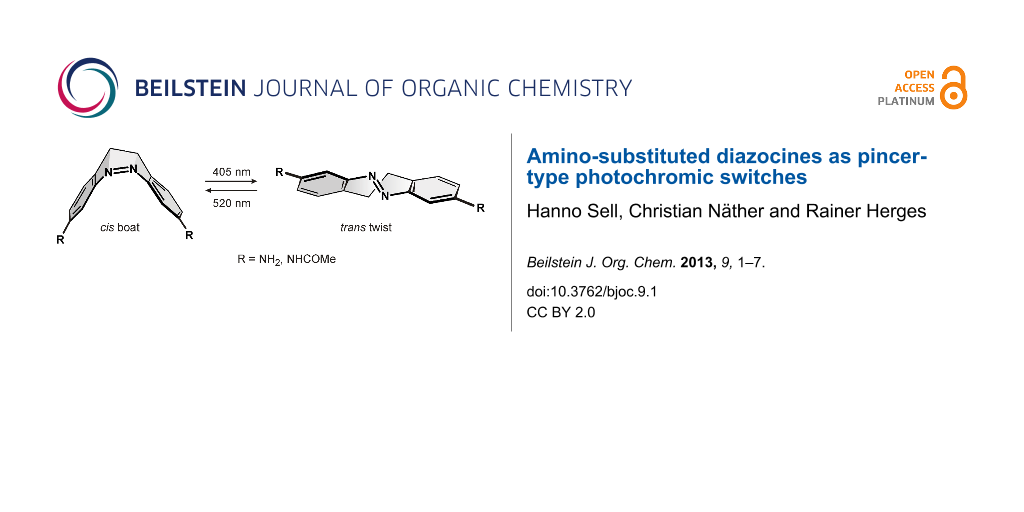
Abstract
Azobenzenes are robust, reliable, and easy to synthesize photochromic switches. However, their high conformational flexibility is a disadvantage in machine-like applications. The almost free rotation of the phenyl groups can be restricted by bridging two ortho positions with a CH2CH2 group, as realized in the dihydrodibenzo diazocine framework. We present the synthesis and properties of 3,3’-amino- and 3,3’-acetamido substituted diazocines. Upon irradiation with light of 405 and 530 nm they isomerize from the cis to the trans configuration and back, and thereby perform a pincer-like motion. In the thermodynamically more stable cis isomer the lone pairs of the amino nitrogen atoms point towards each other, and in the trans form they point in opposite directions. The distance between the amino nitrogen atoms changes between 8 Å (cis) and 11 Å (trans isomer).

Graphical Abstract
Introduction
Azobenzenes probably are the most frequently used photochromic switches in chemistry. They are employed as molecular actuators to drive a number of dynamic machine-like functions [1]. To achieve sophisticated engineering tasks such as directed motion at the molecular level [2,3], the geometry change and the force induced during cis–trans isomerization has to be coupled to the environment. In the macroscopic world, therefore, machines are made from stiff materials. Azobenzene, however, is a rather floppy molecule. Both phenyl rings can rotate with a low activation barrier, and isomerization of the trans form can occur in two different directions, forming two different isomers (enantiomers in the parent system) [4]. Power transmission to neighbouring molecules is inefficient because of energy transfer to internal conformational motion. The first and probably most simple measure to make azobenzene stiffer would be to prevent the phenyl groups from rotating. Connecting both rings with each other via an alkane bridge is probably the most straightforward way to achieve that. Such a molecule, 5,6-dihydrodibenzo[c,g][1,2]diazocine (1), has been known for more than a hundred years [5]. However, only recently we discovered that the photophysical properties of 1 (quantum yields, photostationary states) are superior to those of parent azobenzene, and most of its derivatives [6-8]. In contrast to azobenzene, diazocine 1 is most stable in its cis configuration, which has a boat conformation. The trans isomer comes in two conformations: a chair and a twist form, of which the twist is more stable (Figure 1).
![[1860-5397-9-1-1]](/bjoc/content/figures/1860-5397-9-1-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Configurations and conformations of 5,6-dihydrodibenzo[c,g][1,2]diazocine (1), and DFT (B3LYP/6-31G*) calculated energies (kcal mol−1) relative to the most stable cis boat form. Dipol moments are given in Debye.
