Abstract
A synthetic route to the bench-stable fluorinated masked carbene reagent diethyl 2-diazo-1,1,3,3,3-pentafluoropropylphosphonate, bearing a trifluoromethyl and a difluoromethyl group is reported for the first time. Its application in CuI-catalyzed cyclopropanation reactions with aromatic and aliphatic terminal alkenes under mild reaction conditions is demonstrated. In total, sixteen new cyclopropanes were synthesized in good to very good yields.

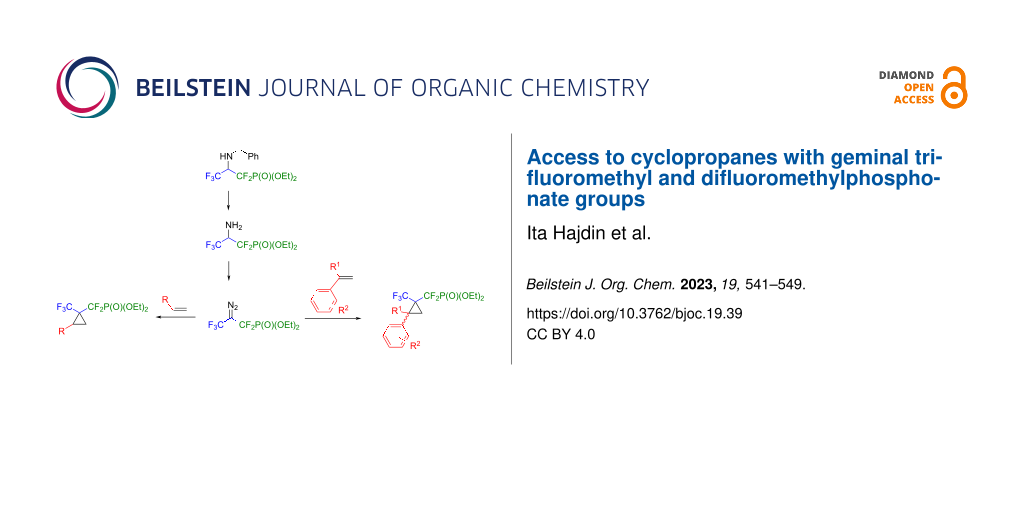
Graphical Abstract
Introduction
Cyclopropanes constitute a fascinating class of organic compounds due to their unique structure and bond properties [1]. However, their synthetic utility is closely linked to the substitution pattern of the cyclopropane unit [2]. The prevalence of the biologically active cyclopropyl derivatives, either isolated from natural sources or rationally designed as pharmaceutical agents, has inspired chemists to find efficient methods for their preparation. Among them, trifluoromethyl- and difluoromethyl-substituted cyclopropanes are of great interest in pharmaceutical, materials and agricultural chemistry [3]. Due to the unique properties of fluorine, such as highest electronegativity, small atomic radius, or low polarizability, the strategic placement of fluorinated moieties within cyclopropyl rings imparts useful properties to these molecules. Thus, it is not surprising that difluoromethyl- or trifluoromethyl-substituted cyclopropanes serve as important structural motifs in many biologically active molecules as well as in special materials [4-6]. Therefore, as relevant building blocks, tremendous efforts have been made to develop reliable methods for their synthesis. Transition-metal-catalyzed cyclopropanation of alkenes with trifluoromethyldiazoalkanes is a commonly used synthetic strategy for the construction of trifluoromethylcyclopropanes. Recently, also regio- and diastereoselective carbometalation of easily accessible trifluoromethyl-substituted cyclopropenes to access trifluoromethylcyclopropanes has been reported (Scheme 1A) [7-31]. In contrast, the synthesis of difluoromethylcyclopropanes utilizing difluoromethyldiazo reagents remains rather unexplored. Nevertheless, some difluorocarbene reagents, including HCF2CH(N2) [32], Ph2S+CH2CF2H TfO− [33], and difluoroacetaldehyde N-triftosylhydrazone (DFHZTfs) [34], have been developed (Scheme 1B). In addition, few examples of difluoromethylphosphonate containing cyclopropanes have been reported to date. These compounds were synthesized either by cyclopropanation of CF2P(O)(OEt)2-containing alkenoates using a Corey–Chaykovsky reagent (sulfaniumyl or oxosulfaniumyl methanides) and diazomethane or by photolysis of pyrazolines bearing difluoromethylphosphonate moieties (Scheme 1C) [35-38]. Notably, among the various CF2-containing functionalities, difluoromethylphosphonates represent a distinct class of compounds whose potential as nonhydrolyzable phosphate mimics is unquestionable. Phosphonates containing CF2 groups can sterically and electronically mimic oxygen, enabling the second dissociation constant, pKa2, to closer mirror those of the phosphates due to the electron-withdrawing effect of fluorine [39]. As a result, improved lipophilicity, metabolic stability or bioavailability of the difluoromethylphosphonate derivatives relative to their nonfluorinated analogues have been observed [40].
![[1860-5397-19-39-i1]](/bjoc/content/inline/1860-5397-19-39-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previous works (A–D) and the extension (this work).
Scheme 1: Previous works (A–D) and the extension (this work).
