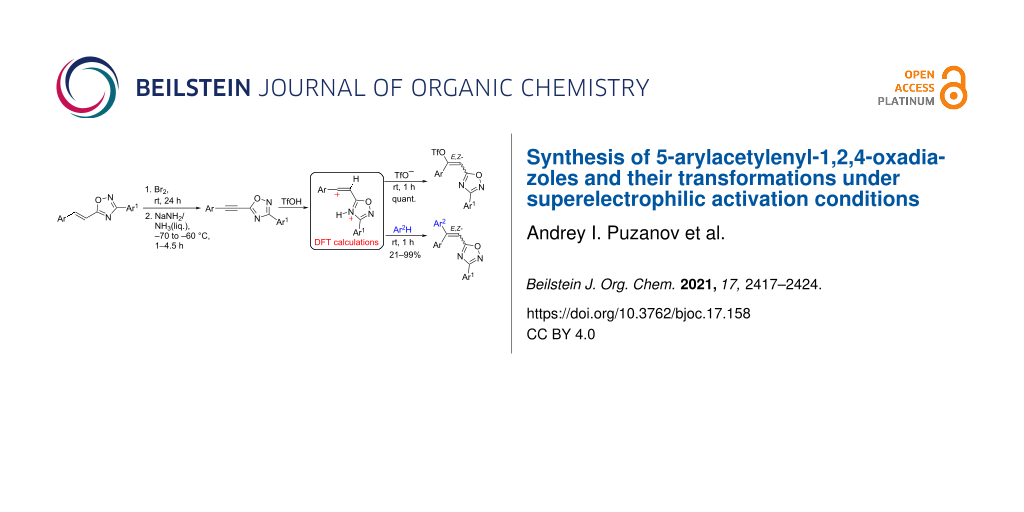
Abstract
Acetylene derivatives of 1,2,4-oxadiazoles, i.e., 5-(2-arylethynyl)-3-aryl-1,2,4-oxadiazoles, have been obtained, for the first time reported, from 5-(2-arylethenyl)-3-aryl-1,2,4-oxadiazoles by their bromination at the carbon–carbon double bond followed by di-dehydrobromination with NaNH2 in liquid NH3. The reaction of the acetylenyl-1,2,4-oxadiazoles with arenes in neat triflic acid TfOH (CF3SO3H) at room temperature for 1 h resulted in the formation of E/Z-5-(2,2-diarylethenyl)-3-aryl-1,2,4-oxadiazoles as products of regioselective hydroarylation of the acetylene bond. The addition of TfOH to the acetylene bond of these oxadiazoles quantitatively resulted in E/Z-vinyl triflates. The reactions of the cationic intermediates have been studied by DFT calculations and the reaction mechanisms are discussed.

Graphical Abstract
Introduction
1,2,4-Oxadiazoles have a great importance in chemistry, biology and medicine. Many drugs contain an 1,2,4-oxadiazole ring, such as butalamine [1], libexin [2], ataluren [3], oxolamine [4], and pleconaril [5]. Various oxadiazole derivatives show different kinds of activity against cancer [6,7], tuberculosis [8], Gram-positive bacteria [9], and they are used in treatment of epilepsy [10] and Alzheimer disease [11-13]. The synthesis of compounds of the 1,2,4-oxadiazole series is an actual task in organic and medicinal chemistry (see selected reviews on this topic [14-22]). However, among all the varieties of 1,2,4-oxadiazoles, their acetylenic derivatives are quite rare. To the best of our knowledge, there is only one example of 1,2,4-oxadiazole conjugated with an acetylene bond, which is 3-phenylethynyl-1,2,4-oxadiazole [23]. Up to the moment, there are no data on the preparation of 1,2,4-oxadiazoles containing a conjugated acetylenic substituent in the position 5 of the heterocyclic ring.
Based on our previous works on the chemistry of 1,2,4-oxadiazoles in superacids [24,25], we undertook this study on further investigation of the transformations of these heterocyclic compounds in electrophilic media. The main goals of this work were the synthesis of 5-arylacetylenyl-1,2,4-oxadiazoles and the study of their reactions with/without arenes under the conditions of superelectrophilic activation by the Brønsted superacid CF3SO3H (TfOH), the strong Lewis acids AlX3 (X = Cl, Br), or the acidic zeolite CBV-720.
