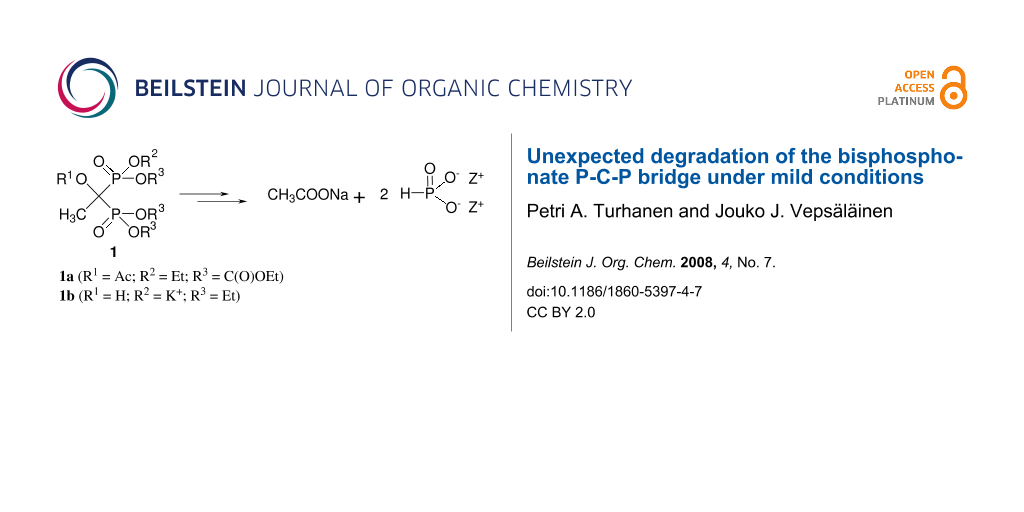
Abstract
Unexpected degradation of the P-C-P bridge from novel bisphosphonate derivative 1a and known etidronate trimethyl ester (1b) has been observed under mild reaction conditions. A proposed reaction mechanism for the unexpected degradation of 1a and 1b is also reported.

Graphical Abstract
Background
Bisphosphonates (BPs) are analogs of naturally occurring pyrophosphate, where the chemically and enzymatically labile P-O-P bridge has been replaced with a P-C-P bridge, making these compounds relatively resistant to chemical hydrolysis and completely resistant to enzymatic hydrolysis (Figure 1) [1-5]. These BP compounds bind strongly to calcium phosphate and inhibit its formation, aggregation and dissolution [6]. The affinity for the bone mineral represents the basis for their use in the treatment of many diseases associated with increased bone resorption, such as metastatic bone disease, Paget's disease and osteoporosis [1-6]. As described above, the BPs have been used for decades in the therapy of bone diseases but recently these compounds have been found to be active in many other fields, such as in the treatment of parasitic diseases [7-11] and atherosclerosis [12]. Furthermore, the BPs have been shown to be effective against calcifying nanoparticles (CNPs, known also as nanobacteria) which may be responsible for several human diseases where calcium phosphate deposition is a hallmark, e.g. cardiovascular diseases, kidney stones, urological diseases, e.g. prostatitis, many cancers and various forms of autoimmune diseases [13,14]. Therefore it is very important to understand the chemistry of BPs in detail.
![[1860-5397-4-7-1]](/bjoc/content/figures/1860-5397-4-7-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of etidronate, pyrophosphate and general structure of bisphosphonates.
Figure 1: Structures of etidronate, pyrophosphate and general structure of bisphosphonates.
Etidronate, (1-hydroxyethylidene)-1,1-bisphosphonic acid (HEBPA) disodium salt, is one of the earliest synthesized and is the most extensively investigated BP compound, still being in clinical use today (Figure 1) [1-6,15]. Our group has designed, synthesized and studied in vitro several different etidronate and alendronate derivatives to act as biodegradable prodrugs of these drugs [16-23]. During our ongoing study to prepare new, possibly bioreversible BP derivatives, we observed unexpected degradation of the P-C-P bridge under mild reaction conditions in two of the prepared etidronate derivatives. Earlier, Szymczak et. al. [24] have described the formation of H-phosphonate (also known as phosphite) and phosphate components from a phosphonate-phosphate compound (same kind of structure as 8 in Scheme 1) either in CH3CN/Et3N/H2O (v/v) or phosphate buffer, pH 7.4 at 37 °C. Szajnman et. al. [25] has reported loss of two molecules of phosphite in tetraethyl oxirane-2,2-diylbis(phosphonate); however the kind of degradation which we will discuss in this paper has not been previously reported.
