Abstract
Novel nitrogen-bridged diazocines (triazocines) were synthesized that carry a formyl or an acetyl group at the CH2NR-bridge and bromo- or iodo-substituents at the distant phenyl ring. The photophysical properties were investigated in acetonitrile and water. As compared to previous approaches the yields of the intramolecular azo cyclizations were increased (from ≈40 to 60%) using an oxidative approach starting from the corresponding aniline precursors. The Z→E photoconversion yields in acetonitrile are 80–85% and the thermal half-lives of the metastable E configurations are 31–74 min. Particularly, the high photoconversion yields (≈70%) of the water-soluble diazocines are noteworthy, which makes them promising candidates for applications in photopharmacology. The halogen substituents allow further functionalization via cross-coupling reactions.
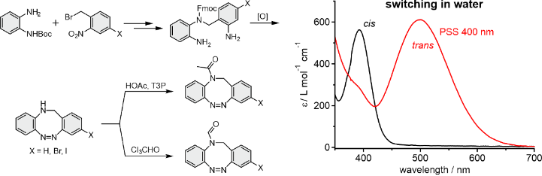
Graphical Abstract
Introduction
Diazocines (bridged azobenzenes) are frequently used photoswitches with outstanding photophysical properties. Parent diazocine (CH2–CH2-bridged) exhibits well-separated n–π* transitions, which allow excellent photoconversion between the Z and E configurations ((Z→E)385 nm = 92%, (E→Z)525 nm > 99% in n-hexane) with light in the visible region [1]. Moreover, the Z-boat configuration is the thermodynamically stable isomer [2-9]. The latter property (i.e., the inverted stability compared to azobenzenes) makes them promising candidates for applications in photopharmacology. In the majority of azobenzene-based photopharmacophores, the bent Z configuration is biologically inactive [10-12]. Hence, (and in contrast to azobenzenes) the thermodynamically stable and biologically inactive Z-isomer can be administered and switched on with light at the site of interest with spatiotemporal resolution. Moreover, the photoconversion yield for the E→Z isomerization is quantitative (within the detection limit of UV and NMR spectroscopy). A high efficiency in switching the biological activity off is important to avoid side effects of residual concentrations of the active form [13].
Water solubility and high Z→E switching efficiencies in water are additional important criteria for applications in biological environments [14]. Our previously published NAc-bridged diazocine 10c (Scheme 1, Table 1) exhibits both properties [15]. This is in stark contrast to the CH2–CH2 and S–CH2-bridged diazocines and the majority of azobenzenes [9,16-20]. Spurred by the promising properties of CH2–NR-bridged diazocines (triazocines), we now explored this class of photoswitches and developed synthetic access to these photochromes (Figure 1).
![[1860-5397-17-107-1]](/bjoc/content/figures/1860-5397-17-107-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bridged diazocines synthesized and investigated in this work.
Figure 1: Bridged diazocines synthesized and investigated in this work.
Results and Discussion
Synthesis
The first three stages of the synthesis of CH2–NR-bridged diazocines are analogous to the previously described synthesis of CH2–NH-bridged diazocine [15]. The single Boc-protected 1,2-phenylenediamine (2, Scheme 1) is reacted with halogen-substituted 2-nitrobenzyl bromides 3 [21] forming N-benzylanilines 4, which were protected with Fmoc chloride to accomplish an orthogonal protective group strategy. The removal of the Boc groups from compounds 5 with TFA gave the mixed aniline and nitro precursors 6.
![[1860-5397-17-107-i1]](/bjoc/content/inline/1860-5397-17-107-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 3-bromo- and 3-iodo-acetylated CH2NR diazocines 10 (R = Ac) and formylated diazocines 11 (R = CHO).
Scheme 1: Synthesis of 3-bromo- and 3-iodo-acetylated CH2NR diazocines 10 (R = Ac) and formylated diazocines ...
In previous approaches, the nitro groups were reduced to hydroxylamines with zinc and oxidized to the corresponding nitroso compounds with iron(III) to perform an intramolecular Baeyer–Mills reaction [15,21]. We found that a complete reduction of the nitro group to aniline 7 and oxidation with mCPBA is increasing the yield of the intramolecular cyclization from 39% to 62% (over two steps) for the unsubstituted diazocine 8c as compared to the pathway via the hydroxylamine. The 3-bromo 8a and 3-iodo 8b compounds were obtained in 56% yield using the oxidative method of Trauner [22] with mCPBA. The Fmoc groups were removed with NEt3 to yield the NH-diazocines 9. The acetylated diazocines 10a–c were synthesized using a mixed anhydride of acetic acid and T3P (propanephosphonic acid anhydride). The formylation of NH-diazocines 9a–c was accomplished with chloral [23] under non-acidic conditions.
