Abstract
The cyclic peptide core of the antifungal and antibiotic cyclic depsipeptide LI-F04a was synthesised by using a modified Yamaguchi macrolactonization approach. Alternative methods of macrolactonization (e.g., Corey–Nicolaou) resulted in significant epimerization of the C-terminal amino acid during the cyclization reaction. The D-stereochemistry of the alanine residue in the naturally occurring cyclic peptide may be required for the antifungal activity of this natural product.

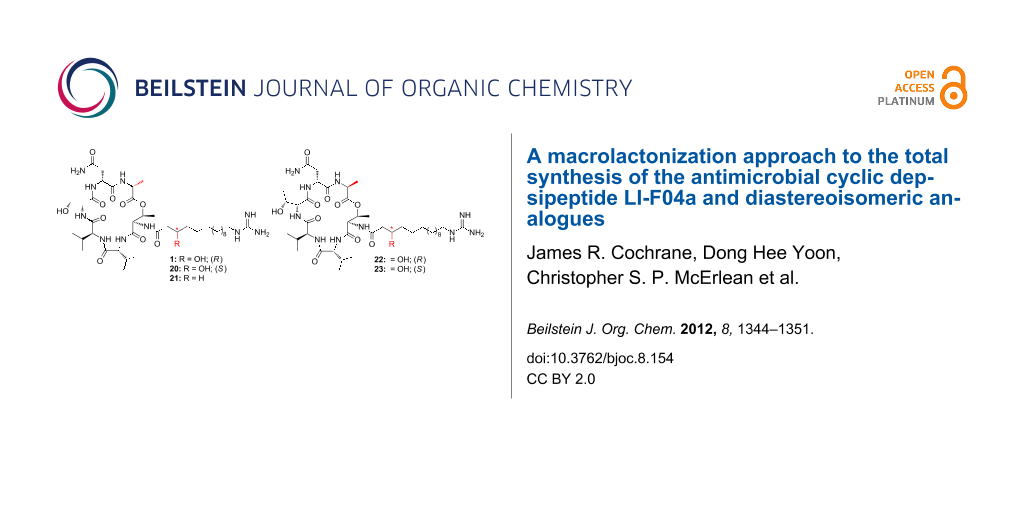
Graphical Abstract
Introduction
The LI-F or fusaricidin class of cyclic depsipeptides are produced by a number of strains of Bacillus (Paenebacillus) and exhibit antifungal and antibacterial activity against a range of clinically relevant species, including Candida albicans, Cryptococcus neoformans, Staphylococcus aureus and Micrococcus luteus [1-7]. These compounds have a cyclic hexadepsipeptide core, in which three amino acids, L-Thr, D-allo-Thr and D-Ala are conserved throughout the series, while there are slight variations in the other three amino acids. In LI-F04a these are D-Asn, L-Val and D-Val. A unique 15-guanidino-3-hydroxypentadecanoyl (GHPD) side chain is appended to the cyclic peptide core through the nitrogen atom of the L-Thr residue. There has been recent interest in the synthesis of the LI-F family of cyclic depsipeptides due to their antifungal activity. Biosynthetic processes have been employed to this end, although these provide mixtures of depsipeptides, which makes it difficult to determine structure–activity relationships [7,8]. More recently, the solid-phase synthesis of a number of analogues of the fusaricidins has been reported. However, in all cases, the side chain 3-hydroxy group was not incorporated into the structure [9]. By total synthesis of both side-chain epimers of this structure we have recently established that the absolute configuration of this side-chain hydroxy group is (R) in the naturally occurring LI-F04a 1 [10]. We employed a late-stage coupling of the cyclic peptide core 2 with the GHDP side chain 3 to enable ready access to both side-chain epimers (Scheme 1). While macrocyclization to give the core 2 could be performed at any of the amide or ester bonds [10], we chose to use a macrolactonization approach to enable ready access to analogues of the LI-F04a core through straightforward Fmoc solid-phase peptide synthesis of the linear precursors. We report here our optimization of these macrolactonization conditions, together with the synthesis of several analogues of LI-F04a using this approach, and an investigation of the antifungal activity of these synthetic lipodepsipeptides.