Figure 1: Configurations and conformations of 5,6-dihydrodibenzo[c,g][1,2]diazocine (1), and DFT (B3LYP/6-31G...
Proper substitution of the diazocine molecular framework is necessary to control the interaction with the environment or with other molecules. Therefore, we explore different approaches to prepare diazocine derivatives. Since the nomenclature is not unambiguous and, hence, potentially confusing, we refer to 5,6-dihydrodibenzo[c,g][1,2]diazocine derivatives as 2,2’-ethylene-bridged azobenzenes (EBABs).
Results and Discussion
Synthesis
Woolley et al. very recently published the synthesis of a 4,4’-diamino-2,2’-ethylene-bridged azobenzene (4,4’-diamino-EBAB), which exhibits excellent photophysical properties [9]. In planning our synthesis (and not yet being aware of the results of the above authors) we were concerned about the fact that amino substituents in the 4-position with respect to the azo group would impair photochemical conversion, as this is known for 4-amino-substituted azobenzenes [10]. We therefore set out to synthesize 3,3’-diamino-EBAB 4 and derivatives thereof. Key steps of the synthesis are the introduction of the substituents and formation of the azo bond. Several approaches were evaluated, changing the order of the steps and the groups from which the azo unit was generated. The preferred procedure starts from commercially available 1,2-bis(4-aminophenyl)ethane (2). Nitration proceeds almost quantitatively in ortho position to the ethylene bridge, forming 1,2-bis(2-nitro-4-aminophenyl)ethane (3) [11]. Intramolecular reductive coupling of the nitro groups to form the azo unit proceeds with notoriously low yields. The most frequently used procedure using Zn as the reducing agent in Ba(OH)2 [12] or NaOH [13] gives irreproducible and low yields varying from 2% to not more than 19%. Glucose, however, in basic ethanolic solution turned out to furnish the azo compound 4 reproducibly in more than 20% yield. The acetamide derivative 5 is formed by treatment of 3,3’-EBAB 4 with acetic anhydride (Scheme 1).
![[1860-5397-9-1-i1]](/bjoc/content/inline/1860-5397-9-1-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 3,3’-diamino-EBAB 4 and its acetamide derivative 5.
Scheme 1: Synthesis of 3,3’-diamino-EBAB 4 and its acetamide derivative 5.
The structures of the products were confirmed by 1H and 13C NMR spectroscopy, as well as X-ray crystallography (Figure 2). As in the parent system 1 the amino and acetamido derivates 4 and 5 are thermodynamically most stable in their cis form.
![[1860-5397-9-1-2]](/bjoc/content/figures/1860-5397-9-1-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structures of the cis isomers of 3,3’-diamino-EBAB 4 and its acetamide derivative 5. The atoms of the ethylene bridge are disordered.
Figure 2: Crystal structures of the cis isomers of 3,3’-diamino-EBAB 4 and its acetamide derivative 5. The at...
Photochromic Properties
trans-Azobenzene exhibits a high-intensity π–π* band at λmax = 316 nm (ε = 22,000 L∙M−1∙cm−1) and a symmetry-forbidden n–π* band at 444 nm with a very low extinction coefficient (ε = 440 L∙M−1∙cm−1) [10]. Irradiation with UV-light of 365 nm converts the trans to the cis isomer. The n–π* absorption of the nonplanar cis-azobenzene is not formally symmetry forbidden. Even though it has a rather low extinction coefficient (1250 L∙M−1∙cm−1), irradiation into the n–π* band leads to complete conversion back to the trans isomer [14]. Photoswitching of 5,6-dihydrodibenzo[c,g][1,2]diazocine (unsubstituted EBAB 1), however, is performed differently [6]. Conversion of cis to trans, as well as isomerization of trans to cis, is achieved by irradiation into the corresponding n–π* bands. This is possible because the n–π* bands appear well separated at different wavelengths (cis: 404 nm, trans: 490 nm) and the transitions are allowed (albeit weak) in both isomers. Since substituents in the meta position are known to interact less efficiently with each other than those in the ortho or para positions we decided to examine EBABs that are 3,3’-substituted, hoping that the excellent switching properties of the parent system would be retained. For determination of the ratio of isomers in photostationary states we used 1H NMR (for details see Supporting Information File 1).
