Abstract
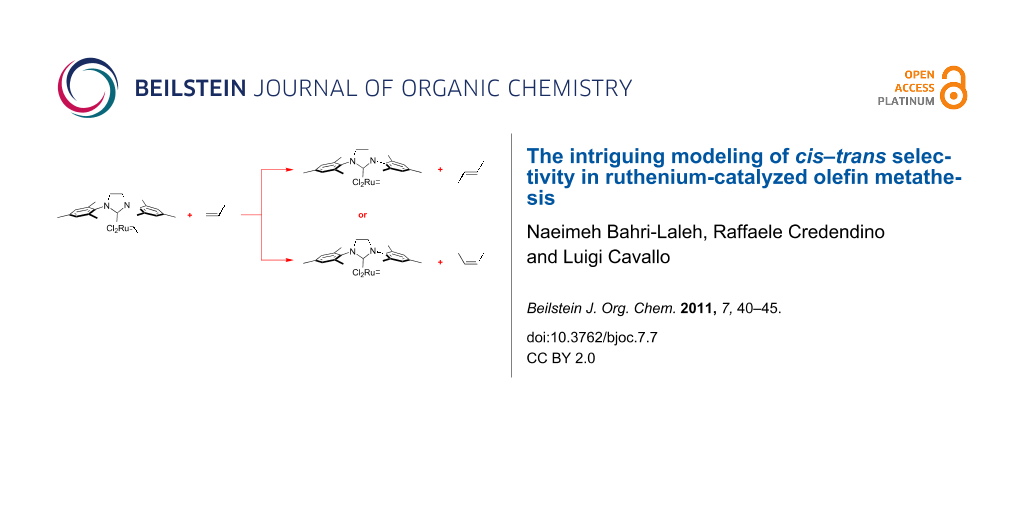
In this study we have investigated computationally the origin of the cis–trans selectivity in the Ru-catalyzed cross metathesis (CM) of a prototype monosubstituted olefin, i.e., propene. Our calculations suggest that the origin of the preferential formation of trans-olefins is in the product release step, which prevents the initially formed cis-olefin from escaping the metal, and returns it to the reaction pool until the trans-olefin is formed.

Graphical Abstract
Introduction
Olefin metathesis is among the most versatile tools when C=C double bonds must be manipulated. For this reaction Ru-based catalysts of various generations are particularly attractive due to their high tolerance for other functional groups [1-4]. Among the most useful possibilities is cross metathesis (CM), see Scheme 1, since it opens the door to the formation of functionalized and/or higher olefins from simpler unsaturated building blocks. Such a wide potential explains why CM applications span from the production of raw materials [5,6] to advanced and challenging organic synthesis [7-10]. On the other hand, the general scope of CM has made a challenging task the comprehension of the working mechanism and the development of rules to control this powerful synthetic tool [11-18]. Among the problems connected with the CM of reactive monosubstituted olefins are the minimization of homodimers and a control over the cis–trans selectivity, see Scheme 1. While minimization of homodimers can be achieved with proper handling of the reaction protocol [15], controlling the cis–trans selectivity is much more complicated, and is usually biased towards the formation of the trans-isomer.
![[1860-5397-7-7-i1]](/bjoc/content/inline/1860-5397-7-7-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Possible products resulting from the CM of terminal olefins.
Scheme 1: Possible products resulting from the CM of terminal olefins.
For this reason, we decided to investigate computationally the cis–trans selectivity in the CM of the simplest terminal olefin, i.e., propene, to yield either cis- and trans-2-butene with the well characterized 2nd generation Ru-catalyst based on the SIMes N-heterocyclic carbene ligand, see Scheme 2. Although it is well known that the steric hindrance of the olefin substituent has a remarkable role on both reactivity and products distribution, propene can still be considered as a prototype of terminal olefins, and can provide insights into the energetics of the basic CM reaction. Steric or electronic effects that would arise from more complex olefins would add to the prototype energy profile investigated here. Finally, for the sake of brevity, we will focus only on the productive CM of propene with the Ru–propylidene bond, while degenerate propene metathesis with the Ru–propylidene moiety, or propene reactivity with the Ru–methylidene moiety, which would be the other Ru–alkylidene bonds present in the reaction mixture after complete activation of the starting precatalyst, are not considered.
