Abstract
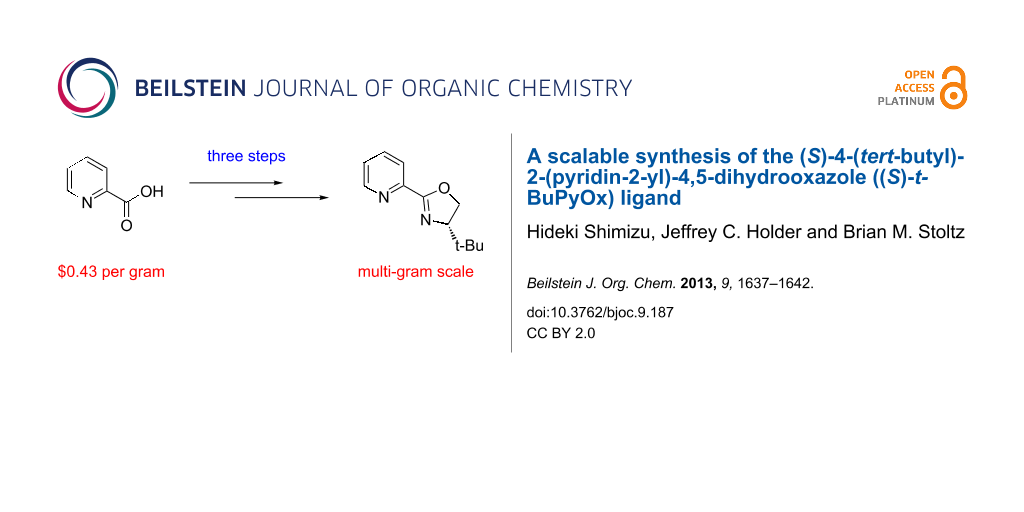
An efficient method for the synthesis of the (S)-4-(tert-butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole ((S)-t-BuPyOx) ligand has been developed. Inconsistent yields and tedious purification in known routes to (S)-t-BuPyOx suggested the need for an efficient, dependable, and scalable synthetic route. Furthermore, a route suitable for the synthesis of PyOx derivatives is desirable. Herein, we describe the development of a three-step route from inexpensive and commercially available picolinic acid. This short procedure is amenable to multi-gram scale synthesis and provides the target ligand in 64% overall yield.

Graphical Abstract
Introduction
Pyridinooxazoline (PyOx) ligands represent a growing class of bidentate dinitrogen ligands used in asymmetric catalysis [1-23]. Recently, our laboratory reported the catalytic asymmetric conjugate addition of arylboronic acids to cyclic, β,β-disubstituted enones utilizing (S)-t-BuPyOx (1) as the chiral ligand (Figure 1) [24]. This robust reaction is insensitive to oxygen atmosphere, highly tolerant of water [25], and provides cyclic ketones bearing β-benzylic quaternary stereocenters in high yields and enantioselectivities. While the reaction itself proved to be amenable to multi-gram scale, the ligand is not yet commercially available and no reliable method for the large-scale synthesis of (S)-t-BuPyOx was known (a number of syntheses are known, including [26]). We sought to address this shortcoming by developing an efficient route starting from a cheap, commercially available precursor to pyridinooxazoline ligands. Herein, we report an efficient, highly scalable synthesis of (S)-t-BuPyOx.
![[1860-5397-9-187-1]](/bjoc/content/figures/1860-5397-9-187-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Initial PyOx synthesis and revised plan.
Figure 1: Initial PyOx synthesis and revised plan.
Results and Discussion
Initially, (S)-t-BuPyOx (1) was synthesized by methanolysis of 2-cyanopyridine (2) to afford methoxyimidate 3, and subsequent acid-catalyzed cyclization to afford the (S)-t-BuPyOx ligand (Figure 1) [27]. We found the yields of this reaction sequence to be highly variable, and the purification by silica gel chromatography to be tedious. In the revised retrosynthesis, picolinic acid (5) was identified as a comparably priced, commonly available surrogate for cyanopyridine 2. Amidation of (S)-tert-leucinol (6) and picolinic acid (5) would generate amide 4, which upon cyclization would generate the ligand framework.