The UV spectrum of cis-3,3’-diamino-EBAB 4 exhibits a π–π* transition at 350 nm, and a shoulder at approximately 400 nm, which arises from the n–π* transition of the azobenzene substructure (Figure 3). Irradiation of a solution of 4 in acetonitrile with light of 405 nm leads to isomerization to the trans isomer with 34% trans form in the photostationary state (determined by 1H NMR). The rather low conversion rate is probably due to the overlap of the π–π* and n–π* transitions in the cis form. In the trans isomer the n–π* transition is shifted to longer wavelengths (~500 nm) and is clearly separated from any other absorption. Irradiation with light at 450–600 nm therefore converts the trans isomer completely back to the cis form. As compared to the bisamino-substituted EBAB 4 the π–π* and the n–π* bands of the cis isomer of the acetamido derivate 5 are better separated (shoulder at 320 nm and 400 nm) (Figure 3). The photostationary state upon irradiation with 405 nm therefore rises to 54% trans form. As in the bisamino-substituted system 4, conversion of 5 back to the cis isomer is quantitative within the error limits of 1H NMR and UV–vis spectroscopy (Figure 4).
![[1860-5397-9-1-3]](/bjoc/content/figures/1860-5397-9-1-3.png?scale=2.3&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of the diazocine derivatives 3,3’-diamino-EBAB 4 and its bisamide derivative 5 in acetonitrile.
Figure 3: UV–vis spectra of the diazocine derivatives 3,3’-diamino-EBAB 4 and its bisamide derivative 5 in ac...
![[1860-5397-9-1-4]](/bjoc/content/figures/1860-5397-9-1-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Absorbances of solutions of 4 and 5 in acetonitrile at 405 nm (red) and 485 nm (blue) in the corresponding photostationary states upon alternating irradiation at 405 and 530 nm.
Figure 4: Absorbances of solutions of 4 and 5 in acetonitrile at 405 nm (red) and 485 nm (blue) in the corres...
Thermal stability of the trans isomers
Amino and alkylamino substituents in para position to the azo group reduce the lifetime of the cis states of azobenzenes [15,16]. A dramatically shortened half-life of an ortho-dimethylamino-substituted cis-phenylazopyridine (cis-4-N,N-dimethylamino(3-phenylazo)pyridine) has also been observed [17]. The reverse effect was observed in our meta-substituted trans-3,3’-diamino-EBAB 4. While the unsubstituted trans-EBAB 1 exhibits a half-life of 4.5 h in n-hexane solution at room temperature, trans-3,3’-EBAB 4 isomerizes to the more stable cis form with a half-life of 74 h. The corresponding half-life of trans-3,3’-acetamido-EBAB 5 is 46 h (Table 1).
Table 1: Half-life and photostationary states of EBAB 1 and derivatives.
| compound | half life [h] |
PSS 405 nm
% trans |
PSS 520 nm
% trans |
|---|---|---|---|
| EBAB 1 | 4.5a | 92% | <1% |
| 3,3’-diamino-EBAB 4 | 74b | 34% | <1% |
| 3,3’-diacetamido-EBAB 5 | 46b | 54% | <1% |
| 4,4’-diacetamido-EBAB | 4.8c | 70% | <1% |
Application as a molecular pincer
In the 1H NMR spectra of cis-4 and cis-5 the four protons of the ethylene bridge yield a centred multiplet. This symmetry of the fine structure shows that they divide into two chemically unequal groups of two chemically equal protons. Hence, there is no boat inversion at room temperature on the NMR time scale. According to the X-ray crystal structures the amino nitrogen lone pairs of cis-3,3’-diamino EBAB 4 point towards each other and (if extended to more than 6.5 Å) would intersect with an angle of about 65°. If the acetamido groups in 5 are rotated appropriately, the N–H bonds (extended to 10.5 Å) intersect with an angle of about 45°. Thus, the EBAB derivatives 4 and 5 should be suitable as molecular pincers.
To demonstrate this property we studied the binding of ethylenediamine to the two isomers of the acetamido derivative 5 (Figure 5).
![[1860-5397-9-1-5]](/bjoc/content/figures/1860-5397-9-1-5.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 5: DFT-calculated structure (B3LYP/6-31+G**) of a complex of 5 with ethylenediamine as a conceivable model of the binding mode of 3,3’-diacetamido-EBAB 5.