![[1860-5397-8-154-i1]](/bjoc/content/inline/1860-5397-8-154-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
The required linear precursor for the synthesis of 2 by a macrolactonization approach is 4 (Scheme 1), in which the N-terminal amino group of the L-Thr residue is protected while the side-chain hydroxy group is free. The Cbz group was chosen as a suitable protecting group for the N-terminus. The D-Asn and D-allo-Thr residues were the only amino acids requiring side-chain protection. Given previous reports that 2,2-dimethylated pseudoprolines (ΨMe,Me'Pros) [11-13] are useful turn-inducers for improving yields of macrolactamization reactions [14-17], we chose to prepare two linear precursors, 5 and 6 (Scheme 2), in which the D-allo-Thr protecting group was either tert-butyl or ΨMe,Me'Pro, respectively, to investigate whether a turn-inducer might assist the macrolactonization reaction. Linear peptides 5 and 6 were prepared by standard Fmoc solid-phase peptide synthesis protocols using PyBOP/Hünigs base as the activation reagent and 2-chlorotritylchloride resin to allow cleavage of the peptide from the solid support with side-chain protecting groups intact. In the case of 6, the ΨMe,Me'Pro was introduced by coupling the known dipeptide Fmoc-Val-D-a-Thr(ΨMe,Me'Pro)-OH [18] into the growing peptide chain.
![[1860-5397-8-154-i2]](/bjoc/content/inline/1860-5397-8-154-i2.svg?scale=1.9&max-width=1024&background=FFFFFF)
Scheme 2: Macrolactonization reactions of seco acids 5 and 6 (for reagents and yields see Table 1 and Table 2).
Scheme 2: Macrolactonization reactions of seco acids 5 and 6 (for reagents and yields see Table 1 and Table 2).
While there are a large number of methods available for macrolactonization reactions, those most commonly employed include the Corey–Nicolaou [19], Boden–Keck [20] and Yamaguchi [21] lactonization procedures. We initially chose to screen these three procedures for the macrolactonization of 5. In all three cases (Table 1, entries 1–3), analysis of the crude product mixtures showed that mixtures of cyclic diastereoisomers were obtained, indicating that the C-terminal amino acid underwent epimerization during the macrolactonization reactions (see Supporting Information File 1 for full experimental details) [22]. However, the ratio of the two diastereoisomers differed significantly under the three sets of conditions, with the major diastereomer formed under the Yamaguchi conditions differing from the major product obtained in the other reactions.
Table 1: Reaction conditions for macrocyclization of 5.
| entry | reaction conditions | yield of major isomera | ratio of 7:8b |
|---|---|---|---|
| 1 | dithiopyridine, triphenylphosphine, MeCN, 80 °C | 56% | 13:87 |
| 2 | DCC, DMAP, camphorsulfonic acid, CH2Cl2 | 14% | 45:55 |
| 3 | 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, toluene, 110 °C | 20% | 61:39 |
| 4 | 2,4,6-trichlorobenzoyl chloride, DMAP, iPr2NEt, toluene, 80 °C | 33% | 69:31 |
| 5 | 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, toluene, 25 °C | 33% | 89:11 |
| 6 | 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, THF, 25 °C | 16% | 82:18 |
| 7 | 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, toluene, 25 °C, slow addition of 5 | 58% | 92:8c |
| 8 | 2-methyl-6-nitrobenzoic anhydride, DMAP, NEt3, toluene, 25 °C, slow addition of 5 | 52% | 94:6 |
| 9 | cyanuric chloride, MeCN, 25 °C | 30% | 80:20 |
aIsolated yield; bas determined by integration of analytical HPLC traces of the crude reaction mixtures; caverage value (5–12% epimerization was observed upon repeat reactions).