However, the extension of this chemistry to highly fluorinated diazo derivatives has not been reported to date. Instead, cyclopropanes with geminal trifluoromethyl substituents were mostly obtained by the deoxofluorination of the corresponding dicarboxylic acids with sulfur tetrafluoride, thiophilic ring-opening reactions with nucleophiles or reactions of donor-substituted furans with bis(trifluoromethyl)-substituted ethylenes (Scheme 1D) [41-43]. The lack of application of highly fluorinated diazo compounds might be rationalized by their low accessibility and high volatility [44].
As a part of our continuing investigations in fluorinated carbene chemistry [45-49], we hypothesized that the synthesis of cyclopropanes with geminal trifluoromethyl and difluoromethylphosphonate groups at the ring might be possible by using our newly developed bench-stable diazo reagent CF3C(N2)CF2P(O)(OEt)2. Therefore, we report herein our preliminary results toward this goal via copper iodide-catalyzed cyclopropanation reaction of an acceptor carbene precursor with selected terminal alkenes. We believe that the synergism of both fluorinated substituents could open new synthetic possibilities in the field of fluorinated organic materials.
Results and Discussion
We began our studies by preparing the fluorinated carbene precursor, diethyl 2-diazo-1,1,3,3,3-pentafluoropropylphosphonate (5). This compound was synthesized smoothly within three steps in 16.6% overall yield. The synthetic route commenced from the preparation of N-protected amine 3, followed by the deprotection of benzylamine 3 to furnish 4 and ended with the diazotization of 4 using tert-butyl nitrite. As a result, the target diazo reagent 5, which contains both a trifluoromethyl and a difluoromethylphosphonate moiety, was isolated as a stable, non-volatile liquid (Scheme 2).
![[1860-5397-19-39-i2]](/bjoc/content/inline/1860-5397-19-39-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of diethyl 2-diazo-1,1,3,3,3-pentafluoropropylphosphonate (5).
Scheme 2: Synthesis of diethyl 2-diazo-1,1,3,3,3-pentafluoropropylphosphonate (5).
To highlight the synthetic utility of the novel carbene precursor 5, subsequent [2 + 1] cycloaddition reactions with selected terminal aromatic and aliphatic olefins were examined. The reaction conditions were first optimized using 4-methylstyrene as a model substrate. The results are summarized in Table 1.
Table 1: Optimization of the reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-39-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | t (h) | T (°C) | Solvent | Conversion rateb (%) |
| 1 | Rh2(OAc)4 | 5 | 111 | toluene | n.r. |
| 2 | Rh2(OAc)4 | 3 | 40 | DCM | n.r. |
| 3 | CuI | 3 | 40 | DCM | n.r. |
| 4 | CuI | 1 | 111 | toluene | 42 |
| 5 | CuI | 2 | 111 | toluene | 59 |
| 6 | CuI | 3.5 | 111 | toluene | 100c |
| 7d | – | 24 | 40 | DCM | n.r. |
aReaction conditions: alkene (0.15 mmol), diazo compound 5 (0.1 mmol), CuI (1 mol %), dry toluene, 111 °C, Ar atmosphere; bDetermined by 19F NMR spectroscopy; cIsolated yield 74%. dUnder UV irradiation.
Initially, dirhodium tetraacetate (Rh2(OAc)4), the most common catalyst for the preparation of cyclopropanes, was applied. To our surprise, the application of Rh2(OAc)4 did not lead to the desired product neither in dichloromethane nor in toluene (Table 1, entries 1 and 2). Switching the catalyst to copper(I) iodide in refluxing DCM, did not result in the formation of product 6a, as well (Table 1, entry 3). However, when CuI was used in boiling toluene, 42% of the diazo reagent 5 was converted to 6a after only 1 hour of stirring. Thus, to increase the conversion rate, the reaction time was prolonged up to 3.5 h. Indeed, after this time, complete conversion of the diazo reagent 5 was observed and cyclopropane 6a was isolated in 74% yield (Table 1, entry 6). In addition, in a catalyst-free reaction under UV irradiation, no reaction occurred and the starting diazo compound 5 was recovered (Table 1, entry 7).
Having identified the optimal catalyst and conditions, we then examined the scope of the cyclopropanation reaction by reacting selected styrenes with the diazo reagent 5. As evident from Scheme 3, this method is widely applicable to a range of styrenes that are either not activated or substituted with electron-donating (Me, OMe) or electron-withdrawing (Cl) groups on the aromatic ring. All reactions were carried out successfully and the corresponding cyclopropanes 6a–i were isolated in moderate to good yields. As expected, the best results were obtained with styrenes bearing electron-donating groups in the para-position furnishing cyclopropanes 6a and 6d in 74% and 67% yield, respectively. The presence of substituents in ortho and meta-positions on the phenyl ring (6e, 6f, 6h, 6i), decelerated the process. In the case of α-methylstyrene, complete conversion of diazo reagent 5 to compound 6b was observed only after 48 hours of heating, which might result from the steric hindrance around the reaction centre.
![[1860-5397-19-39-i3]](/bjoc/content/inline/1860-5397-19-39-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the cyclopropanation. Reaction conditions: alkene (0.15 mmol), diazo compound 5 (0.1 mmol), CuI (1 mol %), dry toluene, 111 °C, Ar atmosphere. aYields refer to isolated products; bdr ratio determined by 19F NMR spectroscopy.