Investigation of the photophysical properties
The UV–vis spectra of diazocines 10a–c, and 11a–c were recorded in acetonitrile at 25 °C. All compounds exhibit an n–π* transition at about 400 nm (Z→E conversion) and an n–π* transition at about 520 nm (E→Z conversion, Figure 2, Table 1).
![[1860-5397-17-107-2]](/bjoc/content/figures/1860-5397-17-107-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis spectra of 3-bromo and 3-iodo, and unsubstituted CH2NAc-bridged (10a–c) and CH2NCHO-bridged (11a–c) diazocines. The spectra of Z-isomers are given in black, the photostationary states at 400 nm are represented as dashed red lines and the extrapolated spectra of the pure E-isomers are in blue.
Figure 2: UV–vis spectra of 3-bromo and 3-iodo, and unsubstituted CH2NAc-bridged (10a–c) and CH2NCHO-bridged (...
Table 1: Photophysical properties of N-diazocines 10a–c and 11a–c in acetonitrile.
| acetonitrile | ||||||||
| λmax(Z) | λmax(E) | ελmax(Z) | ελmax(E) | ΓZ→Ea | t1/2 (25 °C) | ΕΑ | ln(A) | |
| nm | L mol−1 cm−1 | % | min | kJ mol−1 | ||||
| Br-NAc 10a | 397 | 515 | 495 | 791 | 81 | 30.9 | 93.4 | 29.8 |
| I-NAc 10b | 397 | 517 | 480 | 778 | 82 | 28.6 | 87.0 | 27.3 |
| NCHO 11c | 397 | 509 | 502 | 760 | 85 | 74.0 | 88.4 | 26.9 |
| Br-NCHO 11a | 397 | 509 | 469 | 784 | 82 | 49.9 | 93.9 | 29.5 |
| I-NCHO 11b | 398 | 511 | 483 | 798 | 80 | 48.1 | 90.9 | 28.3 |
| NAc 10c | 397 | 513 | 495 | 759 | 88 | 29.5 | 87.6 | 27.5 |
aExtrapolated values (for details, see Supporting Information File 1, section IV).
Irradiation with 400 nm gives the metastable E-isomers of the acetylated and formylated derivatives 10 and 11 with good photoconversion yields (Γ) of 80–85% (Table 1) in acetonitrile. A complete E→Z conversion (>99%) can be achieved with light between 520 and 600 nm. The unsubstituted acetylated and formylated diazocines 10c and 11c exhibit similar conversion yields (88% and 85%) and halogenation as well does not have a significant influence. However, thermal half-lives (t1/2) of the metastable E-isomers of the 3-bromo and 3-iodo N-acetyl diazocines 10a and 10b (≈30 min) are significantly smaller than the half-lives of the corresponding bromo and iodo N-formyl derivatives 11a and 11b (≈50 min). In general, halogenation decreases the half-lives compared to unsubstituted diazocines 10c and 11c. The activation barrier (EA) of the E→Z isomerization (obtained by an Arrhenius plot) is higher in formylated compounds 11 compared to acetylated compounds 10 and is further increased by halogenation.
The unsubstituted N-formyl diazocine 11c and brominated NAc-diazocine 10a were also investigated in pure water since they are water-soluble (11c: ≈250 µM, 10a: ≈150 µM). The highest Z→E conversion yields are observed by irradiation with 400 nm in water and the back-isomerization E→Z can be accomplished by irradiation with light in the range of 525 and 600 nm (Figure 3, Table 2).
![[1860-5397-17-107-3]](/bjoc/content/figures/1860-5397-17-107-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of 3-bromo-NAc-diazocine 10a and N-formyl-diazocine 11c in water. Spectra of Z-isomers (black curve), the photostationary states at 400 nm (dashed red line), and the extrapolated spectra of the pure E-isomers (blue).
Figure 3: UV–vis spectra of 3-bromo-NAc-diazocine 10a and N-formyl-diazocine 11c in water. Spectra of Z-isome...