Figure 5: DFT-calculated structure (B3LYP/6-31+G**) of a complex of 5 with ethylenediamine as a conceivable m...
We carried out 1H NMR titrations of cis-5 as well as of the photostationary mixture of cis-5 and trans-5 upon irradiation with light of a wavelength of 405 nm with ethylenediamine in acetonitrile. The spectra showed a significant chemically induced shift (CIS) of the acetamide protons of both isomers upon addition of ethylenediamine. Equilibrium analysis with respect to the CIS binding isotherms by means of nonlinear least-squares methods (for details see Supporting Information File 1) [18,19] yielded a binding constant for the 1:1 ethylenediamine complex of the cis isomer of Ka,cis = 0.88 ± 0.03 M−1. For the corresponding complex of the trans isomer a slightly lower binding constant of Ka,trans = 0.61 ± 0.05 M−1 was determined in acetonitrile-d3 at 25 °C.
Conclusion
We prepared ethylene-bridged azobenzene (EBAB) derivatives with amino and acetamido substituents in the meta position with respect to the azo group (3,3’-diamino-EBAB 4, and 3,3’-diacetamido-EBAB 5). In contrast to azobenzene, and in agreement with the parent EBAB, the cis isomer is more stable than the trans form. Compared to the parent EBAB, which is a very efficient photoswitch, the conversion from the cis to the trans isomer upon irradiation with 405 nm is reduced to 34% (diamino derivative 4) and 54% (diacetamido derivative 5, cf. 92% in the parent compound). The thermal half-life of the trans isomer, however, is drastically increased (3,3’-diamino-EBAB 4: 74 h, 3,3’-diacetamido-EBAB 5: 46 h). The EBAB derivatives upon photoisomerization perform a pincer-like motion. Diacetamido derivative 5 binds ethylenediamine better in its cis (closed) form than in its trans configuration (open form).
Experimental
General remarks
All chemicals were purchased from commercial sources and used without further purification. All NMR spectra were recorded with instruments of the company Bruker (AC 200, DRX 500, and AV 600). The assignments of the NMR signals were confirmed by the evaluation of COSY, HSQC and HMBC spectra. The chemical shifts of the signals were all referenced to residual solvent peaks. The mass spectra were recorded on a Finnigan MAT 8230 instrument. IR spectra were recorded with a Spectrum 100 instrument from Perkin-Elmer equipped with an ATR unit from the company Loriot-Oriel. Melting points were taken without correction. UV–vis spectra were recorded on a Perkin-Elmer Lambda 14 spectrometer.
Synthesis
1,2-Bis(2-nitro-4-aminophenyl)ethane (3): A solution of 5.0 g 1,2-bis(4-aminophenyl)ethane (24.0 mmol) in 40 mL of sulfuric acid was warmed up to 60 °C and a solution of 4.4 g (52.0 mmol) finely grounded sodium nitrate in 45 mL of sulfuric acid was added dropwise. The mixture was stirred at 60 °C for 6 h and afterwards poured into 200 mL of ice–water. The resulting suspension was neutralised by the addition of an aqueous ammonia solution (32%). The red precipitate was filtered off, washed with water and dried in vacuum over CaCl2. Yield: 7.2 g (23.6 mmol, 99%). Mp 247–249 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.06 (d, 4J6,2 = 2.0 Hz, 2H, 6-H), 6.99 (d, 3J3,2 = 8.3 Hz, 2H, 3-H), 6.78 (dd, 3J2,3 = 8.3 Hz, 4J2,6 = 2.1 Hz, 2H, 2-H), 5.58 (s, 4H, 1-NH2), 2.86 (s, 4H, 7-H); 13C NMR (126 MHz, DMSO-d6) δ 149.90 (Cq, C-5), 148.58 (Cq, C-1), 132.85 (d, C-3), 121.85 (Cq, C-4), 119.21 (d, C-2), 108.48 (d, C-6), 33.25 (t, C-7); IR (ATR): 3444 (m), 3363 (s), 3234 (w), 3061 (w), 2947 (w), 2877 (w), 1622 (s), 1513 (vs), 1495 (vs), 1324 (vs), 1272 (s), 1263 (s), 829 (s), 818 (s) cm−1; EIMS (70 eV) m/z (% relative intensity): 302 (16) [M]+, 151 (100); CIMS (isobutane) m/z (% relative intensity): 303 (100) [M + H]+.