Scheme 3: Scope of the cyclopropanation. Reaction conditions: alkene (0.15 mmol), diazo compound 5 (0.1 mmol)...
In almost all cases, a mixture of two diastereoisomers was obtained, with a slight preference for one diastereoisomer for compounds 6a, 6c–i. Furthermore, we were able to isolate both the main isomer as well as a mixture of two isomers for cyclopropanes 6a, 6c, 6d, 6g, 6h and 6i. To determine the relative stereochemistry of the main isomer, the 19F,1H-HOESY NMR spectrum of compound 6c was recorded. The spectrum shows direct correlation of one fluorine nucleus of the difluoromethyl phosphonate group with two cyclopropane protons at 1.75 and 2.91 ppm, respectively. Thus, it is conceivable that the major diastereoisomer adopts trans configuration (Figure 1).
![[1860-5397-19-39-1]](/bjoc/content/figures/1860-5397-19-39-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 19F,1H-HOESY spectrum of compound 6c.
Figure 1: 19F,1H-HOESY spectrum of compound 6c.
Furthermore, terminal aliphatic alkenes as well as N-Boc-allylamine were subjected to [2 + 1] cycloaddition reactions with the carbene precursor 5. The results are summarized in Scheme 4. As observed, the procedure was successful with a series of terminal olefins with different aliphatic chains. The total conversion of the diazo compound 5 was achieved after 2.5–3.5 hours of heating and the corresponding cyclopropanes 7a–g were obtained in moderate to good yield (28–53%). The highest yield (53%) as well as the best diastereoselectivity were recorded for the reaction with tert-butyl-N-allyl carbamate 7g (1:6) while the reaction with allylcyclohexane resulted with the lowest yield (7f, 28%), probably due to the steric hindrance of the reagents. In the case of N-Boc-allylamine, improved diastereoselectivity might result from the coordination of the amino group to the copper centre of an intermediately produced metallocarbene, thus favouring the formation of one diastereoisomer of 7g [50]. In addition, the cyclopropanes 7a–g were always obtained as mixtures of two diastereoisomers in different ratios and all attempts to separate them using column chromatography were unsuccessful. In addition, alkenes such as allylpentafluorobenzene, diethyl allylmalonate, allylbenzene and ethyl acrylate did not react with 5. Instead, the diazo compound 5 decomposed immediately under the reaction conditions described in Scheme 4.
![[1860-5397-19-39-i4]](/bjoc/content/inline/1860-5397-19-39-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Scope of the cyclopropanation. Reaction conditions: alkene (0.15 mmol), diazo compound 5 (0.1 mmol), CuI (1 mol %), dry toluene, 111 °C, Ar atmosphere. aYields refer to isolated products; bdr ratio determined by 19F NMR spectroscopy.
Scheme 4: Scope of the cyclopropanation. Reaction conditions: alkene (0.15 mmol), diazo compound 5 (0.1 mmol)...
To better understand the mechanism and to confirm the lack of selectivity during the cyclopropanation process with terminal alkenes, the reaction mechanism between the diazo reagent 5 and styrene as a model substrate in the presence of CuI catalyst was investigated by density functional theory (DFT) calculations (Table 2).
Table 2: Change in Gibbs free energy ΔG (kcal∙mol−1) from the CuI-catalyzed cyclopropanation of the diazo compound with styrene for possible stereoisomers Pr1 to Pr4.
| Isomer | Int1 | TS1 | Int2 | TS2a | Int3 | TS3 | Int4 | TS4 | Pr |
| 1 | 5.2 | 16.4 | −8.7 | −26.4 | −22.1 | −52.3 | −41.7 | −44.6 | |
| 2 | 8.5 | −27.6 | −22.5 | −50.5 | −42.0 | −42.5 | |||
| 3 | – | – | −51.1 | −36.0 | −44.1 | ||||
| 4 | – | – | −51.3 | −35.9 | −43.3 | ||||
aExemplarily calculated for one option due to flat potential.
In the first step, CuI adds to the diazo compound 5 under formation of zwitterionic Int1 (ΔG = 5.2 kcal/mol). Afterwards, copper carbene complex Int2 is formed after extrusion of nitrogen. The transition state of this metal carbene formation TS1 was calculated with an activation free energy of 16.4 kcal/mol (Scheme 5).
![[1860-5397-19-39-i5]](/bjoc/content/inline/1860-5397-19-39-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Addition of CuI to the diazo compound 5.
Scheme 5: Addition of CuI to the diazo compound 5.
Int2 adds to styrene to the carbon atom in 2-position of the ethenyl group. At this point, four different orientations of the phenyl group are conceivable (Scheme 6 and Scheme 7, TS2_1 to TS2_4).
![[1860-5397-19-39-i6]](/bjoc/content/inline/1860-5397-19-39-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Possible addition of styrene to Int2 yielding Int4_1 and Int4_2 through Int3_1 and Int3_2.
Scheme 6: Possible addition of styrene to Int2 yielding Int4_1 and Int4_2 through Int3_1 and Int3_2.