![[1860-5397-7-7-i2]](/bjoc/content/inline/1860-5397-7-7-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Representation of the reactions investigated.
Scheme 2: Representation of the reactions investigated.
Results and Discussion
The free energy profile in CH2Cl2 for the formation of both cis- and trans-2-butene from the starting 14e Ru–propylidene species is shown in Figure 1. Coordination of propene to the Ru center of 14e1, leading to the coordination intermediate Co1, is favored by 3.4 kcal·mol−1. In this intermediate the C=C double bond of the coordinated propene molecule is roughly perpendicular to the Ru–propylidene bond. From intermediate Co1, two transition states corresponding to productive metathesis, TS1-cis and TS1-trans, can be reached via rather low energy barriers (3.1 and 2.5 kcal·mol−1 for TS1-cis and TS1-trans, respectively). The two transition states differ in the relative orientation of the Me groups of the propene group and of the propylidene moiety, syn in TS1-cis, and anti in TS1-trans, with TS1-trans favored by 0.6 kcal/mol The experimentally proved free and fast rotation around the Ru–alkylidene bond allows both TS1-cis and TS1-trans to be reached from Co1 [19,20]. The two transition states collapse into the corresponding MCy-cis and MCy-trans metallacycles, at 5.6 and 6.3 kcal·mol−1 below the starting 14e1 intermediate, respectively. The two metallacycles are the most stable structures along the reaction pathway. Release of cis- and trans-butene requires the breaking of the MCy-cis and MCy-trans metallacycles through the transition states TS2-cis and TS2-trans, respectively, with formation of the two coordination intermediates Co2-cis and Co2-trans, from which cis- and trans-butene are released. In this case, breaking the metallacycles is energetically quite expensive, around 9–10 kcal·mol−1, and transition states TS2-cis and TS2-trans, in agreement with other computational studies, are higher in energy than the transition states for metallacycle formation TS1-cis and TS1-trans [21]. The Co2-cis and Co2-trans coordination intermediates are slightly more stable than the corresponding TS2-cis and TS2-trans transition states, which means that the energy barrier for the backward reaction that would return the Co2-cis and Co2-trans coordination intermediates back into the corresponding metallacycle is rather low (2.1 and 3.2 kcal·mol−1 for Co2-cis and Co2-trans, respectively).
![[1860-5397-7-7-1]](/bjoc/content/figures/1860-5397-7-7-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Reaction profile for the formation of cis- and trans-2-butene.
Figure 1: Reaction profile for the formation of cis- and trans-2-butene.
This scenario indicates a rather surprising and unexpected result. First, cis versus trans selectivity is not determined at metallacycle formation, since both the cis and trans TS1 transition states are roughly 3–4 kcal·mol−1 lower in energy that the TS2 transition states. Second, neither is selectivity determined at metallacycle breaking. In fact, the TS2-cis and TS2-trans transition states, within the accuracy of this type of calculation, are of the same energy. On the other hand, and in line with expectations, the MCy-trans metallacycle is more stable than the MCy-cis metallacycle by roughly 1 kcal·mol−1, and the Co2-trans coordination intermediate is similarly more stable than the Co2-cis coordination intermediate. Finally, our computational approach also reproduces well the experimental higher stability, by 1.0 kcal·mol−1, of trans-2-butene relative to cis-2-butene in the gas-phase [22].