![[1860-5397-4-7-i2]](/bjoc/content/inline/1860-5397-4-7-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Degradation of trimethyl ester of etidronate (1b) and stability of the tetramethyl ester (1c, R=R`=Me) and P,P`-dimethyl ester (1d, R=Me, R`=Na+) of etidronate. Reagents and conditions: i) 1 drop of 6 M NaOH, H2O, 1 h, rt, (measured pH was ≥ 11); ii) 5 equiv triethylamine, H2O, 1 h, 60 °C; iii) when R=R`=Me (1c), 1 eq, triethylamine, H2O, 10 min. ca. 98% conversion. iv) when R=Me, R`=Na+ (1d), 5 equiv NaOH, H2O, overnight, reflux.
Scheme 1: Degradation of trimethyl ester of etidronate (1b) and stability of the tetramethyl ester (1c, R=R`=...
Results and Discussion
As mentioned in the introduction, the P-C-P bridge of BPs has been reported to be relatively stable against chemical hydrolysis, however here we report the unexpectedly easy degradation of two etidronate derivatives into acetate and phosphite moieties. In our ongoing study to prepare novel biodegradable BP derivatives, a new carbonate derivative of etidronate was synthesised. The synthesis was started from the known acetylated etidronic acid [21] (5, see Scheme 2) by treating it with ethyl chloroformate and sodium carbonate. The NMR spectroscopy results were surprising since they pointed to the formation of a novel etidronate derivative 1a (see Scheme 2). In the 31P NMR spectrum, there were four doublets (1:1:1:1) due to the presence of two diastereomers. The 1H NMR spectrum contained two complicated splitting patterns at approx. 4.46 and 4.28 ppm, their integral ratio was 1 to 3, respectively, indicating two different kinds of -OCH2 groups in ratio 1:3. After inspection of 13C NMR spectra and the ESI-MS results, we concluded that the prepared molecule had the unanticipated structure of 1a and not the expected structure where R2=R3=C(O)OEt (see Scheme 2). To confirm the selective formation of 1a, the synthesis was repeated several times, but the result was always the same (formation of 1a was observed in all experiments), though in some experiments a transesterification of the acetyl group to C(O)OEt group was observed in yields of 0–13% as confirmed by the 1H and 31P NMR spectra. We were unable to provide any direct explanation for the variation in the transesterification proportion. Etidronic acid was also tested as a starting material to prepare a derivative such as 1a [C(O)OEt group instead of Ac group], but the reaction did not occur under the same conditions as those used in the preparation of 1a. Our subsequent studies with derivative 1a led us to another very surprising result, which occurred when 4 equiv of NaOH (40% NaOH in H2O) were added to the solution of 1a in MeOH and stirred for 30 minutes at room temperature. After evaporation of the reaction mixture to dryness, the residue contained almost exclusively (>95% degradation was observed) sodium acetate 6 and phosphites 2–4 (compound 4 can be also called phosphorous acid monosodium salt) as can be seen in Scheme 2. Compounds 2–4 were readily characterized by their P-H chemical shifts and characteristic 1JHP coupling constants (ca. 600 Hz). In the 31P NMR spectrum, there were three different monophosphorus components confirmed to be compounds 2–4. Two moles of acetate 6 were detected compared to one mole of the total amount of phosphites 2–4 which was the expected result. Interestingly, the decomposition mixture of 1a contained not only monoethyl phosphite 2 and phosphite 4 but also monomethyl derivative 3 (according the 31P NMR spectrum, the ratio was approx.: 1:0.86:1, respectively, see Supporting Information File 1, S13). The formation of this monomethyl phosphite 3 under the conditions used (see Scheme 2, procedure ii) can be explained based on: 1) partial transesterification of bisphosphonate 1a before degradation of P-C-P bridge, 2) partial esterification of phosphonate group after the carbonate groups (R3) decomposition from compound 1a (this is proposed to occur rapidly after the addition of 40% NaOH) and before the degradation of P-C-P bridge, 3) partial transesterification of 2, and 4) esterification of 4 (see Scheme 2).