![[1860-5397-19-39-i7]](/bjoc/content/inline/1860-5397-19-39-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Possible addition of styrene to Int2 yielding Int4_3 and Int4_4 without further intermediates.
Scheme 7: Possible addition of styrene to Int2 yielding Int4_3 and Int4_4 without further intermediates.
After an early transition state with hardly any activation barrier, the addition of styrene to Int2 proceeds in the case of TS2_1 and TS2_2 to the intermediates Int3_1 and Int3_2. Afterwards, the Cu–C bond is broken, and a Cu–O bond is formed. CuI is transferred to the oxygen atom from the phosphonate group, yielding Int4_1 and Int4_2. In the case of TS2_3 and TS2_4, the addition proceeds smoothly towards Int4_3 and Int4_4 without further intermediates (Scheme 7 and Scheme 8). Extrusion of a catalyst yields Pr1, Pr2, Pr3 and Pr4 as presented in Scheme 8.
![[1860-5397-19-39-i8]](/bjoc/content/inline/1860-5397-19-39-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Formation of the products Pr1 to Pr4.
Scheme 8: Formation of the products Pr1 to Pr4.
As shown in Table 2, the formation of the enantiomeric set Pr1 and Pr3 (ΔG = −44.6 and −44.1 kcal·mol−1) is slightly more favorable than the formation of Pr2 and Pr4 (ΔG = −42.5 and −43.3 kcal·mol−1; cis-6c), respectively. This is consistent with the experimental results, since two sets of signals corresponding to the diastereoisomers trans-6c and cis-6c are always observed in the 19F NMR spectrum of the crude mixture, with a slight preference for the trans isomer (diastereomeric ratio 1.4:1). Moreover, although Int4 is the overall thermodynamic minimum of the reaction for all stereoisomers, it is still unsurprising that the reaction proceeds towards the product since the abstraction of the catalyst is the last step. Once abstracted, the catalyst is then involved in the next catalytic cycle and thus removed from the equilibrium between Int4 and Pr + CuI. In addition, since TS2 is an early transition state and the potential is concomitantly very flat, only TS2_2 was found by means of regular optimization towards a first order saddle point. For the other three possible reaction pathways, this step was examined by relaxed potential energy scans along the indicated C–C bond (see Supporting Information File 1 for full experimental data).
Conclusion
In summary, a convenient method for the synthesis of a highly fluorinated diazo reagent, diethyl 2-diazo-1,1,3,3,3-pentafluoropropylphosphonate (5), was presented for the first time. This compound is a bench-stable, non-volatile, and non-explosive liquid, which facilitated the handling of this reagent. Furthermore, our research highlights the possibility for facile and efficient syntheses of difluoromethylphosphonate-containing cyclopropanes in reactions with selected olefins, carried out under mild conditions with good to very good yields, in the presence of CuI, an inexpensive catalyst. As confirmed by quantum mechanical calculations and experimental results, the cyclopropane formation occurs always with a slight preference for one diastereoisomer. The presented results turn of a new page for the future of diazo chemistry.
Supporting Information
| Supporting Information File 1: Experimental section and characterization of synthesized compounds. | ||
| Format: PDF | Size: 7.0 MB | Download |
References
-
de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/anie.197908093
Return to citation in text: [1] -
Faust, R. Fascinating Natural and Artificial Cyclopropane Architectures. Organic Synthesis Set; Wiley-VCH: Weinheim, Germany, 2003; pp 428–434. doi:10.1002/9783527620784.ch39d