Initial efforts focused on the amidation reaction between (S)-tert-leucinol and picolinic acid (5) via acid chloride 7 (Table 1), which was generated in situ by treatment of acid 5 with a number of chlorinating agents. Oxalyl chloride (Table 1, entries 1,2) provided reasonable yields of amide 4, however bis-acylation of (S)-tert-leucinol was observed as a common side product. Importantly, temperature control of this reaction (Table 1, entry 2) allowed the isolation of 75% of desired alcohol 4 in acceptable purity without the use of column chromatography. Use of diphenyl chlorophosphate (Table 1, entries 3,5,6) also resulted in noticeable quantities of over-acylation products, as well as the generation of a small amount of phosphorylation of amide 4. These results encouraged us to explore alternative activation strategies to generate the desired amide bond. Adapting a procedure from Sigman, activation of acid 5 by treatment with isobutylchloroformate and N-methylmorpholine (anhydride 8) facilitated the desired transformation with the highest overall yield, with amide 4 being isolated in 92% yield, albeit requiring column chromatography [28].
Table 1: Amidation reactions of picolinic acid.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-187-i1.svg?max-width=637&scale=1.0)
|
||||||
| entry | reagent | solvent 1/2 | temp (°C)a | base | time (h)b | yield (%)c |
|---|---|---|---|---|---|---|
| 1 | (COCl)2 | THF/THF | 50 | Et3N | 1 | 55 |
| 2 | (COCl)2 | THF/THF | 0 to rt | Et3N | 7 | 75d |
| 3 | DPCP | THF/THF | 0 to rt | Et3N | 6 | 72 |
| 4 | SOCl2 | toluene/THF | rt | none | 5 | trace |
| 5 | DPCP | THF/THF | 50 | none | 2 | 30 |
| 6 | DPCP | THF/THF | 0 to rt | Et3N | 3 | 65d |
| 7 | iBuOCOCl, NMM | CH2Cl2/CH2Cl2 | 0 to rt | NMM | 3 | 92 |
DPCP = diphenyl chlorophosphate, NMM = N-methylmorpholine. aTemperature for second step; bTime for second step; cIsolated yield; dPurification by flash chromatography not required.
Satisfied with our ability to generate amide 4 on gram-scale with good yield, we turned our attention to the completion of the synthesis. The cyclization of amide 4 to (S)-t-BuPyOx (1) proved more challenging than anticipated. Activation of alcohol 4 as mesylate 9 (Table 2, entries 1,2) and tosylate 10 (Table 2, entry 3) followed by in situ cyclization gave the desired product in low yield and incomplete conversion. This could potentially result from ligand hydrolysis under the given reaction conditions [29]. As an alternative to insitu cyclization of an activated intermediate, alcohol 4 was reacted with thionyl chloride (Table 2, entries 4–10) to yield chloride 11, which was isolated as the hydrochloric acid salt and dried under vacuum. This compound proved to be bench stable and was spectroscopically unchanged after being left open to oxygen atmosphere and adventitious moisture for more than one week. Furthermore, chloride 11 proved to be a superior cyclization substrate. A series of bases were screened. Organic amine bases (Table 2, entries 4,5) and sodium hydride (Table 2, entry 6) provided inadequate conversion and low yields, whereas hydroxide and alkoxide bases proved superior (Table 2, entries 7–10). Finally, sodium methoxide was chosen to be optimal, as slower rates of hydrolysis of chloride 11 were observed when compared to the use of potassium hydroxide.
Table 2: Cyclization screen.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-187-i2.svg?max-width=637&scale=1.0)
|
||||||
| entry | conditions | R | temp (°C) | base | time (h) | yield (%)a |
|---|---|---|---|---|---|---|
| 1 | MsCl, Et3N, CH2Cl2 | OMs | 0 to 40 | Et3N | 12 | N.D.b |
| 2 | MsCl, Et3N, ClCH2CH2Cl | OMs | 0 to 80 | Et3N | 12 | N.D.b |
| 3 | TsCl, DMAP, Et3N, ClCH2CH2Cl | OTs | 0 to 80 | Et3N | 12 | N.D.b |
| 4 | SOCl2 | Clc | rt | DABCO | 18 | 38 |
| 5 | SOCl2 | Clc | 50 | DBU | 12 | 59 |
| 6 | SOCl2 | Clc | 0 to 50 | NaH, THF | 18 | 60 |
| 7 | SOCl2 | Clc | 50 | 5% KOH/EtOH | 11 | 58 |
| 8 | SOCl2 | Clc | 50 | 5% KOH/MeOH | 11 | 62 |
| 9 | SOCl2 | Clc | 50 | 25% NaOMe/MeOH | 10 | 71 |
| 10 | SOCl2 | Clc | 50 | 25% NaOMe/MeOH | 3 | 72 |
MsCl = methanesulfonyl chloride, TsCl = 4-toluenesulfonyl chloride, DMAP = 4-dimethylaminopyridine, DABCO = 1,4-diazabicyclo[2.2.2]octane, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. aIsolated yield; bIncomplete conversion; cIntermediate 11 isolated as HCl salt and dried under high vacuum before use in cyclization reactions.