Results and Discussion
The synthesis of 5-arylethynyl-1,2,4-oxadiazoles 3 was based on transformations of the corresponding 5-styryloxadiazoles, i.e., 5-(2-arylethenyl)-3-aryl-1,2,4-oxadiazoles 1a–g (Scheme 1). Bromination of the side chain carbon–carbon double bond in oxadiazoles 1a–g led to pairs of diastereomers of dibromo derivatives 2a–g. Then, several bases were tested for the di-dehydrobromination of compounds 2a–g. However, treatment of 2a–g in the following systems, KOH–EtOH (reflux, 2 h), BuLi–THF (−40 °C, 2 h), t-BuOK–THF (reflux, 2 h), or LiN(iPr)2–THF (−40 °C, 2 h), afforded complex mixtures of reaction products without desired acetylenyloxadiazoles 3. We succeeded to get compounds 3a–e by the reaction of 2a–e with sodium amide in liquid ammonia [NaNH2–NH3(liq.)] only at low temperature −70 to −60 °C (Scheme 1). However, the yields of target compounds were moderate 32–54% (for 3a–c,e) or even low 9% (for 3d). Running this reaction at higher temperature –50 to –40 °C led to a decrease of the yields of compounds 3. Apart from that, compounds 2f,g containing a 3-para-bromophenyl moiety in the heterocyclic core gave no corresponding 5-acetylenyloxadiazoles 3 in the system NaNH2–NH3(liq.), only mixtures of oligomeric materials were formed. Moreover, compound 3e was obtained as an inseparable mixture with styryloxadiazole 1e. The latter may be formed from 3e under the reduction by the solution that contained NaNH2. All these data point out the instability of 5-acetylenyloxadiazoles 3 in strong basic and nucleophilic media. Oxadiazoles 3, which were initially formed from compounds 2 in the system NaNH2–NH3(liq.), underwent further secondary transformations under nucleophilic reaction conditions, even at very low temperature –70 to –60 °C, that resulted in low to moderate yields of the target acetylene derivatives.
![[1860-5397-17-158-i1]](/bjoc/content/inline/1860-5397-17-158-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 5–arylethynyl-3-aryl-1,2,4-oxadiazoles 3a–e.
Scheme 1: Synthesis of 5–arylethynyl-3-aryl-1,2,4-oxadiazoles 3a–e.
Then, electrophilic reactions of 5-acetylenyloxadiazoles 3a–d in different acids were studied. In our recent study on the electrophilic activation of 5-styryl-1,2,4-oxadiazoles 1 [24], it was shown by means of NMR spectroscopy and DFT calculation that the protonation of these oxadiazoles in Brønsted superacids TfOH and FSO3H gave reactive N,C-diprotonated species. The protonation of oxadiazoles 1 takes place at the nitrogen N4 and the α-carbon of the side chain C=C bond. One would expect the formation of similar dications at the protonation of acetylenyloxadiazoles 3 in Brønsted superacids (see Table 1). Table 1 contains data on DFT calculations of cations Aa–d (N-protonated forms) and Ba–d (N,C-diprotonated forms) derived at the protonation of oxadiazoles 3a–d. Charge delocalization, contribution of atomic orbital into LUMO, global electrophilicity indices ω [26,27], and Gibbs free energies of protonation reactions with hydroxonium ion (H3O+) ΔG298 were calculated.