In order to establish which set of conditions gave the highest ratio of the desired product 7, the mixture of cyclic depsipeptides 7 and 8 obtained from macrolactonization under the highest yielding conditions (Corey–Nicolaou) was separated, and the major diastereomer was subjected to hydrolysis upon treatment with MeOH/H2O/aq NH4OH (4:1:1 v/v/v). Comparison of the crude peptide, so obtained, to authentic samples of both 5 and the C-terminal epimer 9 (which was independently prepared by solid-phase peptide synthesis), indicated that the major product obtained upon macrolactonization of 5 under the Corey–Nicolaou conditions was the undesired cyclic depsipeptide 8, in which the C-terminal Ala residue had epimerized (Figure 1). Since the Yamaguchi conditions gave improved ratios of the desired/epimerized cyclic depsipeptides, subsequent optimization of the macrolactonization conditions focussed on this and related procedures (Table 1). The best yields of the desired cyclic depsipeptide 7 (58% isolated yield, with 5–12% of epimer 8 also observed) were obtained using a modification of Yonemitsu’s conditions [23] in which linear peptide 5 was added slowly to a solution of DMAP, 2,4,6-trichlorobenzoyl chloride and triethylamine in toluene at room temperature. Similar yields and low epimerization (52% isolated yield, 6% epimer observed) were obtained using 2-methyl-6-nitrobenzoic anhydride (MNBA) [24] as the activating agent in place of 2,4,6-trichlorobenzoyl chloride.
![[1860-5397-8-154-1]](/bjoc/content/figures/1860-5397-8-154-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Analytical HPLC traces of linear peptides. (a) Compound 9 (retention time = 31.5 min); (b) Compound 5 (retention time = 30.7 min); (c) co-injection of mixture of 9 and 5 (2:1); (d) Crude reaction mixture after hydrolysis of major cyclic peptide from Corey–Nicolaou cyclization, indicating that the major product of hydrolysis is 9; ▲ indicates the unreacted cyclic peptide (retention time = 34.0 min) and ● is a peak attributed to hydrolysis of both the cyclic peptide ester bond and Cbz protecting group (LCMS m/z = 902 [M + H]+).
Figure 1: Analytical HPLC traces of linear peptides. (a) Compound 9 (retention time = 31.5 min); (b) Compound ...
To investigate whether the ΨMe,Me'Pro would assist macrolactonization, linear precursor 6 was subject to cyclization under a range of conditions (Table 2). However, in all cases, the cyclization yields were found to be lower for the ΨMe,Me'Pro-containing peptide than for the tert-butyl protected precursor 5, with higher amounts of C-terminal epimerization also observed, except in the case of cyclization using cyanuric chloride [25]. Unfortunately, the yield could not be improved above 17% in this case, so all further reactions were performed using the Thr(O-t-Bu) protected cyclic peptides 7 and 8. With cyclic peptides 7 and 8 in hand, we chose to attach the GHPD side chain to both compounds to enable the effect of peptide stereochemistry on the biological activity of the LI-F cyclic peptides to be assessed. Additionally, to investigate the effect of the 3-hydroxy group of the GHPD side chain on the biological activity of this class of cyclic peptide, we prepared the dehydroxy side-chain analogue 12 for attachment to the cyclic peptide core (Scheme 3). Notably, a previously synthesised LI-F04a analogue with a twelve-carbon side chain lacking the hydroxy group has been observed to have antimicrobial activity [9]. Thus, hydrolysis of pentadecanolide 13 [26] was followed by esterification to give the methyl ester 14 in excellent yield. Reaction of 14 with di(tert-butoxycarbonyl)guanidine under Mitsunobu conditions [27] proceeded smoothly to give 15 in 86% yield. Hydrolysis of the methyl ester followed by acidic work up to enable extraction of the resulting carboxylic acid gave 12, in which one of the guanidino Boc protecting groups was also removed.