To test whether the relative stability of the four transition states could depend on the chosen HMGGA M06 functional [23], we localized the transition states for metallacycle formation and breaking with the popular GGA BP86 [24-26] and HGGA B3LYP [27-29] functionals, as well as with the more recent MGGA M06L functional [30]. All these tests are summarized in Table 1. First, in all the cases TS1-cis is somewhat higher in energy than TS1-trans but, more importantly, both the TS1 metallacycle forming transition states are 3–5 kcal·mol−1 lower in energy than the TS2 metallacycle breaking transition states. Considering that all the functionals we considered indicate that the metallacycle forming transition state is not rate (selectivity) determining, we will not discuss this point any further.
Table 1: Energy difference, in kcal·mol-1, between the cis and trans transition states TS1 and TS2 of Figure 1, calculated with different computational approaches. In all cases TS1-trans is taken as reference at 0 kcal·mol−1.
| Functional |
Transition
State |
ΔE≠cis–trans
gas-phase |
ΔG≠cis–trans
gas-phase |
ΔG≠cis–trans
CH2Cl2 |
|---|---|---|---|---|
| BP86 | TS1-trans | 0.0 | 0.0 | 0.0 |
| TS1-cis | 0.2 | 1.2 | 1.4 | |
| TS2-trans | 4.5 | 4.1 | 4.1 | |
| TS2-cis | 4.1 | 3.8 | 3.8 | |
| B3LYP | TS1-trans | 0.0 | 0.0 | 0.0 |
| TS1-cis | 0.5 | 0.1 | 0.3 | |
| TS2-trans | 4.2 | 3.3 | 3.2 | |
| TS2-cis | 3.7 | 2.1 | 2.0 | |
| M06L | TS1-trans | 0.0 | 0.0 | 0.0 |
| TS1-cis | 0.1 | 2.3 | 2.2 | |
| TS2-trans | 6.0 | 6.5 | 5.6 | |
| TS2-cis | 4.6 | 5.7 | 5.4 | |
| M06 | TS1-trans | 0.0 | 0.0 | 0.0 |
| TS1-cis | 0.6 | 0.6 | 0.5 | |
| TS2-trans | 4.9 | 4.6 | 4.2 | |
| TS2-cis | 4.3 | 4.6 | 4.1 | |
Focusing on the metallacycle breaking transition state, the values in Table 1 indicate that the difference between the TS2-cis and TS2-trans transition states is somewhat dependent on the computational approach used. Indeed, the BP86, B3LYP and M06L results suggest that the TS2-cis transition state is somewhat preferred over the TS2-trans transition state, thus suggesting a small preference for the formation of cis-olefins. The M06 functional, instead, indicates that these two transition states are practically of the same energy. Further, the values given in Table 1 indicate that in terms of gas-phase potential energy the TS2-cis is reasonably favored over TS2-trans also with the M06 functional. It is the inclusion of the vibrational/entropic part that makes the M06 cis- and trans transition states of similar energy, (compare the ΔE≠cis–trans and ΔG≠cis–trans values in the gas-phase in Table 1). Inclusion of solvent effects does not change the gas-phase trends (compare the ΔG≠cis–trans in gas-phase and the ΔG≠cis–trans in CH2Cl2 values in Table 1). In conclusion, if any preference exists at the level of the metallacycle breaking step, the consensus emerging from the comparison of various functionals is that the cis transition state is favored. The fact that the cis transition state is of lower or similar energy to the trans transition state, despite of the higher stability of the forming trans C=C skeleton, indicates that the SIMes ligand is more suitable to host a cis forming C=C bond rather than a trans C=C bond.