![[1860-5397-4-7-i1]](/bjoc/content/inline/1860-5397-4-7-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of BP derivative 1a (R2 = Et, R3 = C(O)OEt) and its degradation to acetate 6 and phosphites 2–4. Reagents and conditions: i) excess ClC(O)OEt, 6 equiv Na2CO3, reflux, overnight, 55%; ii) 4 equiv NaOH (40% NaOH in H2O), MeOH, 30 min, rt.
Scheme 2: Preparation of BP derivative 1a (R2 = Et, R3 = C(O)OEt) and its degradation to acetate 6 and phosph...
These unexpected degradation results we observed for the HEBPA derivative 1a led us to examine what would happen to more simple derivatives of HEBPA, such as trimethyl (1b), tetramethyl (1c) and P,P`-dimethyl (1d) esters of etidronate under the same kinds of conditions (see Scheme 1). Compounds 1b–d were prepared as reported elsewhere [20,26-28]. Again very surprising results were obtained. Trimethyl ester of etidronate (1b) was degraded to the acetate 6 and phosphorous acid salt 4, under even milder conditions than the degradation of 1a (50 mg of 1b in 1 ml H2O and 1 drop of 6 M NaOH was stirred for 1 hour at rt; measured pH was ≥ 11; see Scheme 1). Tetramethyl (1c) or P,P`-dimethyl ester were not degraded under the same conditions, only the formation of phosphonate-phosphate derivative 8 from 1c was observed as expected in the light of the earlier results concern the rearrangement process [17,20,21,29-32]. This rearrangement of 1c to 8 was observed to happen rapidly and almost completely (98% conversion) when 1 equiv of triethylamine was present in water (see Scheme 1).
Compound 1b was selectively degraded to the phosphite 3 and acetyl phosphonate 7 when 5 equiv of triethylamine was used in H2O (see Scheme 1). Dialkyl acetylphosphonates and dialkyl phosphites are common starting materials for the synthesis of tetraalkyl esters of HEBPA [26,27], but this is the first time when the “reverse” synthesis has been reported. P,P`-dimethyl ester 1d did not degrade to compounds 3 and 7 or 6 and 4 even when refluxed overnight with 5 equiv NaOH in H2O.
The decomposition mechanism for 1b can be explained in two ways; either via a decomposition mechanism resembling the reversible route of the formation of tetraesters (see Scheme 3 route a), since e.g., 1c, are prepared from phosphites, H-P(O)(OMe)2, and phosphonates, MeCOP(O)(OMe)2, or route b resembling the rearrangement process [19]. The driving force in both reactions is the formation of three charged molecules from one P-C-P compound since this is a highly entropically favoured process. Decomposition of the first P-C bond starts with deprotonation of the hydroxyl group followed by elimination of the methyl phosphite and the formation of ketone (route a) or by nucleophilic attack of the oxygen of the ionized phosphate on the bridging carbon to release dimethyl phosphite and the oxirane ring containing derivative (route b). In route a, water or hydroxide ion attacks the carbonyl carbon and P-C bond cleavage occurs giving rise to acetic acid and dimethyl phosphite which can undergo a further reaction with water or hydroxide to give methyl phosphite. In route b, the attack of water on the carbon of oxirane ring yields hydrate followed by elimination of methyl phosphite and acetic acid. We believe that route a is more probable, since during the reaction with a weaker base, such as triethylamine, only the first P-C bond is cleaved and products 3 and 7 are observed. On the other hand, decomposition of 1a is more likely to follow route b.
![[1860-5397-4-7-i3]](/bjoc/content/inline/1860-5397-4-7-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed reaction mechanism for 1b decomposition.
Scheme 3: Proposed reaction mechanism for 1b decomposition.
The proposed decomposition mechanism for 1a (see Scheme 4) is more complicated. The reaction starts with the hydrolysis of one carbonate ester leading to a monoanion comparable to 1b. After this step, the decomposition can continue following routes that are similar to either route a or route b in Scheme 3. The other possibility, route b (in Scheme 4), is a nucleophilic attack of oxygen to the bridging carbon and the formation of an oxirane ring containing derivative, since the adjacent acetate group is a rather good leaving group. Subsequently, P-C-bond decomposition will follow the same mechanism as reported in Scheme 3.
![[1860-5397-4-7-i4]](/bjoc/content/inline/1860-5397-4-7-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed reaction mechanism for 1a decomposition.
Scheme 4: Proposed reaction mechanism for 1a decomposition.