Table 2: Reaction conditions for macrocyclization of 6.
| entry | reaction conditions | yield of major isomera | ratio of 10:11b |
|---|---|---|---|
| 1 | dithiopyridine, triphenylphosphine, MeCN, 80 °C | 24% | <5 to >95 |
| 2 | 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, toluene, 110 °C | 19% | 50:50 |
| 3 | 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, toluene, 25 °C | 7% | 80:20 |
| 4 | 2,4,6-trichlorobenzoyl chloride, DMAP, NEt3, toluene, 25 °C, slow addition of 5 | 24% | 79:21 |
| 5 | cyanuric chloride, MeCN, 25 °C | 17% | >95 to <5 |
aIsolated yield; bas determined by integration of analytical HPLC traces of the crude reaction mixtures.
![[1860-5397-8-154-i3]](/bjoc/content/inline/1860-5397-8-154-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the dehydroxy side chain 12.
Scheme 3: Synthesis of the dehydroxy side chain 12.
Hydrogenolysis of the Cbz protecting groups of 7 and 8 gave the corresponding amines 16 and 17, respectively (Scheme 4). These were coupled with the previously synthesised side chains 18, 19 [10] and 12, by using HATU as the coupling agent. The resulting compounds were not isolated but immediately subjected to global deprotection upon treatment with trifluoroacetic acid/CH2Cl2/H2O (90:5:5 v/v/v) to give LI-F04a (1), side-chain epimer 20, dehydroxy analogue 21 and the two side-chain epimers of the L-Ala derivative, 22 and 23.
![[1860-5397-8-154-i4]](/bjoc/content/inline/1860-5397-8-154-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of LI-F04a (1) and analogues 20–23.
Scheme 4: Synthesis of LI-F04a (1) and analogues 20–23.
The antifungal activity of 1 and 20–23 was evaluated by using a standardised serial dilution sensitivity assay [28] against reference strains of Candida albicans, Cryptococcus neoformans and Aspergillus fumigatus (Table 3). Synthetic LI-F04a was found to exhibit good activity against both C. albicans and C. neoformans, but only modest activity against A. fumigatus, consistent with the previously reported activity of the natural product [1,3,5]. The side-chain epimer 20 exhibited significantly lower activity than 1 against C. albicans and C. neoformans, indicating that the stereochemistry of the side-chain hydroxy group is important for the antifungal activity of the compounds. Removal of the hydroxy group, as in 21, resulted in a further small decrease in activity against these species. Notably, compounds 22 and 23, prepared from the C-terminal-epimerised cyclic peptide did not exhibit antifungal activity against any of the species tested. This suggests that the conformation of the cyclic peptide core is important in determining the antifungal activity of these compounds, since inversion of the stereocentre of one of the amino acids in the macrocycle is expected to result in a significantly different peptide conformation [29]. Modelled structures of the side-chain-acylated cyclic peptides 24 and 25 obtained by using Monte Carlo conformational searches in Macromodel [30] suggest that these cyclic peptides adopt significantly different conformations with different arrangements of hydrogen bonds (Figure 2). The temperature dependence of the chemical shifts of the signals attributable to the amide NHs of 1 in d6-DMSO was determined experimentally and confirmed the involvement of the D-Ala and D-Asn amide protons in hydrogen bonds [31] as suggested by the modelling studies of the acetamide analogue (see Supporting Information File 1 for details). This indicates that these hydrogen bonds may be important in locking the cyclic depsipeptide into a biologically active conformation.
Table 3: Antifungal activity.
| compound |
C. albicans
ATCC 10231 (MIC µM)a |
C. neoformans
ATCC 90112 (MIC µM)a |
A. fumigatus
ATCC 204305 (MIC µM)a |
|---|---|---|---|
| 1 | 5.5 | 2.8 | 44 |
| 20 | 22 | 11 | 22 |
| 21 | 44 | 22 | 22 |
| 22 | >88 | >88 | >88 |
| 23 | >88 | >88 | >88 |
aMICs for amphotericin B: C. albicans 0.35 µM; C. neoformans 0.35 µM; A. fumigatus 0.70 µM.
![[1860-5397-8-154-2]](/bjoc/content/figures/1860-5397-8-154-2.svg?scale=2.1&max-width=1024&background=FFFFFF)
Figure 2: Structures and lowest-energy conformers of 24 (left) and 25 (right) obtained using Macromodel. Hydrogen bonding is highlighted in yellow.