Return to citation in text: [1] -
Pons, A.; Delion, L.; Poisson, T.; Charette, A. B.; Jubault, P. Acc. Chem. Res. 2021, 54, 2969–2990. doi:10.1021/acs.accounts.1c00261
Return to citation in text: [1] -
Bourlière, M.; Pietri, O.; Castellani, P.; Oules, V.; Adhoute, X. Ther. Adv. Gastroenterol. 2018, 11, 175628481881235. doi:10.1177/1756284818812358
Return to citation in text: [1] -
Abutaleb, A.; Kottilil, S.; Wilson, E. Hepatol. Int. 2018, 12, 214–222. doi:10.1007/s12072-018-9873-y
Return to citation in text: [1] -
Davies, J. C.; Moskowitz, S. M.; Brown, C.; Horsley, A.; Mall, M. A.; McKone, E. F.; Plant, B. J.; Prais, D.; Ramsey, B. W.; Taylor-Cousar, J. L.; Tullis, E.; Uluer, A.; McKee, C. M.; Robertson, S.; Shilling, R. A.; Simard, C.; Van Goor, F.; Waltz, D.; Xuan, F.; Young, T.; Rowe, S. M. N. Engl. J. Med. 2018, 379, 1599–1611. doi:10.1056/nejmoa1807119
Return to citation in text: [1] -
Le Maux, P.; Juillard, S.; Simonneaux, G. Synthesis 2006, 1701–1704. doi:10.1055/s-2006-926451
Return to citation in text: [1] -
Mykhailiuk, P. K.; Afonin, S.; Ulrich, A. S.; Komarov, I. V. Synthesis 2008, 1757–1760. doi:10.1055/s-2008-1067041
Return to citation in text: [1] -
Mykhailiuk, P. K.; Afonin, S.; Palamarchuk, G. V.; Shishkin, O. V.; Ulrich, A. S.; Komarov, I. V. Angew. Chem., Int. Ed. 2008, 47, 5765–5767. doi:10.1002/anie.200801022
Return to citation in text: [1] -
Artamonov, O. S.; Mykhailiuk, P. K.; Voievoda, N. M.; Volochnyuk, D. M.; Komarov, I. V. Synthesis 2010, 443–446. doi:10.1055/s-0029-1217141
Return to citation in text: [1] -
Artamonov, O. S.; Slobodyanyuk, E. Y.; Shishkin, O. V.; Komarov, I. V.; Mykhailiuk, P. K. Synthesis 2013, 45, 225–230. doi:10.1055/s-0032-1316831
Return to citation in text: [1] -
Artamonov, O. S.; Slobodyanyuk, E. Y.; Volochnyuk, D. M.; Komarov, I. V.; Tolmachev, A. A.; Mykhailiuk, P. K. Eur. J. Org. Chem. 2014, 3592–3598. doi:10.1002/ejoc.201402158
Return to citation in text: [1] -
Morandi, B.; Carreira, E. M. Angew. Chem. 2010, 122, 4390–4392. doi:10.1002/ange.201000787
Return to citation in text: [1] -
Morandi, B.; Carreira, E. M. Angew. Chem. 2010, 122, 950–953. doi:10.1002/ange.200905573
Return to citation in text: [1] -
Morandi, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2011, 50, 9085–9088. doi:10.1002/anie.201103526
Return to citation in text: [1] -
Morandi, B.; Carreira, E. M. Org. Lett. 2011, 13, 5984–5985. doi:10.1021/ol202423s
Return to citation in text: [1] -
Künzi, S. A.; Morandi, B.; Carreira, E. M. Org. Lett. 2012, 14, 1900–1901. doi:10.1021/ol300539e
Return to citation in text: [1] -
Hamilton, J. Y.; Morandi, B.; Carreira, E. M. Synthesis 2013, 45, 1857–1862. doi:10.1055/s-0033-1338485
Return to citation in text: [1] -
Wolf, J. R.; Hamaker, C. G.; Djukic, J.-P.; Kodadek, T.; Woo, L. K. J. Am. Chem. Soc. 1995, 117, 9194–9199. doi:10.1021/ja00141a011
Return to citation in text: [1] -
Morandi, B.; Cheang, J.; Carreira, E. M. Org. Lett. 2011, 13, 3080–3081. doi:10.1021/ol200983s
Return to citation in text: [1] -
Mori, T.; Ujihara, K.; Matsumoto, O.; Yanagi, K.; Matsuo, N. J. Fluorine Chem. 2007, 128, 1174–1181. doi:10.1016/j.jfluchem.2007.07.016
Return to citation in text: [1] -
Morandi, B.; Mariampillai, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2011, 50, 1101–1104. doi:10.1002/anie.201004269
Return to citation in text: [1] -
Chen, Y.; Ruppel, J. V.; Zhang, X. P. J. Am. Chem. Soc. 2007, 129, 12074–12075. doi:10.1021/ja074613o
Return to citation in text: [1] -
Liu, C.-B.; Meng, W.; Li, F.; Wang, S.; Nie, J.; Ma, J.-A. Angew. Chem., Int. Ed. 2012, 51, 6227–6230. doi:10.1002/anie.201202372
Return to citation in text: [1] -