Table 2: Photophysical properties of water-soluble N-diazocines 10a, 10c, and 11c in H2O.
| H2O | ||||||||
| λmax(Z) | λmax(E) | ελmax(Z) | ελmax(E) | ΓZ→Ea | t1/2 (25 °C) | ΕΑ | ln(A) | |
| nm | L mol−1 cm−1 | % | min | kJ mol−1 | ||||
| Br-NAc 10a | 394 | 502 | 534 | 975 | 70 | 69.6 | 99.9 | 31.6 |
| NCHO 11c | 393 | 500 | 567 | 871 | 69 | 198 | 97.8 | 29.7 |
| NAc 10c | 393 | 502 | 564 | 850 | 72 | 72.8 | 90.4 | 27.7 |
aExtrapolated values (for details, see Supporting Information File 1, section IV).
The photoconversion yields (Z→E) of N-formyl diazocine 11c in water and bromo-NAc diazocine 10a are about 70%, which do not differ significantly from unsubstituted NAc diazocine 10c (72%) [15]. It is interesting to note that the half-lives and activation barriers (E→Z) are increasing (t1/2 ≈ 2–2.5-fold) in water as compared to the less polar acetonitrile.
Conclusion
Five nitrogen-bridged diazocines (triazocines) were synthesized and characterized. Formyl (R = CHO) and acetyl groups (R = Ac) were introduced at the CH2NR bridge and the distant phenyl rings are Br and I substituted. In contrast to previous approaches, the azo cyclization (ring closure) was achieved via the oxidation of the bis-anilines 7 with mCPBA (≈60% yield). Among the nitrogen-bridged diazocines compounds 10a and 11c are water soluble and retained their high switching efficiency (≈70%) also in water. The half-lives of the metastable E-isomers are larger for the N-formyl diazocines 11a–c compared to the acetylated compounds 10a–c and generally, the half-lives are larger in water than in acetonitrile. Halogen atoms Br and I at the phenyl rings in 3-position as in 10a,b, and 11a,b are a good starting point for further functionalization [17,20,24]. We conclude that CH2NAc and CH2NCHO bridged diazocines (triazocines) are promising candidates for applications in biological environments and particularly as photoswitches in light-activatable drugs.
Supporting Information
| Supporting Information File 1: Analytical equipment, experimental procedures, NMR and UV–vis spectra. | ||
| Format: PDF | Size: 4.7 MB | Download |
References
-
Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d
Return to citation in text: [1] -
Siewertsen, R.; Schönborn, J. B.; Hartke, B.; Renth, F.; Temps, F. Phys. Chem. Chem. Phys. 2011, 13, 1054–1063. doi:10.1039/c0cp01148g
Return to citation in text: [1] -
Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685
Return to citation in text: [1] -
Moormann, W.; Langbehn, D.; Herges, R. Beilstein J. Org. Chem. 2019, 15, 727–732. doi:10.3762/bjoc.15.68
Return to citation in text: [1] -
Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem., Int. Ed. 2012, 51, 6452–6455. doi:10.1002/anie.201202383
Return to citation in text: [1] -
Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1
Return to citation in text: [1] -
Li, S.; Eleya, N.; Staubitz, A. Org. Lett. 2020, 22, 1624–1627. doi:10.1021/acs.orglett.0c00122
Return to citation in text: [1] -
Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846
Return to citation in text: [1] -
Heintze, L.; Schmidt, D.; Rodat, T.; Witt, L.; Ewert, J.; Kriegs, M.; Herges, R.; Peifer, C. Int. J. Mol. Sci. 2020, 21, 8961–8981. doi:10.3390/ijms21238961
Return to citation in text: [1] [2] -
Beharry, A. A.; Woolley, G. A. Chem. Soc. Rev. 2011, 40, 4422–4437. doi:10.1039/c1cs15023e
Return to citation in text: [1] -