Figure 2: Structures and lowest-energy conformers of 24 (left) and 25 (right) obtained using Macromodel. Hydr...
Conclusion
In summary, macrolactonization to form the cyclic depsipeptide core of LI-F04a was achieved in good yields by using either modified Yonemitsu conditions or similar conditions, in which the 2,4,6-trichlorobenzoyl chloride activating agent was replaced with MNBA. Slow addition of the linear seco-acid to the activating agents was found to be the key factor to minimizing epimerization of the C-terminal amino acid during the macrolactonization reaction. Synthetic LI-F04a was found to exhibit similar antifungal activity to that reported for the naturally occurring material. The antifungal activity of 1 was reduced upon either inversion of the stereochemistry, or deletion of the side-chain hydroxy group. Inversion of the D-Ala residue in the cyclic depsipeptide core resulted in complete loss of antifungal activity, indicating that the cyclic peptide conformation may be important in the biological activity of this class of cyclic lipodepsipeptide.
Acknowledgements
We thank the University of Sydney for financial support and the award of a postgraduate scholarship to JRC; and Prof. Tania Sorrell and Dr. Julianne Djordjevic (Centre for Infectious Diseases and Microbiology, Westmead Hospital, University of Sydney) for assistance with the antifungal assays.
References
-
Kurusu, K.; Ohba, K.; Arai, T.; Fukushima, K. J. Antibiot. 1987, 40, 1506–1514. doi:10.7164/antibiotics.40.1506
Return to citation in text: [1] [2] -
Kajimura, Y.; Sugiyama, M.; Kaneda, M. J. Antibiot. 1995, 48, 1095–1103. doi:10.7164/antibiotics.48.1095
Return to citation in text: [1] -
Kajimura, Y.; Kaneda, M. J. Antibiot. 1996, 49, 129–135. doi:10.7164/antibiotics.49.129
Return to citation in text: [1] [2] -
Kajimura, Y.; Kaneda, M. J. Antibiot. 1997, 50, 220–228. doi:10.7164/antibiotics.50.220
Return to citation in text: [1] -
Kuroda, J.; Fukai, T.; Konishi, M.; Uno, J.; Kurusu, K.; Nomura, T. Heterocycles 2000, 53, 1533–1549. doi:10.3987/COM-00-8922
Return to citation in text: [1] [2] -
Kaneda, M.; Kajimura, Y. Yakugaku Zasshi 2002, 122, 651–671.
Return to citation in text: [1] -
Choi, S.-K.; Park, S.-Y.; Kim, R.; Lee, C.-H.; Kim, J. F.; Park, S.-H. Biochem. Biophys. Res. Commun. 2008, 365, 89–95. doi:10.1016/j.bbrc.2007.10.147
Return to citation in text: [1] [2] -
Li, J.; Beatty, P. K.; Shah, S.; Jensen, S. E. Appl. Environ. Microbiol. 2007, 73, 3480–3489. doi:10.1128/AEM.02662-06
Return to citation in text: [1] -
Bionda, N.; Stawikowski, M.; Stawikowska, R.; Cudic, M.; López-Vallejo, F.; Treitl, D.; Medina-Franco, J.; Cudic, P. ChemMedChem 2012, 7, 871–882. doi:10.1002/cmdc.201200016
Return to citation in text: [1] [2] -
Cochrane, J. R.; McErlean, C. S. P.; Jolliffe, K. A. Org. Lett. 2010, 12, 3394–3397. doi:10.1021/ol101254m
Return to citation in text: [1] [2] [3] -