Table 1: Selected electronic characteristics for cations Aa–d and Ba–d calculated by DFT from protonation of oxadiazoles 3a–d.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-158-i6.svg?max-width=637&scale=1.0)
|
||||||||
| Species | EHOMO, eV | ELUMO, eV | ω,a eV | q(Cβ),b e | k(Cβ)LUMO,c % | q(N2),b e | q(N4),b e | ΔG298 of protonation, kJ/mol |
| Cations A (N-protonated species) | ||||||||
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-158-i7.svg?max-width=637&scale=1.0)
|
−7.44 | −3.56 | 3.9 | 0.23 | 7.2 | −0.13 | −0.49 | −80.8 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-158-i8.svg?max-width=637&scale=1.0)
|
−7.23 | −3.49 | 3.8 | 0.23 | 7.4 | −0.13 | −0.50 | −83.4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-158-i9.svg?max-width=637&scale=1.0)
|
−7.28 | −3.62 | 4.0 | 0.23 | 7.4 | −0.13 | −0.49 | −79.2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-158-i10.svg?max-width=637&scale=1.0)
|
−6.84 | −3.38 | 3.8 | −0.30 | 6.1 | −0.14 | −0.53 | −86.6 |
| Dications B (N,C-diprotonated species) | ||||||||
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-158-i11.svg?max-width=637&scale=1.0)
|
−7.83 | −5.28 | 8.4 | 0.47 | 30.0 | −0.11 | −0.47 | +18.0 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-158-i12.svg?max-width=637&scale=1.0)
|
−7.82 | −5.04 | 7.4 | 0.44 | 29.0 | −0.11 | −0.48 | −1.2 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-158-i13.svg?max-width=637&scale=1.0)
|
−7.82 | −5.25 | 8.3 | 0.45 | 24.3 | 0.1 | 0.4 | +18.3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-158-i14.svg?max-width=637&scale=1.0)
|
−7.80 | −4.63 | 6.1 | 0.40 | 16.7 | −0.11 | −0.48 | −28.8 |
aGlobal electrophilicity index ω = (EHOMO + ELUMO)2/8(ELUMO − EHOMO). bNatural charges. cContribution of atomic orbital into molecular orbital.
Big negative values of ΔG298 (−86.6 to −79.2 kJ/mol) of the first protonation step show that the formation of N-protonated species Aa–d is extremely energetically favorable. For the second protonation (reaction A→B) leading to dications Ba–d, the ΔG298 values vary from −28.6 to 18.3 kJ/mol. Although the second protonations are sometimes mildly endergonic (and hence there would be an unfavourable equilibrium between species A and B), the capture of the diaction B by a nucleophile is likely to be very exergonic and this can drive the reaction through to the products. Calculated electronic characteristic of these dications reveal their high electrophilicity, the indexes ω are 6.1–8.4 eV. Carbon Cβ bears a large positive charge (0.40–0.47 e) and gives a big contribution into LUMO (16.7–30%), pointing out that this carbon is a reactive electrophilic center by charge and orbital factors.
Thus, according our previous data on reactions of 5-styryl-1,2,4-oxadiazoles 1 [24] and results of the DFT calculations for protonation of 5-acetylenyl-1,2,4-oxadiazoles 3 (Table 1), one would propose the following reaction pathways for compounds 3 in Brønsted superacids (Scheme 2). Protonation of oxadiazole 3 affords dication B, which may react with counter anion of acid X− giving rise to vinyl derivatives 4. In the presence of nucleophilic arene molecules, species B should afford substances 5 as products of hydroarylation of the acetylene bond of the starting compounds 3.
![[1860-5397-17-158-i2]](/bjoc/content/inline/1860-5397-17-158-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible reaction mechanism for transformations of 5-acetylenyl-1,2,4-oxadiazoles 3 in Brønsted superacids.
Scheme 2: Plausible reaction mechanism for transformations of 5-acetylenyl-1,2,4-oxadiazoles 3 in Brønsted su...
Indeed, reaction of 5-acetylenyl-1,2,4-oxadiazoles 3a–c with excess of TfOH at room temperature for 1 h resulted in the quantitative preparation of E/Z-isomers of vinyl triflates 4a–c with a predominant formation of Z-isomers as product of an anti-addition of TfOH to the acetylene bond (Scheme 3). E/Z-Stereochemistry of compounds 4a–c was determined by H,F-NOESY correlation between vinyl proton (>C=CH–) and the CF3 group from the TfO substituent (see Supporting Information File 1). It should be noted that attempts of chromatographic separation of triflates 4a–c into individual E- and Z-isomers on silica gel led to a decrease of their yields and a change in E/Z-ratio. That reveals instability of these compounds on silica gel.
![[1860-5397-17-158-i3]](/bjoc/content/inline/1860-5397-17-158-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Quantitative formation of E/Z-vinyl triflates 4a–c from 5-acetylenyl-1,2,4-oxadiazoles 3a–c in TfOH.
Scheme 3: Quantitative formation of E/Z-vinyl triflates 4a–c from 5-acetylenyl-1,2,4-oxadiazoles 3a–c in TfOH....