The initial reaction in Scheme 3 also explains the formation of rearranged product 8 from tetraester 1c (this rearrangement is proposed to happen via oxirane ring) [19], since the charged oxygen is a good nucleophile compared to OH-group and far better than oxygen bound to phosphorus with a double bond (P=O).
All of the compounds were easily identified by their 1H, 13C and 31P NMR spectra. In 31P NMR signals for the phosphites 2, 3 and 4 were 6.26 ppm, 8.47 ppm and 5.81 ppm, respectively and 0.10 ppm for acetylphosphonate 7. These values were comparable to those reported earlier [27].
Conclusion
In conclusion, a novel carbonate derivative of etidronate (1a) was prepared by the reaction of acetylated etidronic acid with ethyl chloroformate and sodium carbonate. Compound 1a was found to undergo remarkably facile cleavage of the P-C bond under mild basic conditions. The trimethyl ester of etidronate (1b) was also found to undergo readily P-C bond cleavage under similar conditions. The trimethyl ester of etidronate (1b) was also observed to be degraded to phosphite 3 and acetylphosphonate 7 when mixed in H2O containing 5 equiv of triethylamine. Some mechanisms to explain these behaviors have been proposed though further investigations will be necessary to confirm the proposed degradation pathways.
Supporting Information
| Supporting Information File 1: Unexpected degradation of bisphosphonate P-C-P bridge under mild conditions. Experimental procedures, full spectroscopic data and NMR spectra for the novel compound 1a and NMR spectra for the degradation studies of 1a and 1b. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Fleisch, H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient; The Parthenon Publishing Group Inc.: New York, 1995.
Return to citation in text: [1] [2] [3] -
Papapoulos, S. E.; Landman, J. O.; Bijvoet, O. L. M.; Löwik, C. W. G. M.; Valkema, R.; Pauwels, E. K. J.; Vermeij, P. Bone 1992, 13 (Suppl. 1), S41–S49.
Return to citation in text: [1] [2] [3] -
Yates, A. J.; Rodan, G. A. Drug Discovery Today 1998, 3, 69–78. doi:10.1016/S1359-6446(97)01134-3
Return to citation in text: [1] [2] [3] -
Papapoulos, S. E. Am. J. Med. 1993, 95 (5, Suppl. 1), S48–S52. doi:10.1016/0002-9343(93)90383-Z
Return to citation in text: [1] [2] [3] -
Giannini, S.; D'Angelo, A.; Sartori, L.; Passeri, G.; Dalle Carbonare, L.; Crepaldi, G. Obstet. Gynecol. 1996, 88, 431–436. doi:10.1016/0029-7844(96)00171-8
Return to citation in text: [1] [2] [3] -
Fleisch, H. Drugs 1991, 42, 919–944.
Return to citation in text: [1] [2] [3] -
Szajnman, S. H.; Montalvetti, A.; Wang, Y.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2003, 13, 3231–3235. doi:10.1016/S0960-894X(03)00663-2
Return to citation in text: [1] -
Szajnman, S. H.; Bailey, B. N.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2001, 11, 789–792. doi:10.1016/S0960-894X(01)00057-9
Return to citation in text: [1] -
Martin, M. B.; Sanders, J. M.; Kendrick, H.; de Luca-Fradley, K.; Lewis, J. C.; Grimley, J. S.; Van Brussel, E. M.; Olsen, J. R.; Meints, G. A.; Burzynska, A.; Kafarski, P.; Croft, S. L.; Oldfield, E. J. Med. Chem. 2002, 45, 2904–2914. doi:10.1021/jm0102809
Return to citation in text: [1] -
Garzoni, L. R.; Caldera, A.; Nazareth L. Meirelles, M.; Castro, S. L.; Docampo, R.; Meints, G. A.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 273–285. doi:10.1016/j.ijantimicag.2003.07.020
Return to citation in text: [1] -
Garzoni, L. R.; Waghabi, M. C.; Baptista, M. M.; Castro, S. L.; Nazareth L. Meirelles, M.; Britto, C. C.; Docampo, R.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 286–290. doi:10.1016/j.ijantimicag.2003.07.019
Return to citation in text: [1] -
Ylitalo, R. Gen. Pharmacol. 2000, 35, 287–296. doi:10.1016/S0306-3623(01)00121-5
Return to citation in text: [1] -
Kajander, E. O. Lett. Appl. Microbiol. 2006, 42, 549–552. doi:10.1111/j.1472-765X.2006.01945.x
Return to citation in text: [1] -
Aho, K.; Soininen, T.; Turhanen, P. A.; Kajander, E. O.; Vepsäläinen, J. J. Unpublished results.