(Z)-11,12-Dihydrodibenzo[c,g][1,2]diazocine-3,8-diamine (4): A suspension of 1,2-bis(2-nitro-4-aminophenyl)ethane (3) (1.059 g, 3.5 mmol) in a mixture of 140 mL ethanol and a solution of 8.8 g (220 mmol) sodium hydroxide in 35 mL water was heated to 70 °C. A solution of 6.5 g (36 mmol) glucose in 20 mL water was added, and the reaction mixture was stirred overnight. After cooling to room temperature, 500 mL water was added, and the resulting mixture was extracted three times with 100 mL of ethyl acetate. The organic phase was separated and dried over sodium sulfate, and the solvent was evaporated in vacuum. From the obtained residue the product was isolated by flash chromatography (silica gel, cyclohexane/ethyl acetate 1:1) (254 mg, 1.1 mmol, 30%). Mp 193–196 °C; 1H NMR (600 MHz, DMSO-d6) δ 6.67 (d, 3J6,5 = 8.2 Hz, 2H, 6-H), 6.24 (dd, 3J5,6 = 8.2 Hz, 4J5,3 = 2.3 Hz, 2H, 5-H), 5.97 (d, 4J3,5 = 2.3 Hz, 2H, 3-H), 5.15 (br.s, 4H, 4-NH2), 2.63 (mc, 4H, 7-Ha, 7-Hb,); 13C NMR (150 MHz, DMSO-d6) δ 155.87 (Cq, C-1), 146.76 (Cq, C-2), 130.15 (d, C-6), 115.18 (Cq, C-4), 112.88 (d, C-5), 103.11 (d, C-3), 30.44 (t, C-7); IR (ATR): 3433 (m), 3344 (m), 3433 (m), 2952 (m), 2850 (m), 1703 (w), 1609 (vs), 1570 (m), 1497 (vs), 1455 (s), 1435 (m), 1303 (s), 1272 (s), 1170 (m), 1142 (m), 1094 (m), 1020 (m), 930 (w), 900 (m), 856 (m), 808 (vs) cm−1; EIMS (70 eV) m/z (% relative intensity): 238 (100) [M]+, 209 (92), 193 (46); CIMS (isobutane) m/z (% relative intensity): 239 (100) [M + H]+.
(Z)-N,N'-(11,12-Dihydrodibenzo[c,g][1,2]diazocine-3,8-diyl)diacetamide (5): In acetic acid anhydride (25 mL), (Z)-11,12-dihydrodibenzo[c,g][1,2]diazocine-3,8-diamine (4) (5 mg, 20 mmol) was dissolved. The solution was stirred at room temperature overnight. Afterwards the solvent was evaporated in vacuum, and the product was obtained as a pale yellow solid (7 mg, 20 mmol, 100%). Mp 220–221 °C; 1H NMR (500 MHz, MeCN-d3) δ 8.25 (s, 2H, 5-NH), 7.06 (d, 4J3,5 = 2.2 Hz, 2H, 3-H), 7.03 (dd, 3J5,6 = 8.2 Hz, 4J5,3 = 2.2 Hz, 2H, 5-H), 6.87 (d, 3J3,5 = 2.2 Hz, 2H, 3-H), 2.73 (m, 2H, 7-Ha,), 2.70 (mc, 4H, 7-Ha, 7-Hb), 2.00 (s, 6H, 9-H); 13C NMR (125 MHz, MeCN-d3) δ 168.36 (Cq, C-8), 155.27 (Cq, C-1), 137.45 (Cq, C-2), 129.94 (d, C-6), 123.17 (Cq, C-4), 117.44 (d, C-5), 108.59 (d, C-3), 30.29 (t, C-7), 22.96 (q, C-9); IR (ATR): 3253 (m), 3174 (m), 3102 (m), 3048 (m), 2924 (m), 1711 (m), 1680 (m), 1300 (s), 1260 (s), 1020 (s), 980 (m), 957 (m), 899 (m), 883 (m), 814 (s), 763 (m) cm−1. EIMS (70 eV) m/z (% relative intensity): 322 (50) [M]+, 252 (92), 209 (100); CIMS (isobutane): m/z (% relative intensity) 323 (100) [M + H]+.