Szymański, W.; Beierle, J. M.; Kistemaker, H. A. V.; Velema, W. A.; Feringa, B. L. Chem. Rev. 2013, 113, 6114–6178. doi:10.1021/cr300179f
Return to citation in text: [1] -
Hüll, K.; Morstein, J.; Trauner, D. Chem. Rev. 2018, 118, 10710–10747. doi:10.1021/acs.chemrev.8b00037
Return to citation in text: [1] -
Fuchter, M. J. J. Med. Chem. 2020, 63, 11436–11447. doi:10.1021/acs.jmedchem.0c00629
Return to citation in text: [1] -
Lubbe, A. S.; Szymanski, W.; Feringa, B. L. Chem. Soc. Rev. 2017, 46, 1052–1079. doi:10.1039/c6cs00461j
Return to citation in text: [1] -
Lentes, P.; Stadler, E.; Röhricht, F.; Brahms, A.; Gröbner, J.; Sönnichsen, F. D.; Gescheidt, G.; Herges, R. J. Am. Chem. Soc. 2019, 141, 13592–13600. doi:10.1021/jacs.9b06104
Return to citation in text: [1] [2] [3] [4] -
Lentes, P.; Frühwirt, P.; Freißmuth, H.; Moormann, W.; Kruse, F.; Gescheidt, G.; Herges, R. J. Org. Chem. 2021, 86, 4355–4360. doi:10.1021/acs.joc.1c00065
Return to citation in text: [1] -
Cabré, G.; Garrido-Charles, A.; González-Lafont, À.; Moormann, W.; Langbehn, D.; Egea, D.; Lluch, J. M.; Herges, R.; Alibés, R.; Busqué, F.; Gorostiza, P.; Hernando, J. Org. Lett. 2019, 21, 3780–3784. doi:10.1021/acs.orglett.9b01222
Return to citation in text: [1] [2] -
Trads, J. B.; Hüll, K.; Matsuura, B. S.; Laprell, L.; Fehrentz, T.; Görldt, N.; Kozek, K. A.; Weaver, C. D.; Klöcker, N.; Barber, D. M.; Trauner, D. Angew. Chem., Int. Ed. 2019, 58, 15421–15428. doi:10.1002/anie.201905790
Return to citation in text: [1] -
Preußke, N.; Moormann, W.; Bamberg, K.; Lipfert, M.; Herges, R.; Sönnichsen, F. D. Org. Biomol. Chem. 2020, 18, 2650–2660. doi:10.1039/c9ob02442e
Return to citation in text: [1] -
Thapaliya, E. R.; Zhao, J.; Ellis-Davies, G. C. R. ACS Chem. Neurosci. 2019, 10, 2481–2488. doi:10.1021/acschemneuro.8b00734
Return to citation in text: [1] [2] -
Schehr, M.; Hugenbusch, D.; Moje, T.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2018, 14, 2799–2804. doi:10.3762/bjoc.14.257
Return to citation in text: [1] [2] -
Maier, M. S.; Hüll, K.; Reynders, M.; Matsuura, B. S.; Leippe, P.; Ko, T.; Schäffer, L.; Trauner, D. J. Am. Chem. Soc. 2019, 141, 17295–17304. doi:10.1021/jacs.9b08794
Return to citation in text: [1] -
Liebig, J. Ann. Pharm. (Lemgo, Ger.) 1832, 1, 182–230. doi:10.1002/jlac.18320010203
Return to citation in text: [1] -
Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Chem. Rev. 2018, 118, 2249–2295. doi:10.1021/acs.chemrev.7b00443
Return to citation in text: [1]
| 1. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 14. | Lubbe, A. S.; Szymanski, W.; Feringa, B. L. Chem. Soc. Rev. 2017, 46, 1052–1079. doi:10.1039/c6cs00461j |
| 13. | Fuchter, M. J. J. Med. Chem. 2020, 63, 11436–11447. doi:10.1021/acs.jmedchem.0c00629 |
| 10. | Beharry, A. A.; Woolley, G. A. Chem. Soc. Rev. 2011, 40, 4422–4437. doi:10.1039/c1cs15023e |
| 11. | Szymański, W.; Beierle, J. M.; Kistemaker, H. A. V.; Velema, W. A.; Feringa, B. L. Chem. Rev. 2013, 113, 6114–6178. doi:10.1021/cr300179f |
| 12. | Hüll, K.; Morstein, J.; Trauner, D. Chem. Rev. 2018, 118, 10710–10747. doi:10.1021/acs.chemrev.8b00037 |
| 15. | Lentes, P.; Stadler, E.; Röhricht, F.; Brahms, A.; Gröbner, J.; Sönnichsen, F. D.; Gescheidt, G.; Herges, R. J. Am. Chem. Soc. 2019, 141, 13592–13600. doi:10.1021/jacs.9b06104 |
| 2. | Siewertsen, R.; Schönborn, J. B.; Hartke, B.; Renth, F.; Temps, F. Phys. Chem. Chem. Phys. 2011, 13, 1054–1063. doi:10.1039/c0cp01148g |