Return to citation in text: [1] -
Major, P. P.; Lipton, A.; Berenson, J.; Hortobagyi, G. Cancer (N. Y.) 2000, 88, 6–14. doi:10.1002/(SICI)1097-0142(20000101)88:1<6::AID-CNCR3>3.0.CO;2-D
Return to citation in text: [1] -
Turhanen, P. A.; Niemi, R.; Peräkylä, M.; Järvinen, T.; Vepsäläinen, J. J. Org. Biomol. Chem. 2003, 1, 3223–3226. doi:10.1039/b305979k
Return to citation in text: [1] -
Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 2119–2121. doi:10.1055/s-2005-869984
Return to citation in text: [1] [2] -
Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 3063–3066. doi:10.1055/s-2005-916032
Return to citation in text: [1] -
Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3
Return to citation in text: [1] [2] [3] -
Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Synthesis 2001, 633–637. doi:10.1055/s-2001-12353
Return to citation in text: [1] [2] [3] -
Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2004, 992–994. doi:10.1055/s-2004-822345
Return to citation in text: [1] [2] [3] -
Turhanen, P. A.; Vepsäläinen, J. J. Beilstein J. Org. Chem. 2006, 2, No. 2. doi:10.1186/1860-5397-2-2
Return to citation in text: [1] -
Vepsäläinen, J. J. Curr. Med. Chem. 2002, 9, 1201–1208.
Return to citation in text: [1] -
Szymczak, M.; Szymańska, A.; Stawiński, J.; Boryski, J.; Kraszewski, A. Org. Lett. 2003, 5, 3571–3573. doi:10.1021/ol035166u
Return to citation in text: [1] -
Szajnman, S. H.; García Liñares, G.; Moro, P.; Rodriguez, J. B. Eur. J. Org. Chem. 2005, 3687–3696. doi:10.1002/ejoc.200500097
Return to citation in text: [1] -
Nicholson, D. A.; Vaughn, H. J. Org. Chem. 1971, 36, 3843–3845. doi:10.1021/jo00823a621
Return to citation in text: [1] [2] -
Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Phosphorus, Sulfur Silicon Relat. Elem. 2001, 170, 115–133. doi:10.1080/10426500108040589
Return to citation in text: [1] [2] [3] -
Van Gelder, J. M.; Breuer, E.; Ornoy, A.; Schlossman, A.; Patlas, N.; Golomb, G. Bone 1995, 16, 511–520. doi:10.1016/8756-3282(95)00081-N
Return to citation in text: [1] -
Vachal, P.; Hale, J. J.; Lu, Z.; Streckfuss, E. C.; Mills, S. G.; MacCoss, M.; Yin, D. H.; Algayer, K.; Manser, K.; Kesisoglou, F.; Ghosh, S.; Alani, L. L. J. Med. Chem. 2006, 49, 3060–3063. doi:10.1021/jm060398v
Return to citation in text: [1] -
Fitch, S. J.; Moedritzer, K. J. Am. Chem. Soc. 1962, 84, 1876–1879. doi:10.1021/ja00869a022
Return to citation in text: [1] -
Ruel, R.; Bouvier, J.-P.; Young, R. N. J. Org. Chem. 1995, 60, 5209–5213. doi:10.1021/jo00121a044
Return to citation in text: [1] -
Tromelin, A.; El Manouni, D.; Burgada, R. Phosphorus Sulfur Relat. Elem. 1986, 27, 301–312. doi:10.1080/03086648608072784
Return to citation in text: [1]
| 27. | Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Phosphorus, Sulfur Silicon Relat. Elem. 2001, 170, 115–133. doi:10.1080/10426500108040589 |
| 1. | Fleisch, H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient; The Parthenon Publishing Group Inc.: New York, 1995. |
| 2. | Papapoulos, S. E.; Landman, J. O.; Bijvoet, O. L. M.; Löwik, C. W. G. M.; Valkema, R.; Pauwels, E. K. J.; Vermeij, P. Bone 1992, 13 (Suppl. 1), S41–S49. |