Based on these calculations, the conclusion emerging from the energy profiles of Figure 1 and the values given in Table 1 is that, in the framework of a dissociative mechanism, the key step determining the experimentally observed preferential formation of trans-olefins is product release with formation of the methylidene Ru 14e intermediate 14e2 and a free cis- or trans-2-butene molecule from Co2-cis and Co2-trans (in this regard it must be noted that an associative mechanism has been proposed to be operative for ethylene degenerate metathesis at low temperature [31,32], and for the activation step in Hoveyda/Grela type catalysts [33]). Product release is endoergonic due to the coordination energy of 2-butene that amounts to roughly 6 kcal·mol−1 both for the cis- and trans-isomers, and is clearly higher in energy than both TS2 transition states that would revert the just formed 2-butenes into the most stable metallacycle. For this reason, the energy profile of Figure 1 suggests that the most likely event from Co2-cis and Co2-trans is not product release, but rather their transformation into MCy2-cis and MCy2-trans. The escape from Co2-cis and Co2-trans is controlled by the free energy difference between 14e2 and transition states TS2-cis and TS2-trans, which amounts to 3.6 and 2.5 kcal·mol−1 for the cis and trans pathways, respectively. These numbers indicate that release of trans-2-butene is favored by the higher stability of the trans-olefin relative to the cis-isomer.
Of course, considering the high reactivity of both trans- and cis-butene towards metathesis, it is clear that secondary metathesis of the produced butenes, whose energetic can be still derived from Figure 1, will result in a statistical distribution of the products according to their thermodynamic stability [15,18]. Thus, a high trans/cis-butene ratio would be reached, even if a lower trans/cis ratio was initially produced. This conclusion is in qualitative agreement with CM experiments using terminal olefins, which resulted in a trans/cis ratio of around 2–4 at low conversions, that increased to 9–10 at higher conversions [18]. Finally, it is clear that the steric and electronic properties of more complex olefins can have a strong impact on the energy profile of Figure 1, which however, remains the base energy profile to be modified. We are currently working in this direction.
Conclusion
In this study we have investigated computationally the origin of the cis–trans selectivity in the CM of the prototype monosubstituted olefin, i.e., propene. Our calculations suggest that the origin of the preferential formation of trans-olefins is not the energy difference at the transition states corresponding to either metallacycle formation of breaking. Actually, focusing on the transition state higher in energy (the one corresponding to metallacycle breaking), we found that the transition state leading to the formation of cis-butene is of similar energy, or even favored, relative to that leading to the formation of trans-butene. Thus, CM of propene (and by consequence of simple linear 1-olefins) should kinetically lead first to the formation of cis-olefins, followed by gradual conversion to the more stable trans-isomer. Our calculations suggest that the key step to rationalize the preferential formation of trans-olefins, even at low conversion, is in the product release step, since trans-olefins have a higher tendency to be released from the catalyst at the end of the CM reaction. Conversely, the initially formed cis-olefins have a minor tendency to be released from the catalyst, and thus they have a higher chance to return to the reaction pool until the trans-olefin is formed.
Experimental
Computational Details
The DFT calculations of the full energy profile of Figure 1 were performed at the HMGGA level with the Gaussian09 package [34], using the M06 functional of Truhlar [23]. Free energies in CH2Cl2 were deduced from the gas-phase free energies plus the solvation energy term estimated in single point calculations on the gas-phase optimized structures, based on the polarizable continuum solvation model PCM using CH2Cl2 as the solvent [35]. In case of olefin coordination, we assumed a –TΔS contribution of 10 kcal·mol−1, since the gas-phase rotational/translational entropy of coordination from classical statistical thermodynamics is generally considered to overestimate the coordination entropy in solution. The –TΔS contribution of 10 kcal·mol−1 is the experimental coordination entropy of C2H4 to a Pd-complex [36]. The test calculations in Table 1 were performed with the GGA BP86 functional of Becke and Perdew [24-26], with the HGGA B3LYP functional of Becke, Lee, Parr, and Yang [27-29], and with the MGGA M06L functional of Truhlar [30]. In all cases the electronic configuration of the molecular systems was described with the standard split-valence basis set with a polarization function of Ahlrichs and co-workers for H, C, N, O and Cl (SVP keyword in Gaussian03) [37]. For Ru we used the small-core, quasi-relativistic Stuttgart/Dresden effective core potential, with an associated (8s7p6d)/[6s5p3d] valence basis set (SDD keywords in Gaussian03) [38]. Characterization of the located stationary points as minima or transition state was performed by frequency calculations.