Wöhr, T.; Wahl, F.; Nefzi, A.; Rohwedder, B.; Sato, T.; Sun, X.; Mutter, M. J. Am. Chem. Soc. 1996, 118, 9218–9227. doi:10.1021/ja961509q
Return to citation in text: [1] -
Dumy, P.; Keller, M.; Ryan, D. E.; Rohwedder, B.; Wöhr, T.; Mutter, M. J. Am. Chem. Soc. 1997, 119, 918–925. doi:10.1021/ja962780a
Return to citation in text: [1] -
Keller, M.; Sager, C.; Dumy, P.; Schutkowski, M.; Fischer, G. S.; Mutter, M. J. Am. Chem. Soc. 1998, 120, 2714–2720. doi:10.1021/ja973966s
Return to citation in text: [1] -
Skropeta, D. S.; Jolliffe, K. A.; Turner, P. J. Org. Chem. 2004, 69, 8804–8809. doi:10.1021/jo0484732
Return to citation in text: [1] -
Sayyadi, N.; Skropeta, D.; Jolliffe, K. A. Org. Lett. 2005, 7, 5497–5499. doi:10.1021/ol0522891
Return to citation in text: [1] -
Fairweather, K. A.; Sayyadi, N.; Luck, I. J.; Clegg, J. K.; Jolliffe, K. A. Org. Lett. 2010, 12, 3136–3139. doi:10.1021/ol101018w
Return to citation in text: [1] -
Wong, M. S. Y.; Jolliffe, K. A. Aust. J. Chem. 2010, 63, 797–801. doi:10.1071/CH09643
Return to citation in text: [1] -
Clegg, J. K.; Cochrane, J. R.; Sayyadi, N.; Skropeta, D.; Turner, P.; Jolliffe, K. A. Aust. J. Chem. 2009, 62, 711–719. doi:10.1071/CH09151
Return to citation in text: [1] -
Corey, E. J.; Nicolaou, K. C. J. Am. Chem. Soc. 1974, 96, 5614–5616. doi:10.1021/ja00824a073
Return to citation in text: [1] -
Boden, E. P.; Keck, G. E. J. Org. Chem. 1985, 50, 2394–2395. doi:10.1021/jo00213a044
Return to citation in text: [1] -
Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. doi:10.1246/bcsj.52.1989
Return to citation in text: [1] -
Atherton, E.; Sheppard, R. C. Solid Phase Peptide Synthesis: A Practical Approach; IRL Press: Oxford, New York, 1989.
Return to citation in text: [1] -
Hikota, M.; Sakurai, Y.; Horita, K.; Yonemitsu, O. Tetrahedron Lett. 1990, 31, 6367–6370. doi:10.1016/S0040-4039(00)97066-7
Return to citation in text: [1] -
Shiina, I.; Kubota, M.; Ibuka, R. Tetrahedron Lett. 2002, 43, 7535–7539. doi:10.1016/S0040-4039(02)01819-1
Return to citation in text: [1] -
Venkataraman, K.; Wagle, D. R. Tetrahedron Lett. 1980, 21, 1893–1896. doi:10.1016/S0040-4039(00)92809-0
Return to citation in text: [1] -
Nesmeyanov, A. N.; Zakharkin, L. I.; Kost, T. A.; Friedlina, R. K. Russ. Chem. Bull. 1960, 9, 195–199. doi:10.1007/BF00942889
Return to citation in text: [1] -
Dodd, D. S.; Kozikowski, A. P. Tetrahedron Lett. 1994, 35, 977–980. doi:10.1016/S0040-4039(00)79943-6
Return to citation in text: [1] -
Reference Method for Broth Dilution Susceptibility Testing of Yeasts: Approved Standard. NCCLS document M27-A (ISBN 1-56238-186-5). National Committee for Clinical Laboratory Standards, Pennsylvania, USA, 1997; Reference Method for Broth Dilution Susceptibility Testing of Filamentous Fungi: Approved Standard. NCCLS document M38-A (ISBN 1-56238-186-5). National Committee for Clinical Laboratory Standards, Pennsylvania, USA, 2002.