In the same reaction in H2SO4 (Scheme 4), oxadiazole 3a gave the product of hydration of the acetylene bond (4d, yield of 65%) existing in solution as equilibrium between ketone and enol forms in a ratio of 1.2:1 according to NMR data (see Supporting Information File 1).
![[1860-5397-17-158-i4]](/bjoc/content/inline/1860-5397-17-158-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Formation of compound 4d from 5-acetylenyl-1,2,4-oxadiazole 3a in H2SO4.
Scheme 4: Formation of compound 4d from 5-acetylenyl-1,2,4-oxadiazole 3a in H2SO4.
Then, reactions of 5-acetylenyl-1,2,4-oxadiazole 3a–d with arenes (benzene and o-, m-, p-xylenes) in TfOH at room temperature for 1 h leading to products of hydroarylation of the acetylene bond, compounds (E/Z)-5a–g, were carried out (Scheme 5). This reaction gave E/Z-isomers 5b–g, their stereochemical configuration was determined by H,H NOESY correlations between the vinyl proton and aromatic protons (see Supporting Information File 1). In the case of the reaction with o-xylene, pairs of E/Z-isomers of two regioisomers, (E/Z)-5b and (E/Z)-5b1, were obtained.
![[1860-5397-17-158-i5]](/bjoc/content/inline/1860-5397-17-158-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Hydroarylation of 5-acetylenyl-1,2,4-oxadiazole 3a–d by arenes in TfOH leading to compounds E/Z-5a–g.
Scheme 5: Hydroarylation of 5-acetylenyl-1,2,4-oxadiazole 3a–d by arenes in TfOH leading to compounds E/Z-5a–g...
We also checked the reaction of oxadiazole 3a with benzene under the action of Lewis acids AlCl3, AlBr3 and acidic zeolite CBV-720 (Table 2). However, these Lewis acids showed unsatisfactory results leading to oligomeric materials (Table 2, entries 1 and 2). Probably, due to some secondary reactions of the formed compound 5a with AlCl3, AlBr3. The yield of target compound 5a in the reaction with zeolite was lower than in the same reaction in TfOH (compare entry 3 in Table 2 with data shown in Scheme 5). Thus, among the tested acidic reagents, TfOH showed better results for the hydroarylation of compounds 3.
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-158-i15.svg?max-width=637&scale=1.0)
Additionally, the reaction of oxadiazole 3a with benzene in TfOH (rt, 1 h) in the presence of cyclohexane, as a hydride ion source, was conducted to achieve the ionic hydrogenation of intermediate cationic species. However, no products of ionic hydrogenation were obtained, only the product of the hydrophenylation of the acetylene bond 5a was quantitatively isolated (compare with data shown in Scheme 5).
Conclusion
For the first time reported, we have synthesized 5-arylacetylene derivatives of 1,2,4-oxadiazoles, i.e., 5-(2-arylethynyl)-3-aryl-1,2,4-oxadiazoles. In the Brønsted superacid TfOH, these oxadiazoles react in a way of electrophilic addition to the acetylene bond. They give products of hydroarylation of the acetylene bond in the reaction with arenes or vinyl triflates in reaction with TfOH without arenes.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and 1H and 13C NMR spectra of compounds, as well as data of DFT calculations. | ||
| Format: PDF | Size: 5.7 MB | Download |
References
-
Palazzo, G.; Corsi, G. Arzneim. Forsch. 1962, 12, 545–549.
Return to citation in text: [1] -
Coupar, I. M.; Hedges, A.; Metcalfe, H. L.; Turner, P. J. Pharm. Pharmacol. 1969, 21, 474–475. doi:10.1111/j.2042-7158.1969.tb08294.x
Return to citation in text: [1] -
Jones, A. M.; Helm, J. M. Drugs 2009, 69, 1903–1910. doi:10.2165/11318500-000000000-00000
Return to citation in text: [1] -
Silvestrini, B. Minerva Med. 1960, 51, 4091–4094.