| 3. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 4. | Moormann, W.; Langbehn, D.; Herges, R. Beilstein J. Org. Chem. 2019, 15, 727–732. doi:10.3762/bjoc.15.68 |
| 5. | Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem., Int. Ed. 2012, 51, 6452–6455. doi:10.1002/anie.201202383 |
| 6. | Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1 |
| 7. | Li, S.; Eleya, N.; Staubitz, A. Org. Lett. 2020, 22, 1624–1627. doi:10.1021/acs.orglett.0c00122 |
| 8. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 9. | Heintze, L.; Schmidt, D.; Rodat, T.; Witt, L.; Ewert, J.; Kriegs, M.; Herges, R.; Peifer, C. Int. J. Mol. Sci. 2020, 21, 8961–8981. doi:10.3390/ijms21238961 |
| 17. | Cabré, G.; Garrido-Charles, A.; González-Lafont, À.; Moormann, W.; Langbehn, D.; Egea, D.; Lluch, J. M.; Herges, R.; Alibés, R.; Busqué, F.; Gorostiza, P.; Hernando, J. Org. Lett. 2019, 21, 3780–3784. doi:10.1021/acs.orglett.9b01222 |
| 20. | Thapaliya, E. R.; Zhao, J.; Ellis-Davies, G. C. R. ACS Chem. Neurosci. 2019, 10, 2481–2488. doi:10.1021/acschemneuro.8b00734 |
| 24. | Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Chem. Rev. 2018, 118, 2249–2295. doi:10.1021/acs.chemrev.7b00443 |
| 21. | Schehr, M.; Hugenbusch, D.; Moje, T.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2018, 14, 2799–2804. doi:10.3762/bjoc.14.257 |
| 22. | Maier, M. S.; Hüll, K.; Reynders, M.; Matsuura, B. S.; Leippe, P.; Ko, T.; Schäffer, L.; Trauner, D. J. Am. Chem. Soc. 2019, 141, 17295–17304. doi:10.1021/jacs.9b08794 |
| 15. | Lentes, P.; Stadler, E.; Röhricht, F.; Brahms, A.; Gröbner, J.; Sönnichsen, F. D.; Gescheidt, G.; Herges, R. J. Am. Chem. Soc. 2019, 141, 13592–13600. doi:10.1021/jacs.9b06104 |
| 23. | Liebig, J. Ann. Pharm. (Lemgo, Ger.) 1832, 1, 182–230. doi:10.1002/jlac.18320010203 |
| 9. | Heintze, L.; Schmidt, D.; Rodat, T.; Witt, L.; Ewert, J.; Kriegs, M.; Herges, R.; Peifer, C. Int. J. Mol. Sci. 2020, 21, 8961–8981. doi:10.3390/ijms21238961 |
| 16. | Lentes, P.; Frühwirt, P.; Freißmuth, H.; Moormann, W.; Kruse, F.; Gescheidt, G.; Herges, R. J. Org. Chem. 2021, 86, 4355–4360. doi:10.1021/acs.joc.1c00065 |
| 17. | Cabré, G.; Garrido-Charles, A.; González-Lafont, À.; Moormann, W.; Langbehn, D.; Egea, D.; Lluch, J. M.; Herges, R.; Alibés, R.; Busqué, F.; Gorostiza, P.; Hernando, J. Org. Lett. 2019, 21, 3780–3784. doi:10.1021/acs.orglett.9b01222 |
| 18. | Trads, J. B.; Hüll, K.; Matsuura, B. S.; Laprell, L.; Fehrentz, T.; Görldt, N.; Kozek, K. A.; Weaver, C. D.; Klöcker, N.; Barber, D. M.; Trauner, D. Angew. Chem., Int. Ed. 2019, 58, 15421–15428. doi:10.1002/anie.201905790 |
| 19. | Preußke, N.; Moormann, W.; Bamberg, K.; Lipfert, M.; Herges, R.; Sönnichsen, F. D. Org. Biomol. Chem. 2020, 18, 2650–2660. doi:10.1039/c9ob02442e |
| 20. | Thapaliya, E. R.; Zhao, J.; Ellis-Davies, G. C. R. ACS Chem. Neurosci. 2019, 10, 2481–2488. doi:10.1021/acschemneuro.8b00734 |
| 15. | Lentes, P.; Stadler, E.; Röhricht, F.; Brahms, A.; Gröbner, J.; Sönnichsen, F. D.; Gescheidt, G.; Herges, R. J. Am. Chem. Soc. 2019, 141, 13592–13600. doi:10.1021/jacs.9b06104 |
| 15. | Lentes, P.; Stadler, E.; Röhricht, F.; Brahms, A.; Gröbner, J.; Sönnichsen, F. D.; Gescheidt, G.; Herges, R. J. Am. Chem. Soc. 2019, 141, 13592–13600. doi:10.1021/jacs.9b06104 |
| 21. | Schehr, M.; Hugenbusch, D.; Moje, T.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2018, 14, 2799–2804. doi:10.3762/bjoc.14.257 |
© 2021 Lentes et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)