Return to citation in text: [1] -
Haubner, R.; Finsinger, D.; Kessler, H. Angew. Chem., Int. Ed. Engl. 1997, 36, 1374–1389. doi:10.1002/anie.199713741
Return to citation in text: [1] -
Mohamadi, F.; Richards, N. G. J.; Guida, W. C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W. C. J. Comput. Chem. 1990, 11, 440–467. doi:10.1002/jcc.540110405
Return to citation in text: [1] -
Kessler, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 512–523. doi:10.1002/anie.198205121
Return to citation in text: [1]
| 10. | Cochrane, J. R.; McErlean, C. S. P.; Jolliffe, K. A. Org. Lett. 2010, 12, 3394–3397. doi:10.1021/ol101254m |
| 26. | Nesmeyanov, A. N.; Zakharkin, L. I.; Kost, T. A.; Friedlina, R. K. Russ. Chem. Bull. 1960, 9, 195–199. doi:10.1007/BF00942889 |
| 27. | Dodd, D. S.; Kozikowski, A. P. Tetrahedron Lett. 1994, 35, 977–980. doi:10.1016/S0040-4039(00)79943-6 |
| 1. | Kurusu, K.; Ohba, K.; Arai, T.; Fukushima, K. J. Antibiot. 1987, 40, 1506–1514. doi:10.7164/antibiotics.40.1506 |
| 2. | Kajimura, Y.; Sugiyama, M.; Kaneda, M. J. Antibiot. 1995, 48, 1095–1103. doi:10.7164/antibiotics.48.1095 |
| 3. | Kajimura, Y.; Kaneda, M. J. Antibiot. 1996, 49, 129–135. doi:10.7164/antibiotics.49.129 |
| 4. | Kajimura, Y.; Kaneda, M. J. Antibiot. 1997, 50, 220–228. doi:10.7164/antibiotics.50.220 |
| 5. | Kuroda, J.; Fukai, T.; Konishi, M.; Uno, J.; Kurusu, K.; Nomura, T. Heterocycles 2000, 53, 1533–1549. doi:10.3987/COM-00-8922 |
| 6. | Kaneda, M.; Kajimura, Y. Yakugaku Zasshi 2002, 122, 651–671. |
| 7. | Choi, S.-K.; Park, S.-Y.; Kim, R.; Lee, C.-H.; Kim, J. F.; Park, S.-H. Biochem. Biophys. Res. Commun. 2008, 365, 89–95. doi:10.1016/j.bbrc.2007.10.147 |
| 10. | Cochrane, J. R.; McErlean, C. S. P.; Jolliffe, K. A. Org. Lett. 2010, 12, 3394–3397. doi:10.1021/ol101254m |
| 25. | Venkataraman, K.; Wagle, D. R. Tetrahedron Lett. 1980, 21, 1893–1896. doi:10.1016/S0040-4039(00)92809-0 |
| 10. | Cochrane, J. R.; McErlean, C. S. P.; Jolliffe, K. A. Org. Lett. 2010, 12, 3394–3397. doi:10.1021/ol101254m |
| 9. | Bionda, N.; Stawikowski, M.; Stawikowska, R.; Cudic, M.; López-Vallejo, F.; Treitl, D.; Medina-Franco, J.; Cudic, P. ChemMedChem 2012, 7, 871–882. doi:10.1002/cmdc.201200016 |
| 9. | Bionda, N.; Stawikowski, M.; Stawikowska, R.; Cudic, M.; López-Vallejo, F.; Treitl, D.; Medina-Franco, J.; Cudic, P. ChemMedChem 2012, 7, 871–882. doi:10.1002/cmdc.201200016 |
| 23. | Hikota, M.; Sakurai, Y.; Horita, K.; Yonemitsu, O. Tetrahedron Lett. 1990, 31, 6367–6370. doi:10.1016/S0040-4039(00)97066-7 |
| 31. | Kessler, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 512–523. doi:10.1002/anie.198205121 |
| 7. | Choi, S.-K.; Park, S.-Y.; Kim, R.; Lee, C.-H.; Kim, J. F.; Park, S.-H. Biochem. Biophys. Res. Commun. 2008, 365, 89–95. doi:10.1016/j.bbrc.2007.10.147 |