Return to citation in text: [1] -
Rotbart, H. A.; Webster, A. D. Clin. Infect. Dis. 2001, 32, 228–235. doi:10.1086/318452
Return to citation in text: [1] -
Zhang, H.-Z.; Kasibhatla, S.; Kuemmerle, J.; Kemnitzer, W.; Ollis-Mason, K.; Qiu, L.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S. X. J. Med. Chem. 2005, 48, 5215–5223. doi:10.1021/jm050292k
Return to citation in text: [1] -
Shamsi, F.; Hasan, P.; Queen, A.; Hussain, A.; Khan, P.; Zeya, B.; King, H. M.; Rana, S.; Garrison, J.; Alajmi, M. F.; Rizvi, M. M. A.; Zahid, M.; Imtaiyaz Hassan, M.; Abid, M. Bioorg. Chem. 2020, 98, 103754. doi:10.1016/j.bioorg.2020.103754
Return to citation in text: [1] -
Atmaram Upare, A.; Gadekar, P. K.; Sivaramakrishnan, H.; Naik, N.; Khedkar, V. M.; Sarkar, D.; Choudhari, A.; Mohana Roopan, S. Bioorg. Chem. 2019, 86, 507–512. doi:10.1016/j.bioorg.2019.01.054
Return to citation in text: [1] -
Janardhanan, J.; Chang, M.; Mobashery, S. Curr. Opin. Microbiol. 2016, 33, 13–17. doi:10.1016/j.mib.2016.05.009
Return to citation in text: [1] -
Mohammadi-Khanaposhtani, M.; Shabani, M.; Faizi, M.; Aghaei, I.; Jahani, R.; Sharafi, Z.; Shamsaei Zafarghandi, N.; Mahdavi, M.; Akbarzadeh, T.; Emami, S.; Shafiee, A.; Foroumadi, A. Eur. J. Med. Chem. 2016, 112, 91–98. doi:10.1016/j.ejmech.2016.01.054
Return to citation in text: [1] -
Querfurth, H. W.; LaFerla, F. M. N. Engl. J. Med. 2010, 362, 329–344. doi:10.1056/nejmra0909142
Return to citation in text: [1] -
Jiang, C.-S.; Fu, Y.; Zhang, L.; Gong, J.-X.; Wang, Z.-Z.; Xiao, W.; Zhang, H.-Y.; Guo, Y.-W. Bioorg. Med. Chem. Lett. 2015, 25, 216–220. doi:10.1016/j.bmcl.2014.11.068
Return to citation in text: [1] -
Mei, W.-w.; Ji, S.-s.; Xiao, W.; Wang, X.-d.; Jiang, C.-s.; Ma, W.-q.; Zhang, H.-y.; Gong, J.-x.; Guo, Y.-w. Monatsh. Chem. 2017, 148, 1807–1815. doi:10.1007/s00706-017-1993-x
Return to citation in text: [1] -
Hemming, K. J. Chem. Res., Synop. 2001, 209–216. doi:10.3184/030823401103169603
Return to citation in text: [1] -
Kayukova, L. A. Pharm. Chem. J. 2005, 39, 539–547. doi:10.1007/s11094-006-0017-7
Return to citation in text: [1] -
Hemming, K. 1.2.4-Oxadiazoles. In Five-membered Rings: Triazoles, Oxadiazoles, Thiadiazoles and their Fused Carbocyclic Derivatives; Zhdankin, V. V., Ed.; Comprehensive Heterocyclic Chemistry III, Vol. 5; Elsevier, 2008; pp 243–314. doi:10.1016/b978-008044992-0.00504-6
Return to citation in text: [1] -
Pace, A.; Pierro, P. Org. Biomol. Chem. 2009, 7, 4337–4348. doi:10.1039/b908937c
Return to citation in text: [1] -
Bora, R. O.; Dar, B.; Pradhan, V.; Farooqui, M. Mini-Rev. Med. Chem. 2014, 14, 355–369. doi:10.2174/1389557514666140329200745
Return to citation in text: [1] -
Pace, A.; Buscemi, S.; Piccionello, A. P.; Pibiri, I. Adv. Heterocycl. Chem. 2015, 116, 85–136. doi:10.1016/bs.aihch.2015.05.001
Return to citation in text: [1] -