Acknowledgements
We thank M. Barbasiewicz, Warsaw University, for useful discussions. The research leading to these results received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement n° CP-FP 211468-2 EUMET. LC thanks the HPC team of Enea (http://www.enea.it) for use of the ENEA-GRID and the HPC facilities CRESCO (http://www.cresco.enea.it) in Portici, Italy.
References
-
Grubbs, R. H. Handbook of Olefin Metathesis; Wiley-VCH: Weinheim, Germany, 2003.
Return to citation in text: [1] -
Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900–1923. doi:10.1002/anie.200200556
Return to citation in text: [1] -
Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Malacea, R.; Fischmeister, C.; Bruneau, C.; Dubois, J.-L.; Couturier, J.-L.; Dixneuf, P. H. Green Chem. 2009, 11, 152–155. doi:10.1039/b816917a
Return to citation in text: [1] -
Rybak, A.; Meier, M. A. R. Green Chem. 2007, 9, 1356–1361. doi:10.1039/b712293d
Return to citation in text: [1] -
Demchuk, O. M.; Pietrusiewicz, K. M.; Michrowska, A.; Grela, K. Org. Lett. 2003, 5, 3217–3220. doi:10.1021/ol035011m
Return to citation in text: [1] -
Cantrill, S. J.; Grubbs, R. H.; Lanari, D.; Leung, K. C. F.; Nelson, A.; Poulin-Kerstien, K. G.; Smidt, S. P.; Stoddart, J. F.; Tirrell, D. A. Org. Lett. 2005, 7, 4213–4216. doi:10.1021/ol051599g
Return to citation in text: [1] -
Chatterjee, A. K.; Sanders, D. P.; Grubbs, R. H. Org. Lett. 2002, 4, 1939–1942. doi:10.1021/ol0259793
Return to citation in text: [1] -
Boeda, F.; Bantreil, X.; Clavier, H.; Nolan, S. P. Adv. Synth. Catal. 2008, 350, 2959–2966. doi:10.1002/adsc.200800495
Return to citation in text: [1] -
Berlin, J. M.; Goldberg, S. D.; Grubbs, R. H. Angew. Chem., Int. Ed. 2006, 45, 7591–7595. doi:10.1002/anie.200602469
Return to citation in text: [1] -
Stewart, I. C.; Douglas, C. J.; Grubbs, R. H. Org. Lett. 2008, 10, 441–444. doi:10.1021/ol702624n
Return to citation in text: [1] -
Funk, T. W.; Efskind, J.; Grubbs, R. H. Org. Lett. 2005, 7, 187–190. doi:10.1021/ol047929z
Return to citation in text: [1] -
Chatterjee, A. K.; Grubbs, R. H. Org. Lett. 1999, 1, 1751–1753. doi:10.1021/ol991023p
Return to citation in text: [1] -
Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882
Return to citation in text: [1] [2] [3] -
Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::AID-ANIE1277>3.0.CO;2-E
Return to citation in text: [1] -
Smith, A. B., III; Adams, C. M.; Kozmin, S. A. J. Am. Chem. Soc. 2001, 123, 990–991. doi:10.1021/ja003745d
Return to citation in text: [1] -
Anderson, D. R.; Ung, T.; Mkrtumyan, G.; Bertrand, G.; Grubbs, R. H.; Schrodi, Y. Organometallics 2008, 27, 563–566. doi:10.1021/om7008028
Return to citation in text: [1] [2] [3] -
Aagaard, O. M.; Meier, R. J.; Buda, F. J. Am. Chem. Soc. 1998, 120, 7174–7182. doi:10.1021/ja974131k
Return to citation in text: [1] -
van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653
Return to citation in text: [1] -
Rowley, C. N.; van der Eide, E. F.; Piers, W. E.; Woo, T. K. Organometallics 2008, 27, 6043–6045. doi:10.1021/om8008519
Return to citation in text: [1] -
Lide, D. R. Handbook of Chemistry and Physics, 84th ed.; CRC Press Inc.: London, 2004.