Supporting Information
1H NMR spectra of 4 and 5 before and after the irradiation with 405 nm, and 1H NMR binding study of 3,3-acetamido-EBAB (5) with ethylenediamine. cif-Files of X-ray crystal structures of cis-4 and cis-5, and gaussian09 input file of the geometry optimization of the complex of cis-5 and ethylenediamine (DFT B3LYP/6-31+G**).
| Supporting Information File 1: Additional NMR spectra and 1H NMR binding study of 3,3-acetamido-EBAB (5) with ethylenediamine. | ||
| Format: PDF | Size: 662.3 KB | Download |
| Supporting Information File 2: Crystallographic information file of compound cis-4. | ||
| Format: CIF | Size: 11.8 KB | Download |
| Supporting Information File 3: Crystallographic information file of compound cis-5. | ||
| Format: CIF | Size: 10.3 KB | Download |
| Supporting Information File 4: Gaussian09 input file of the geometry optimization of the complex of cis-5 and ethylenediamine. | ||
| Format: GJF | Size: 9.4 KB | Download |
References
-
Merino, E.; Ribagorda, M. Beilstein J. Org. Chem. 2012, 8, 1071–1090. doi:10.3762/bjoc.8.119
Return to citation in text: [1] -
Purcell, E. M. Am. J. Phys. 1977, 45, 3–11. doi:10.1119/1.10903
Return to citation in text: [1] -
Hänggi, P.; Marchesoni, F. Rev. Mod. Phys. 2009, 81, 387–442. doi:10.1103/RevModPhys.81.387
Return to citation in text: [1] -
Haberhauer, G.; Kallweit, C. Angew. Chem. 2010, 122, 2468–2471. doi:10.1002/ange.200906731
Angew. Chem., Int. Ed. 2010, 49, 2418-2421. doi:10.1002/anie.200906731
Return to citation in text: [1] -
Duval, H. Bull. Soc. Chim. Fr. 1910, 7, 727.
Return to citation in text: [1] -
Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d
Return to citation in text: [1] [2] [3] -
Jiang, C.-W.; Xie, R.-H.; Li, F.-L.; Allen, R. E. J. Phys. Chem. A 2011, 115, 244–249. doi:10.1021/jp107991a
Return to citation in text: [1] -
Böckmann, M.; Doltsinis, N. L.; Marx, D. Angew. Chem., Int. Ed. 2010, 49, 3382–3384. doi:10.1002/anie.200907039
Return to citation in text: [1] -
Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383
Angew. Chem., Int. Ed. 2012, 51, 6452–6455. doi:10.1002/anie.201202383
Return to citation in text: [1] [2] -
Rau, H. In Azocompounds in Photochromism, Molecules and Systems; Dürr, H.; Bouas-Laurent, H., Eds.; Elsevier: Amsterdam, 1990; p 165.
The p-Amino substitution leads to a bathochromic shift of the p–p* transition and thus to an overlap of the p–p* and n–p* transitions.
Return to citation in text: [1] [2] -
Matei, S. Rev. Roum. Chim. 1966, 11, 843.
Return to citation in text: [1] -
Paudler, W. W.; Zeiler, A. G. J. Org. Chem. 1969, 34, 3237–3239. doi:10.1021/jo01263a004
Return to citation in text: [1] -
Shine, H. J.; Chamness, J. T. J. Org. Chem. 1963, 28, 1232–1236. doi:10.1021/jo01040a016
Return to citation in text: [1] -
Knoll, H. Photoisomerism of Azobenzenes. CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press LLC: Boca Raton, 2004; 89-1–89-16.
Return to citation in text: [1] -
Schulte-Frohlinde, D. Justus Liebigs Ann. Chem. 1958, 612, 138–152. doi:10.1002/jlac.19586120115
Return to citation in text: [1] -
Mourot, A.; Kienzler, M. A.; Banghart, M. R.; Fehrentz, T.; Huber, F. M. E.; Stein, M.; Kramer, R. H.; Trauner, D. ACS Chem. Neurosci. 2011, 2, 536–543. doi:10.1021/cn200037p
Return to citation in text: [1] -
Thies, S.; Sell, H.; Bornholdt, C.; Schütt, C.; Köhler, F.; Tuczek, F.; Herges, R. Chem.–Eur. J. doi:10.1002/chem.201201698.
