
© 2021 Gorbunova et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)