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] [2] -
Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Return to citation in text: [1] [2] -
Perdew, J. P. Phys. Rev. B 1986, 33, 8822–8824. doi:10.1103/PhysRevB.33.8822
Return to citation in text: [1] [2] -
Perdew, J. P. Phys. Rev. B 1986, 34, 7406. doi:10.1103/PhysRevB.34.7406
Return to citation in text: [1] [2] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] [2] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text: [1] [2] -
Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001
Return to citation in text: [1] [2] -
Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101. doi:10.1063/1.2370993
Return to citation in text: [1] [2] -
Romero, P. E.; Piers, W. E. J. Am. Chem. Soc. 2007, 129, 1698–1704. doi:10.1021/ja0675245
Return to citation in text: [1] -
Webster, C. E. J. Am. Chem. Soc. 2007, 129, 7490–7491. doi:10.1021/ja071588d
Return to citation in text: [1] -
Vorfalt, T.; Wannowius, K.-J.; Plenio, H. Angew. Chem., Int. Ed. 2010, 49, 5533–5536. doi:10.1002/anie.201000581
Return to citation in text: [1] -
Gaussian 03, B. 1; Gaussian, Inc.: Pittsburg PA, 2003.
Return to citation in text: [1] -
Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027–2094. doi:10.1021/cr00031a013
Return to citation in text: [1] -
Rix, F. C.; Brookhart, M. J. Am. Chem. Soc. 1995, 117, 1137–1138. doi:10.1021/ja00108a034
Return to citation in text: [1] -
Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571–2577. doi:10.1063/1.463096
Return to citation in text: [1] -
Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. J. Chem. Phys. 1994, 100, 7535–7542. doi:10.1063/1.466847
Return to citation in text: [1]
| 1. | Grubbs, R. H. Handbook of Olefin Metathesis; Wiley-VCH: Weinheim, Germany, 2003. |
| 2. | Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900–1923. doi:10.1002/anie.200200556 |
| 3. | Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f |
| 4. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 15. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 15. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 18. | Anderson, D. R.; Ung, T.; Mkrtumyan, G.; Bertrand, G.; Grubbs, R. H.; Schrodi, Y. Organometallics 2008, 27, 563–566. doi:10.1021/om7008028 |
| 11. | Berlin, J. M.; Goldberg, S. D.; Grubbs, R. H. Angew. Chem., Int. Ed. 2006, 45, 7591–7595. doi:10.1002/anie.200602469 |
| 12. | Stewart, I. C.; Douglas, C. J.; Grubbs, R. H. Org. Lett. 2008, 10, 441–444. doi:10.1021/ol702624n |
| 13. | Funk, T. W.; Efskind, J.; Grubbs, R. H. Org. Lett. 2005, 7, 187–190. doi:10.1021/ol047929z |
| 14. | Chatterjee, A. K.; Grubbs, R. H. Org. Lett. 1999, 1, 1751–1753. doi:10.1021/ol991023p |
| 15. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 16. | Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::AID-ANIE1277>3.0.CO;2-E |
| 17. | Smith, A. B., III; Adams, C. M.; Kozmin, S. A. J. Am. Chem. Soc. 2001, 123, 990–991. doi:10.1021/ja003745d |
| 18. | Anderson, D. R.; Ung, T.; Mkrtumyan, G.; Bertrand, G.; Grubbs, R. H.; Schrodi, Y. Organometallics 2008, 27, 563–566. doi:10.1021/om7008028 |
| 18. | Anderson, D. R.; Ung, T.; Mkrtumyan, G.; Bertrand, G.; Grubbs, R. H.; Schrodi, Y. Organometallics 2008, 27, 563–566. doi:10.1021/om7008028 |