Return to citation in text: [1] -
del Piero, S.; Melchior, A.; Polese, P.; Portanova, R.; Tolazzi, M. Ann. Chim. (Rome, Italy) 2006, 96, 29–49. doi:10.1002/adic.200690005
Return to citation in text: [1] -
de Levie, R. J. Chem. Educ. 1999, 76, 1594–1598. doi:10.1021/ed076p1594
Return to citation in text: [1]
| 9. |
Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383
Angew. Chem., Int. Ed. 2012, 51, 6452–6455. doi:10.1002/anie.201202383 |
| 18. | del Piero, S.; Melchior, A.; Polese, P.; Portanova, R.; Tolazzi, M. Ann. Chim. (Rome, Italy) 2006, 96, 29–49. doi:10.1002/adic.200690005 |
| 19. | de Levie, R. J. Chem. Educ. 1999, 76, 1594–1598. doi:10.1021/ed076p1594 |
| 1. | Merino, E.; Ribagorda, M. Beilstein J. Org. Chem. 2012, 8, 1071–1090. doi:10.3762/bjoc.8.119 |
| 6. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 7. | Jiang, C.-W.; Xie, R.-H.; Li, F.-L.; Allen, R. E. J. Phys. Chem. A 2011, 115, 244–249. doi:10.1021/jp107991a |
| 8. | Böckmann, M.; Doltsinis, N. L.; Marx, D. Angew. Chem., Int. Ed. 2010, 49, 3382–3384. doi:10.1002/anie.200907039 |
| 17. | Thies, S.; Sell, H.; Bornholdt, C.; Schütt, C.; Köhler, F.; Tuczek, F.; Herges, R. Chem.–Eur. J. doi:10.1002/chem.201201698. |
| 6. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 4. |
Haberhauer, G.; Kallweit, C. Angew. Chem. 2010, 122, 2468–2471. doi:10.1002/ange.200906731
Angew. Chem., Int. Ed. 2010, 49, 2418-2421. doi:10.1002/anie.200906731 |
| 6. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 2. | Purcell, E. M. Am. J. Phys. 1977, 45, 3–11. doi:10.1119/1.10903 |
| 3. | Hänggi, P.; Marchesoni, F. Rev. Mod. Phys. 2009, 81, 387–442. doi:10.1103/RevModPhys.81.387 |
| 15. | Schulte-Frohlinde, D. Justus Liebigs Ann. Chem. 1958, 612, 138–152. doi:10.1002/jlac.19586120115 |
| 16. | Mourot, A.; Kienzler, M. A.; Banghart, M. R.; Fehrentz, T.; Huber, F. M. E.; Stein, M.; Kramer, R. H.; Trauner, D. ACS Chem. Neurosci. 2011, 2, 536–543. doi:10.1021/cn200037p |
| 12. | Paudler, W. W.; Zeiler, A. G. J. Org. Chem. 1969, 34, 3237–3239. doi:10.1021/jo01263a004 |
| 10. |
Rau, H. In Azocompounds in Photochromism, Molecules and Systems; Dürr, H.; Bouas-Laurent, H., Eds.; Elsevier: Amsterdam, 1990; p 165.
The p-Amino substitution leads to a bathochromic shift of the p–p* transition and thus to an overlap of the p–p* and n–p* transitions. |
| 14. | Knoll, H. Photoisomerism of Azobenzenes. CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press LLC: Boca Raton, 2004; 89-1–89-16. |
| 10. |
Rau, H. In Azocompounds in Photochromism, Molecules and Systems; Dürr, H.; Bouas-Laurent, H., Eds.; Elsevier: Amsterdam, 1990; p 165.
The p-Amino substitution leads to a bathochromic shift of the p–p* transition and thus to an overlap of the p–p* and n–p* transitions. |
| 9. |
Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383
Angew. Chem., Int. Ed. 2012, 51, 6452–6455. doi:10.1002/anie.201202383 |
| 13. | Shine, H. J.; Chamness, J. T. J. Org. Chem. 1963, 28, 1232–1236. doi:10.1021/jo01040a016 |
© 2013 Sell et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)