| 3. | Yates, A. J.; Rodan, G. A. Drug Discovery Today 1998, 3, 69–78. doi:10.1016/S1359-6446(97)01134-3 |
| 4. | Papapoulos, S. E. Am. J. Med. 1993, 95 (5, Suppl. 1), S48–S52. doi:10.1016/0002-9343(93)90383-Z |
| 5. | Giannini, S.; D'Angelo, A.; Sartori, L.; Passeri, G.; Dalle Carbonare, L.; Crepaldi, G. Obstet. Gynecol. 1996, 88, 431–436. doi:10.1016/0029-7844(96)00171-8 |
| 12. | Ylitalo, R. Gen. Pharmacol. 2000, 35, 287–296. doi:10.1016/S0306-3623(01)00121-5 |
| 19. | Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3 |
| 7. | Szajnman, S. H.; Montalvetti, A.; Wang, Y.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2003, 13, 3231–3235. doi:10.1016/S0960-894X(03)00663-2 |
| 8. | Szajnman, S. H.; Bailey, B. N.; Docampo, R.; Rodriguez, J. B. Bioorg. Med. Chem. Lett. 2001, 11, 789–792. doi:10.1016/S0960-894X(01)00057-9 |
| 9. | Martin, M. B.; Sanders, J. M.; Kendrick, H.; de Luca-Fradley, K.; Lewis, J. C.; Grimley, J. S.; Van Brussel, E. M.; Olsen, J. R.; Meints, G. A.; Burzynska, A.; Kafarski, P.; Croft, S. L.; Oldfield, E. J. Med. Chem. 2002, 45, 2904–2914. doi:10.1021/jm0102809 |
| 10. | Garzoni, L. R.; Caldera, A.; Nazareth L. Meirelles, M.; Castro, S. L.; Docampo, R.; Meints, G. A.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 273–285. doi:10.1016/j.ijantimicag.2003.07.020 |
| 11. | Garzoni, L. R.; Waghabi, M. C.; Baptista, M. M.; Castro, S. L.; Nazareth L. Meirelles, M.; Britto, C. C.; Docampo, R.; Oldfield, E.; Urbina, J. A. Int. J. Antimicrob. Agents 2004, 23, 286–290. doi:10.1016/j.ijantimicag.2003.07.019 |
| 19. | Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3 |
| 1. | Fleisch, H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient; The Parthenon Publishing Group Inc.: New York, 1995. |
| 2. | Papapoulos, S. E.; Landman, J. O.; Bijvoet, O. L. M.; Löwik, C. W. G. M.; Valkema, R.; Pauwels, E. K. J.; Vermeij, P. Bone 1992, 13 (Suppl. 1), S41–S49. |
| 3. | Yates, A. J.; Rodan, G. A. Drug Discovery Today 1998, 3, 69–78. doi:10.1016/S1359-6446(97)01134-3 |
| 4. | Papapoulos, S. E. Am. J. Med. 1993, 95 (5, Suppl. 1), S48–S52. doi:10.1016/0002-9343(93)90383-Z |
| 5. | Giannini, S.; D'Angelo, A.; Sartori, L.; Passeri, G.; Dalle Carbonare, L.; Crepaldi, G. Obstet. Gynecol. 1996, 88, 431–436. doi:10.1016/0029-7844(96)00171-8 |
| 6. | Fleisch, H. Drugs 1991, 42, 919–944. |
| 17. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 2119–2121. doi:10.1055/s-2005-869984 |
| 20. | Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Synthesis 2001, 633–637. doi:10.1055/s-2001-12353 |
| 21. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2004, 992–994. doi:10.1055/s-2004-822345 |
| 29. | Vachal, P.; Hale, J. J.; Lu, Z.; Streckfuss, E. C.; Mills, S. G.; MacCoss, M.; Yin, D. H.; Algayer, K.; Manser, K.; Kesisoglou, F.; Ghosh, S.; Alani, L. L. J. Med. Chem. 2006, 49, 3060–3063. doi:10.1021/jm060398v |
| 30. | Fitch, S. J.; Moedritzer, K. J. Am. Chem. Soc. 1962, 84, 1876–1879. doi:10.1021/ja00869a022 |
| 31. | Ruel, R.; Bouvier, J.-P.; Young, R. N. J. Org. Chem. 1995, 60, 5209–5213. doi:10.1021/jo00121a044 |
| 32. | Tromelin, A.; El Manouni, D.; Burgada, R. Phosphorus Sulfur Relat. Elem. 1986, 27, 301–312. doi:10.1080/03086648608072784 |