Li, F.; Nie, J.; Sun, L.; Zheng, Y.; Ma, J.-A. Angew. Chem., Int. Ed. 2013, 52, 6255–6258. doi:10.1002/anie.201301870
Return to citation in text: [1] -
Li, S.; Cao, W.-J.; Ma, J.-A. Synlett 2017, 28, 673–678. doi:10.1055/s-0036-1588363
Return to citation in text: [1] -
Bordeaux, M.; Tyagi, V.; Fasan, R. Angew. Chem. 2015, 127, 1764–1768. doi:10.1002/ange.201409928
Return to citation in text: [1] -
Wang, H.-X.; Wan, Q.; Low, K.-H.; Zhou, C.-Y.; Huang, J.-S.; Zhang, J.-L.; Che, C.-M. Chem. Sci. 2020, 11, 2243–2259. doi:10.1039/c9sc05432d
Return to citation in text: [1] -
Lin, J.-H.; Xiao, J.-C. Acc. Chem. Res. 2020, 53, 1498–1510. doi:10.1021/acs.accounts.0c00244
Return to citation in text: [1] -
Duan, Y.; Lin, J.-H.; Xiao, J.-C.; Gu, Y.-C. Org. Lett. 2016, 18, 2471–2474. doi:10.1021/acs.orglett.6b01042
Return to citation in text: [1] -
Myronova, V.; Cahard, D.; Marek, I. Org. Lett. 2022, 24, 9076–9080. doi:10.1021/acs.orglett.2c03714
Return to citation in text: [1] -
Hock, K. J.; Mertens, L.; Koenigs, R. M. Chem. Commun. 2016, 52, 13783–13786. doi:10.1039/c6cc07745e
Return to citation in text: [1] -
Duan, Y.; Lin, J.-H.; Xiao, J.-C.; Gu, Y.-C. Chem. Commun. 2017, 53, 3870–3873. doi:10.1039/c7cc01636k
Return to citation in text: [1] -
Ning, Y.; Zhang, X.; Gai, Y.; Dong, Y.; Sivaguru, P.; Wang, Y.; Reddy, B. R. P.; Zanoni, G.; Bi, X. Angew. Chem., Int. Ed. 2020, 59, 6473–6481. doi:10.1002/anie.202000119
Angew. Chem. 2020, 132, 6535–6543. doi:10.1002/ange.202000119
Return to citation in text: [1] -
Yokomatsu, T.; Sato, M.; Abe, H.; Suemune, K.; Matsumoto, K.; Kihara, T.; Soeda, S.; Shimeno, H.; Shibuya, S. Tetrahedron 1997, 53, 11297–11306. doi:10.1016/s0040-4020(97)00704-7
Return to citation in text: [1] -
Yokomatsu, T.; Abe, H.; Sato, M.; Suemune, K.; Kihara, T.; Soeda, S.; Shimeno, H.; Shibuya, S. Bioorg. Med. Chem. 1998, 6, 2495–2505. doi:10.1016/s0968-0896(98)80023-0
Return to citation in text: [1] -
Yokomatsu, T.; Abe, H.; Yamagishi, T.; Suemune, K.; Shibuya, S. J. Org. Chem. 1999, 64, 8413–8418. doi:10.1021/jo990922o
Return to citation in text: [1] -
Yokomatsu, T.; Yamagishi, T.; Suemune, K.; Abe, H.; Kihara, T.; Soeda, S.; Shimeno, H.; Shibuya, S. Tetrahedron 2000, 56, 7099–7108. doi:10.1016/s0040-4020(00)00620-7
Return to citation in text: [1] -
Shevchuk, M.; Wang, Q.; Pajkert, R.; Xu, J.; Mei, H.; Röschenthaler, G.-V.; Han, J. Adv. Synth. Catal. 2021, 2912–2968. doi:10.1002/adsc.202001464
Return to citation in text: [1] -
Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q
Return to citation in text: [1] -
Dmowski, W.; Wolniewicz, A. J. Fluorine Chem. 2000, 102, 141–146. doi:10.1016/s0022-1139(99)00233-x
Return to citation in text: [1] -
Petrov, V. J. Fluorine Chem. 2018, 212, 1–4. doi:10.1016/j.jfluchem.2018.05.004
Return to citation in text: [1] -
Mloston, G.; Huisgen, R. J. Heterocycl. Chem. 1994, 31, 1279–1282. doi:10.1002/jhet.5570310528
Return to citation in text: [1] -
Linev, V. V.; Kolomiets, A. F.; Fokin, A. V. Russ. Chem. Bull. 1992, 41, 360–362. doi:10.1007/bf00869535
Return to citation in text: [1] -
Mei, H.; Liu, J.; Pajkert, R.; Wang, L.; Röschenthaler, G.-V.; Han, J. Org. Chem. Front. 2021, 8, 767–772. doi:10.1039/d0qo01394c
Return to citation in text: [1] -
Mei, H.; Wang, L.; Pajkert, R.; Wang, Q.; Xu, J.; Liu, J.; Röschenthaler, G.-V.; Han, J. Org. Lett. 2021, 23, 1130–1134. doi:10.1021/acs.orglett.1c00150
Return to citation in text: [1] -
Liu, J.; Xu, J.; Pajkert, R.; Mei, H.; Röschenthaler, G.-V.; Han, J. Acta Chim. Sin. (Chin. Ed.) 2021, 79, 747–750. doi:10.6023/a21030096
Return to citation in text: [1] -