Piccionello, A. P.; Pace, A.; Buscemi, S. Chem. Heterocycl. Compd. 2017, 53, 936–947. doi:10.1007/s10593-017-2154-1
Return to citation in text: [1] -
Aggarwal, S.; Goyal, A.; Kaur, R. Res. J. Pharm. Technol. 2020, 13, 5026–5033. doi:10.5958/0974-360x.2020.00880.x
Return to citation in text: [1] -
Lelyukh, M.; Demchuk, I.; Harkov, S.; Chaban, T.; Drapak, I.; Chaban, I.; Shelepeten, L.; Matiychuk, V. Biointerface Res. Appl. Chem. 2020, 10 (4), 5960–5971. doi:10.33263/briac104.960971
Return to citation in text: [1] -
Claisse, J. A.; Foxton, M. W.; Gregory, G. I.; Sheppard, A. H.; Tiley, E. P.; Warburton, W. K.; Wilson, M. J. J. Chem. Soc., Perkin Trans. 1 1973, 2241–2249. doi:10.1039/p19730002241
Return to citation in text: [1] -
Zalivatskaya, A. S.; Ryabukhin, D. S.; Tarasenko, M. V.; Ivanov, A. Y.; Boyarskaya, I. A.; Grinenko, E. V.; Osetrova, L. V.; Kofanov, E. R.; Vasilyev, A. V. Beilstein J. Org. Chem. 2017, 13, 883–894. doi:10.3762/bjoc.13.89
Return to citation in text: [1] [2] [3] -
Golushko, A. A.; Khoroshilova, O. V.; Vasilyev, A. V. J. Org. Chem. 2019, 84, 7495–7500. doi:10.1021/acs.joc.9b00812
Return to citation in text: [1] -
Parr, R. G.; Szentpály, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x
Return to citation in text: [1] -
Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p
Return to citation in text: [1]
| 5. | Rotbart, H. A.; Webster, A. D. Clin. Infect. Dis. 2001, 32, 228–235. doi:10.1086/318452 |
| 26. | Parr, R. G.; Szentpály, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x |
| 27. | Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p |
| 24. | Zalivatskaya, A. S.; Ryabukhin, D. S.; Tarasenko, M. V.; Ivanov, A. Y.; Boyarskaya, I. A.; Grinenko, E. V.; Osetrova, L. V.; Kofanov, E. R.; Vasilyev, A. V. Beilstein J. Org. Chem. 2017, 13, 883–894. doi:10.3762/bjoc.13.89 |
| 3. | Jones, A. M.; Helm, J. M. Drugs 2009, 69, 1903–1910. doi:10.2165/11318500-000000000-00000 |
| 24. | Zalivatskaya, A. S.; Ryabukhin, D. S.; Tarasenko, M. V.; Ivanov, A. Y.; Boyarskaya, I. A.; Grinenko, E. V.; Osetrova, L. V.; Kofanov, E. R.; Vasilyev, A. V. Beilstein J. Org. Chem. 2017, 13, 883–894. doi:10.3762/bjoc.13.89 |
| 25. | Golushko, A. A.; Khoroshilova, O. V.; Vasilyev, A. V. J. Org. Chem. 2019, 84, 7495–7500. doi:10.1021/acs.joc.9b00812 |
| 2. | Coupar, I. M.; Hedges, A.; Metcalfe, H. L.; Turner, P. J. Pharm. Pharmacol. 1969, 21, 474–475. doi:10.1111/j.2042-7158.1969.tb08294.x |
| 24. | Zalivatskaya, A. S.; Ryabukhin, D. S.; Tarasenko, M. V.; Ivanov, A. Y.; Boyarskaya, I. A.; Grinenko, E. V.; Osetrova, L. V.; Kofanov, E. R.; Vasilyev, A. V. Beilstein J. Org. Chem. 2017, 13, 883–894. doi:10.3762/bjoc.13.89 |
| 10. | Mohammadi-Khanaposhtani, M.; Shabani, M.; Faizi, M.; Aghaei, I.; Jahani, R.; Sharafi, Z.; Shamsaei Zafarghandi, N.; Mahdavi, M.; Akbarzadeh, T.; Emami, S.; Shafiee, A.; Foroumadi, A. Eur. J. Med. Chem. 2016, 112, 91–98. doi:10.1016/j.ejmech.2016.01.054 |