| 7. | Demchuk, O. M.; Pietrusiewicz, K. M.; Michrowska, A.; Grela, K. Org. Lett. 2003, 5, 3217–3220. doi:10.1021/ol035011m |
| 8. | Cantrill, S. J.; Grubbs, R. H.; Lanari, D.; Leung, K. C. F.; Nelson, A.; Poulin-Kerstien, K. G.; Smidt, S. P.; Stoddart, J. F.; Tirrell, D. A. Org. Lett. 2005, 7, 4213–4216. doi:10.1021/ol051599g |
| 9. | Chatterjee, A. K.; Sanders, D. P.; Grubbs, R. H. Org. Lett. 2002, 4, 1939–1942. doi:10.1021/ol0259793 |
| 10. | Boeda, F.; Bantreil, X.; Clavier, H.; Nolan, S. P. Adv. Synth. Catal. 2008, 350, 2959–2966. doi:10.1002/adsc.200800495 |
| 31. | Romero, P. E.; Piers, W. E. J. Am. Chem. Soc. 2007, 129, 1698–1704. doi:10.1021/ja0675245 |
| 32. | Webster, C. E. J. Am. Chem. Soc. 2007, 129, 7490–7491. doi:10.1021/ja071588d |
| 5. | Malacea, R.; Fischmeister, C.; Bruneau, C.; Dubois, J.-L.; Couturier, J.-L.; Dixneuf, P. H. Green Chem. 2009, 11, 152–155. doi:10.1039/b816917a |
| 6. | Rybak, A.; Meier, M. A. R. Green Chem. 2007, 9, 1356–1361. doi:10.1039/b712293d |
| 33. | Vorfalt, T.; Wannowius, K.-J.; Plenio, H. Angew. Chem., Int. Ed. 2010, 49, 5533–5536. doi:10.1002/anie.201000581 |
| 23. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 27. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 28. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 29. | Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001 |
| 22. | Lide, D. R. Handbook of Chemistry and Physics, 84th ed.; CRC Press Inc.: London, 2004. |
| 30. | Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101. doi:10.1063/1.2370993 |
| 21. | Rowley, C. N.; van der Eide, E. F.; Piers, W. E.; Woo, T. K. Organometallics 2008, 27, 6043–6045. doi:10.1021/om8008519 |
| 19. | Aagaard, O. M.; Meier, R. J.; Buda, F. J. Am. Chem. Soc. 1998, 120, 7174–7182. doi:10.1021/ja974131k |
| 20. | van der Eide, E. F.; Piers, W. E. Nat. Chem. 2010, 2, 571–576. doi:10.1038/nchem.653 |
| 24. | Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098 |
| 25. | Perdew, J. P. Phys. Rev. B 1986, 33, 8822–8824. doi:10.1103/PhysRevB.33.8822 |
| 26. | Perdew, J. P. Phys. Rev. B 1986, 34, 7406. doi:10.1103/PhysRevB.34.7406 |
| 35. | Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027–2094. doi:10.1021/cr00031a013 |
| 23. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 37. | Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571–2577. doi:10.1063/1.463096 |
| 38. | Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. J. Chem. Phys. 1994, 100, 7535–7542. doi:10.1063/1.466847 |
| 27. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 28. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 29. | Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001 |
| 30. | Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101. doi:10.1063/1.2370993 |
| 36. | Rix, F. C.; Brookhart, M. J. Am. Chem. Soc. 1995, 117, 1137–1138. doi:10.1021/ja00108a034 |
| 24. | Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098 |
| 25. | Perdew, J. P. Phys. Rev. B 1986, 33, 8822–8824. doi:10.1103/PhysRevB.33.8822 |
| 26. | Perdew, J. P. Phys. Rev. B 1986, 34, 7406. doi:10.1103/PhysRevB.34.7406 |
© 2011 Bahri-Laleh et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)