Wang, Q.; Wang, L.; Pajkert, R.; Hajdin, I.; Mei, H.; Röschenthaler, G.-V.; Han, J. J. Fluorine Chem. 2021, 251, 109899. doi:10.1016/j.jfluchem.2021.109899
Return to citation in text: [1] -
Liu, J.; Pajkert, R.; Wang, L.; Mei, H.; Röschenthaler, G.-V.; Han, J. Chin. Chem. Lett. 2022, 33, 2429–2432. doi:10.1016/j.cclet.2021.10.066
Return to citation in text: [1] -
Ha, T. M.; Guo, W.; Wang, Q.; Zhu, J. Adv. Synth. Catal. 2020, 362, 2205–2210. doi:10.1002/adsc.202000276
Return to citation in text: [1]
| 1. | de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/anie.197908093 |
| 7. | Le Maux, P.; Juillard, S.; Simonneaux, G. Synthesis 2006, 1701–1704. doi:10.1055/s-2006-926451 |
| 8. | Mykhailiuk, P. K.; Afonin, S.; Ulrich, A. S.; Komarov, I. V. Synthesis 2008, 1757–1760. doi:10.1055/s-2008-1067041 |
| 9. | Mykhailiuk, P. K.; Afonin, S.; Palamarchuk, G. V.; Shishkin, O. V.; Ulrich, A. S.; Komarov, I. V. Angew. Chem., Int. Ed. 2008, 47, 5765–5767. doi:10.1002/anie.200801022 |
| 10. | Artamonov, O. S.; Mykhailiuk, P. K.; Voievoda, N. M.; Volochnyuk, D. M.; Komarov, I. V. Synthesis 2010, 443–446. doi:10.1055/s-0029-1217141 |
| 11. | Artamonov, O. S.; Slobodyanyuk, E. Y.; Shishkin, O. V.; Komarov, I. V.; Mykhailiuk, P. K. Synthesis 2013, 45, 225–230. doi:10.1055/s-0032-1316831 |
| 12. | Artamonov, O. S.; Slobodyanyuk, E. Y.; Volochnyuk, D. M.; Komarov, I. V.; Tolmachev, A. A.; Mykhailiuk, P. K. Eur. J. Org. Chem. 2014, 3592–3598. doi:10.1002/ejoc.201402158 |
| 13. | Morandi, B.; Carreira, E. M. Angew. Chem. 2010, 122, 4390–4392. doi:10.1002/ange.201000787 |
| 14. | Morandi, B.; Carreira, E. M. Angew. Chem. 2010, 122, 950–953. doi:10.1002/ange.200905573 |
| 15. | Morandi, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2011, 50, 9085–9088. doi:10.1002/anie.201103526 |
| 16. | Morandi, B.; Carreira, E. M. Org. Lett. 2011, 13, 5984–5985. doi:10.1021/ol202423s |
| 17. | Künzi, S. A.; Morandi, B.; Carreira, E. M. Org. Lett. 2012, 14, 1900–1901. doi:10.1021/ol300539e |
| 18. | Hamilton, J. Y.; Morandi, B.; Carreira, E. M. Synthesis 2013, 45, 1857–1862. doi:10.1055/s-0033-1338485 |
| 19. | Wolf, J. R.; Hamaker, C. G.; Djukic, J.-P.; Kodadek, T.; Woo, L. K. J. Am. Chem. Soc. 1995, 117, 9194–9199. doi:10.1021/ja00141a011 |
| 20. | Morandi, B.; Cheang, J.; Carreira, E. M. Org. Lett. 2011, 13, 3080–3081. doi:10.1021/ol200983s |
| 21. | Mori, T.; Ujihara, K.; Matsumoto, O.; Yanagi, K.; Matsuo, N. J. Fluorine Chem. 2007, 128, 1174–1181. doi:10.1016/j.jfluchem.2007.07.016 |
| 22. | Morandi, B.; Mariampillai, B.; Carreira, E. M. Angew. Chem., Int. Ed. 2011, 50, 1101–1104. doi:10.1002/anie.201004269 |
| 23. | Chen, Y.; Ruppel, J. V.; Zhang, X. P. J. Am. Chem. Soc. 2007, 129, 12074–12075. doi:10.1021/ja074613o |
| 24. | Liu, C.-B.; Meng, W.; Li, F.; Wang, S.; Nie, J.; Ma, J.-A. Angew. Chem., Int. Ed. 2012, 51, 6227–6230. doi:10.1002/anie.201202372 |
| 25. | Li, F.; Nie, J.; Sun, L.; Zheng, Y.; Ma, J.-A. Angew. Chem., Int. Ed. 2013, 52, 6255–6258. doi:10.1002/anie.201301870 |
| 26. | Li, S.; Cao, W.-J.; Ma, J.-A. Synlett 2017, 28, 673–678. doi:10.1055/s-0036-1588363 |
| 27. | Bordeaux, M.; Tyagi, V.; Fasan, R. Angew. Chem. 2015, 127, 1764–1768. doi:10.1002/ange.201409928 |
| 28. | Wang, H.-X.; Wan, Q.; Low, K.-H.; Zhou, C.-Y.; Huang, J.-S.; Zhang, J.-L.; Che, C.-M. Chem. Sci. 2020, 11, 2243–2259. doi:10.1039/c9sc05432d |
| 29. | Lin, J.-H.; Xiao, J.-C. Acc. Chem. Res. 2020, 53, 1498–1510. doi:10.1021/acs.accounts.0c00244 |
| 30. | Duan, Y.; Lin, J.-H.; Xiao, J.-C.; Gu, Y.-C. Org. Lett. 2016, 18, 2471–2474. doi:10.1021/acs.orglett.6b01042 |
| 31. | Myronova, V.; Cahard, D.; Marek, I. Org. Lett. 2022, 24, 9076–9080. doi:10.1021/acs.orglett.2c03714 |