| 26. | Nicholson, D. A.; Vaughn, H. J. Org. Chem. 1971, 36, 3843–3845. doi:10.1021/jo00823a621 |
| 27. | Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Phosphorus, Sulfur Silicon Relat. Elem. 2001, 170, 115–133. doi:10.1080/10426500108040589 |
| 24. | Szymczak, M.; Szymańska, A.; Stawiński, J.; Boryski, J.; Kraszewski, A. Org. Lett. 2003, 5, 3571–3573. doi:10.1021/ol035166u |
| 21. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2004, 992–994. doi:10.1055/s-2004-822345 |
| 16. | Turhanen, P. A.; Niemi, R.; Peräkylä, M.; Järvinen, T.; Vepsäläinen, J. J. Org. Biomol. Chem. 2003, 1, 3223–3226. doi:10.1039/b305979k |
| 17. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 2119–2121. doi:10.1055/s-2005-869984 |
| 18. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2005, 3063–3066. doi:10.1055/s-2005-916032 |
| 19. | Niemi, R.; Turhanen, P.; Vepsäläinen, J.; Taipale, H.; Järvinen, T. Eur. J. Pharm. Sci. 2000, 11, 173–180. doi:10.1016/S0928-0987(00)00099-3 |
| 20. | Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Synthesis 2001, 633–637. doi:10.1055/s-2001-12353 |
| 21. | Turhanen, P. A.; Vepsäläinen, J. J. Synthesis 2004, 992–994. doi:10.1055/s-2004-822345 |
| 22. | Turhanen, P. A.; Vepsäläinen, J. J. Beilstein J. Org. Chem. 2006, 2, No. 2. doi:10.1186/1860-5397-2-2 |
| 23. | Vepsäläinen, J. J. Curr. Med. Chem. 2002, 9, 1201–1208. |
| 20. | Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Synthesis 2001, 633–637. doi:10.1055/s-2001-12353 |
| 26. | Nicholson, D. A.; Vaughn, H. J. Org. Chem. 1971, 36, 3843–3845. doi:10.1021/jo00823a621 |
| 27. | Turhanen, P. A.; Ahlgren, M. J.; Järvinen, T.; Vepsäläinen, J. J. Phosphorus, Sulfur Silicon Relat. Elem. 2001, 170, 115–133. doi:10.1080/10426500108040589 |
| 28. | Van Gelder, J. M.; Breuer, E.; Ornoy, A.; Schlossman, A.; Patlas, N.; Golomb, G. Bone 1995, 16, 511–520. doi:10.1016/8756-3282(95)00081-N |
| 1. | Fleisch, H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient; The Parthenon Publishing Group Inc.: New York, 1995. |
| 2. | Papapoulos, S. E.; Landman, J. O.; Bijvoet, O. L. M.; Löwik, C. W. G. M.; Valkema, R.; Pauwels, E. K. J.; Vermeij, P. Bone 1992, 13 (Suppl. 1), S41–S49. |
| 3. | Yates, A. J.; Rodan, G. A. Drug Discovery Today 1998, 3, 69–78. doi:10.1016/S1359-6446(97)01134-3 |
| 4. | Papapoulos, S. E. Am. J. Med. 1993, 95 (5, Suppl. 1), S48–S52. doi:10.1016/0002-9343(93)90383-Z |
| 5. | Giannini, S.; D'Angelo, A.; Sartori, L.; Passeri, G.; Dalle Carbonare, L.; Crepaldi, G. Obstet. Gynecol. 1996, 88, 431–436. doi:10.1016/0029-7844(96)00171-8 |
| 6. | Fleisch, H. Drugs 1991, 42, 919–944. |
| 15. | Major, P. P.; Lipton, A.; Berenson, J.; Hortobagyi, G. Cancer (N. Y.) 2000, 88, 6–14. doi:10.1002/(SICI)1097-0142(20000101)88:1<6::AID-CNCR3>3.0.CO;2-D |
| 13. | Kajander, E. O. Lett. Appl. Microbiol. 2006, 42, 549–552. doi:10.1111/j.1472-765X.2006.01945.x |
| 14. | Aho, K.; Soininen, T.; Turhanen, P. A.; Kajander, E. O.; Vepsäläinen, J. J. Unpublished results. |
| 25. | Szajnman, S. H.; García Liñares, G.; Moro, P.; Rodriguez, J. B. Eur. J. Org. Chem. 2005, 3687–3696. doi:10.1002/ejoc.200500097 |
© 2008 Turhanen and Vepsäläinen; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)