| 8. | Li, J.; Beatty, P. K.; Shah, S.; Jensen, S. E. Appl. Environ. Microbiol. 2007, 73, 3480–3489. doi:10.1128/AEM.02662-06 |
| 24. | Shiina, I.; Kubota, M.; Ibuka, R. Tetrahedron Lett. 2002, 43, 7535–7539. doi:10.1016/S0040-4039(02)01819-1 |
| 19. | Corey, E. J.; Nicolaou, K. C. J. Am. Chem. Soc. 1974, 96, 5614–5616. doi:10.1021/ja00824a073 |
| 21. | Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. doi:10.1246/bcsj.52.1989 |
| 29. | Haubner, R.; Finsinger, D.; Kessler, H. Angew. Chem., Int. Ed. Engl. 1997, 36, 1374–1389. doi:10.1002/anie.199713741 |
| 18. | Clegg, J. K.; Cochrane, J. R.; Sayyadi, N.; Skropeta, D.; Turner, P.; Jolliffe, K. A. Aust. J. Chem. 2009, 62, 711–719. doi:10.1071/CH09151 |
| 22. | Atherton, E.; Sheppard, R. C. Solid Phase Peptide Synthesis: A Practical Approach; IRL Press: Oxford, New York, 1989. |
| 30. | Mohamadi, F.; Richards, N. G. J.; Guida, W. C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W. C. J. Comput. Chem. 1990, 11, 440–467. doi:10.1002/jcc.540110405 |
| 14. | Skropeta, D. S.; Jolliffe, K. A.; Turner, P. J. Org. Chem. 2004, 69, 8804–8809. doi:10.1021/jo0484732 |
| 15. | Sayyadi, N.; Skropeta, D.; Jolliffe, K. A. Org. Lett. 2005, 7, 5497–5499. doi:10.1021/ol0522891 |
| 16. | Fairweather, K. A.; Sayyadi, N.; Luck, I. J.; Clegg, J. K.; Jolliffe, K. A. Org. Lett. 2010, 12, 3136–3139. doi:10.1021/ol101018w |
| 17. | Wong, M. S. Y.; Jolliffe, K. A. Aust. J. Chem. 2010, 63, 797–801. doi:10.1071/CH09643 |
| 28. | Reference Method for Broth Dilution Susceptibility Testing of Yeasts: Approved Standard. NCCLS document M27-A (ISBN 1-56238-186-5). National Committee for Clinical Laboratory Standards, Pennsylvania, USA, 1997; Reference Method for Broth Dilution Susceptibility Testing of Filamentous Fungi: Approved Standard. NCCLS document M38-A (ISBN 1-56238-186-5). National Committee for Clinical Laboratory Standards, Pennsylvania, USA, 2002. |
| 11. | Wöhr, T.; Wahl, F.; Nefzi, A.; Rohwedder, B.; Sato, T.; Sun, X.; Mutter, M. J. Am. Chem. Soc. 1996, 118, 9218–9227. doi:10.1021/ja961509q |
| 12. | Dumy, P.; Keller, M.; Ryan, D. E.; Rohwedder, B.; Wöhr, T.; Mutter, M. J. Am. Chem. Soc. 1997, 119, 918–925. doi:10.1021/ja962780a |
| 13. | Keller, M.; Sager, C.; Dumy, P.; Schutkowski, M.; Fischer, G. S.; Mutter, M. J. Am. Chem. Soc. 1998, 120, 2714–2720. doi:10.1021/ja973966s |
| 20. | Boden, E. P.; Keck, G. E. J. Org. Chem. 1985, 50, 2394–2395. doi:10.1021/jo00213a044 |
| 1. | Kurusu, K.; Ohba, K.; Arai, T.; Fukushima, K. J. Antibiot. 1987, 40, 1506–1514. doi:10.7164/antibiotics.40.1506 |
| 3. | Kajimura, Y.; Kaneda, M. J. Antibiot. 1996, 49, 129–135. doi:10.7164/antibiotics.49.129 |
| 5. | Kuroda, J.; Fukai, T.; Konishi, M.; Uno, J.; Kurusu, K.; Nomura, T. Heterocycles 2000, 53, 1533–1549. doi:10.3987/COM-00-8922 |
© 2012 Cochrane et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)