| 50. | Ha, T. M.; Guo, W.; Wang, Q.; Zhu, J. Adv. Synth. Catal. 2020, 362, 2205–2210. doi:10.1002/adsc.202000276 |
| 4. | Bourlière, M.; Pietri, O.; Castellani, P.; Oules, V.; Adhoute, X. Ther. Adv. Gastroenterol. 2018, 11, 175628481881235. doi:10.1177/1756284818812358 |
| 5. | Abutaleb, A.; Kottilil, S.; Wilson, E. Hepatol. Int. 2018, 12, 214–222. doi:10.1007/s12072-018-9873-y |
| 6. | Davies, J. C.; Moskowitz, S. M.; Brown, C.; Horsley, A.; Mall, M. A.; McKone, E. F.; Plant, B. J.; Prais, D.; Ramsey, B. W.; Taylor-Cousar, J. L.; Tullis, E.; Uluer, A.; McKee, C. M.; Robertson, S.; Shilling, R. A.; Simard, C.; Van Goor, F.; Waltz, D.; Xuan, F.; Young, T.; Rowe, S. M. N. Engl. J. Med. 2018, 379, 1599–1611. doi:10.1056/nejmoa1807119 |
| 3. | Pons, A.; Delion, L.; Poisson, T.; Charette, A. B.; Jubault, P. Acc. Chem. Res. 2021, 54, 2969–2990. doi:10.1021/acs.accounts.1c00261 |
| 44. | Linev, V. V.; Kolomiets, A. F.; Fokin, A. V. Russ. Chem. Bull. 1992, 41, 360–362. doi:10.1007/bf00869535 |
| 2. | Faust, R. Fascinating Natural and Artificial Cyclopropane Architectures. Organic Synthesis Set; Wiley-VCH: Weinheim, Germany, 2003; pp 428–434. doi:10.1002/9783527620784.ch39d |
| 45. | Mei, H.; Liu, J.; Pajkert, R.; Wang, L.; Röschenthaler, G.-V.; Han, J. Org. Chem. Front. 2021, 8, 767–772. doi:10.1039/d0qo01394c |
| 46. | Mei, H.; Wang, L.; Pajkert, R.; Wang, Q.; Xu, J.; Liu, J.; Röschenthaler, G.-V.; Han, J. Org. Lett. 2021, 23, 1130–1134. doi:10.1021/acs.orglett.1c00150 |
| 47. | Liu, J.; Xu, J.; Pajkert, R.; Mei, H.; Röschenthaler, G.-V.; Han, J. Acta Chim. Sin. (Chin. Ed.) 2021, 79, 747–750. doi:10.6023/a21030096 |
| 48. | Wang, Q.; Wang, L.; Pajkert, R.; Hajdin, I.; Mei, H.; Röschenthaler, G.-V.; Han, J. J. Fluorine Chem. 2021, 251, 109899. doi:10.1016/j.jfluchem.2021.109899 |
| 49. | Liu, J.; Pajkert, R.; Wang, L.; Mei, H.; Röschenthaler, G.-V.; Han, J. Chin. Chem. Lett. 2022, 33, 2429–2432. doi:10.1016/j.cclet.2021.10.066 |
| 35. | Yokomatsu, T.; Sato, M.; Abe, H.; Suemune, K.; Matsumoto, K.; Kihara, T.; Soeda, S.; Shimeno, H.; Shibuya, S. Tetrahedron 1997, 53, 11297–11306. doi:10.1016/s0040-4020(97)00704-7 |
| 36. | Yokomatsu, T.; Abe, H.; Sato, M.; Suemune, K.; Kihara, T.; Soeda, S.; Shimeno, H.; Shibuya, S. Bioorg. Med. Chem. 1998, 6, 2495–2505. doi:10.1016/s0968-0896(98)80023-0 |
| 37. | Yokomatsu, T.; Abe, H.; Yamagishi, T.; Suemune, K.; Shibuya, S. J. Org. Chem. 1999, 64, 8413–8418. doi:10.1021/jo990922o |
| 38. | Yokomatsu, T.; Yamagishi, T.; Suemune, K.; Abe, H.; Kihara, T.; Soeda, S.; Shimeno, H.; Shibuya, S. Tetrahedron 2000, 56, 7099–7108. doi:10.1016/s0040-4020(00)00620-7 |
| 40. | Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q |
| 34. |
Ning, Y.; Zhang, X.; Gai, Y.; Dong, Y.; Sivaguru, P.; Wang, Y.; Reddy, B. R. P.; Zanoni, G.; Bi, X. Angew. Chem., Int. Ed. 2020, 59, 6473–6481. doi:10.1002/anie.202000119
Angew. Chem. 2020, 132, 6535–6543. doi:10.1002/ange.202000119 |
| 41. | Dmowski, W.; Wolniewicz, A. J. Fluorine Chem. 2000, 102, 141–146. doi:10.1016/s0022-1139(99)00233-x |
| 42. | Petrov, V. J. Fluorine Chem. 2018, 212, 1–4. doi:10.1016/j.jfluchem.2018.05.004 |
| 43. | Mloston, G.; Huisgen, R. J. Heterocycl. Chem. 1994, 31, 1279–1282. doi:10.1002/jhet.5570310528 |
| 33. | Duan, Y.; Lin, J.-H.; Xiao, J.-C.; Gu, Y.-C. Chem. Commun. 2017, 53, 3870–3873. doi:10.1039/c7cc01636k |
| 32. | Hock, K. J.; Mertens, L.; Koenigs, R. M. Chem. Commun. 2016, 52, 13783–13786. doi:10.1039/c6cc07745e |
| 39. | Shevchuk, M.; Wang, Q.; Pajkert, R.; Xu, J.; Mei, H.; Röschenthaler, G.-V.; Han, J. Adv. Synth. Catal. 2021, 2912–2968. doi:10.1002/adsc.202001464 |
© 2023 Hajdin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.