| 14. | Hemming, K. J. Chem. Res., Synop. 2001, 209–216. doi:10.3184/030823401103169603 |
| 15. | Kayukova, L. A. Pharm. Chem. J. 2005, 39, 539–547. doi:10.1007/s11094-006-0017-7 |
| 16. | Hemming, K. 1.2.4-Oxadiazoles. In Five-membered Rings: Triazoles, Oxadiazoles, Thiadiazoles and their Fused Carbocyclic Derivatives; Zhdankin, V. V., Ed.; Comprehensive Heterocyclic Chemistry III, Vol. 5; Elsevier, 2008; pp 243–314. doi:10.1016/b978-008044992-0.00504-6 |
| 17. | Pace, A.; Pierro, P. Org. Biomol. Chem. 2009, 7, 4337–4348. doi:10.1039/b908937c |
| 18. | Bora, R. O.; Dar, B.; Pradhan, V.; Farooqui, M. Mini-Rev. Med. Chem. 2014, 14, 355–369. doi:10.2174/1389557514666140329200745 |
| 19. | Pace, A.; Buscemi, S.; Piccionello, A. P.; Pibiri, I. Adv. Heterocycl. Chem. 2015, 116, 85–136. doi:10.1016/bs.aihch.2015.05.001 |
| 20. | Piccionello, A. P.; Pace, A.; Buscemi, S. Chem. Heterocycl. Compd. 2017, 53, 936–947. doi:10.1007/s10593-017-2154-1 |
| 21. | Aggarwal, S.; Goyal, A.; Kaur, R. Res. J. Pharm. Technol. 2020, 13, 5026–5033. doi:10.5958/0974-360x.2020.00880.x |
| 22. | Lelyukh, M.; Demchuk, I.; Harkov, S.; Chaban, T.; Drapak, I.; Chaban, I.; Shelepeten, L.; Matiychuk, V. Biointerface Res. Appl. Chem. 2020, 10 (4), 5960–5971. doi:10.33263/briac104.960971 |
| 9. | Janardhanan, J.; Chang, M.; Mobashery, S. Curr. Opin. Microbiol. 2016, 33, 13–17. doi:10.1016/j.mib.2016.05.009 |
| 23. | Claisse, J. A.; Foxton, M. W.; Gregory, G. I.; Sheppard, A. H.; Tiley, E. P.; Warburton, W. K.; Wilson, M. J. J. Chem. Soc., Perkin Trans. 1 1973, 2241–2249. doi:10.1039/p19730002241 |
| 8. | Atmaram Upare, A.; Gadekar, P. K.; Sivaramakrishnan, H.; Naik, N.; Khedkar, V. M.; Sarkar, D.; Choudhari, A.; Mohana Roopan, S. Bioorg. Chem. 2019, 86, 507–512. doi:10.1016/j.bioorg.2019.01.054 |
| 6. | Zhang, H.-Z.; Kasibhatla, S.; Kuemmerle, J.; Kemnitzer, W.; Ollis-Mason, K.; Qiu, L.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S. X. J. Med. Chem. 2005, 48, 5215–5223. doi:10.1021/jm050292k |
| 7. | Shamsi, F.; Hasan, P.; Queen, A.; Hussain, A.; Khan, P.; Zeya, B.; King, H. M.; Rana, S.; Garrison, J.; Alajmi, M. F.; Rizvi, M. M. A.; Zahid, M.; Imtaiyaz Hassan, M.; Abid, M. Bioorg. Chem. 2020, 98, 103754. doi:10.1016/j.bioorg.2020.103754 |
| 11. | Querfurth, H. W.; LaFerla, F. M. N. Engl. J. Med. 2010, 362, 329–344. doi:10.1056/nejmra0909142 |
| 12. | Jiang, C.-S.; Fu, Y.; Zhang, L.; Gong, J.-X.; Wang, Z.-Z.; Xiao, W.; Zhang, H.-Y.; Guo, Y.-W. Bioorg. Med. Chem. Lett. 2015, 25, 216–220. doi:10.1016/j.bmcl.2014.11.068 |
| 13. | Mei, W.-w.; Ji, S.-s.; Xiao, W.; Wang, X.-d.; Jiang, C.-s.; Ma, W.-q.; Zhang, H.-y.; Gong, J.-x.; Guo, Y.-w. Monatsh. Chem. 2017, 148, 1807–1815. doi:10.1007/s00706-017-1993-x |
© 2021 Puzanov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)