Attempts to purify ligand 1 via salt formation failed due to instability of the generated products [30]. Purification by silica gel chromatography also proved challenging as up to 10% of crude ligand 1 was observed to decompose, even with the addition of triethylamine to the eluent. Finally, the use of neutral silica gel (American International Chemical ZEOprep ECO silica gel, 40–63 micron, $18/kg) allowed isolation of ligand 1 in high purity and with no observed decomposition.
Conclusion
In conclusion, we have developed a concise, highly efficient and scalable synthesis of the chiral ligand (S)-t-BuPyOx (1) (Figure 2). Efforts to further refine the synthesis by telescoping the procedure and removing chromatographic purifications are currently underway.
![[1860-5397-9-187-2]](/bjoc/content/figures/1860-5397-9-187-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Scale-up synthesis of (S)-t-BuPyOx.
Figure 2: Scale-up synthesis of (S)-t-BuPyOx.
Experimental
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-187-i3.svg?max-width=637&scale=1.18182)
(S)-N-(1-hydroxy-3,3-dimethylbutan-2-yl)picolinamide (4): To a 200 mL round bottom flask was added picolinic acid (2.46 g, 20.0 mmol, 1.00 equiv), 50 mL CH2Cl2, and N-methylmorpholine (3.03 g, 30.0 mmol, 1.50 equiv). The reaction mixture was cooled to 0 °C in an ice bath and isobutyl chloroformate (3.14 g, 23.0 mmol, 1.15 equiv) was added dropwise over 30 min. Following complete addition, the reaction mixture was stirred for 30 min at 0 °C. In a separate flask, (S)-tert-leucinol (2.58 g, 22.0 mmol, 1.10 equiv) was dissolved in CH2Cl2 (25 mL), and N-methylmorpholine (2.43 g, 24.0 mmol, 1.20 equiv) was added. This solution was transferred dropwise over the course of 1 h to the cooled reaction mixture using a syringe pump. The cooling bath was removed and the reaction mixture was allowed to warm to room temperature and stirred for 2 h. The mixture was quenched with an aqueous solution of NH4Cl (10 g in 50 mL H2O) and the aqueous phase was extracted with CH2Cl2 (20 mL). The combined organic phase was dried over Na2SO4 (5 g), filtered, and concentrated under reduced pressure. The residue was purified with flash silica gel column chromatography (4:1 hexanes/acetone) to afford amide alcohol 4 as a white solid (4.10 g, 92% yield). Rf 0.32 with 3:2 hexanes/acetone; mp 79.6–79.9 °C; 1H NMR (500 MHz, CDCl3) δ 8.56 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H), 8.32 (br d, J = 8.9 Hz, -NH), 8.19 (dt, J = 7.8, 1.1 Hz, 1H), 7.85 (td, J = 7.7, 1.7 Hz, 1H), 7.43 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 4.02–3.96 (m, 2H), 3.69 (m, 1H), 2.72 (br t, J = 6.5 Hz, -OH), 1.05 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 165.6, 149.7, 148.2, 137.6, 126.4, 122.6, 63.7, 60.6, 33.9, 27.1; IR (neat film, NaCl): 3375, 2962, 1669, 1591, 1570, 1528, 1465, 1434, 1366, 1289, 1244, 1088, 1053, 998 cm−1; HRMS (MultiMode ESI/APCI) m/z: [M + H]+ calcd for C12H19N2O2, 223.1447; found, 223.1448; [α]25D −8.7 (c 1.17, CHCl3, >99% ee).
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-187-i4.svg?max-width=637&scale=1.18182)
(S)-N-(1-chloro-3,3-dimethylbutan-2-yl)picolinamide hydrochloride (11): A 500 mL 3-neck round bottom flask was charged with a stir bar, amide alcohol 4 (8.89 g, 40.0 mmol, 1.00 equiv) and toluene (140 mL). The resulting clear solution was warmed to 60 °C. In a separate flask, SOCl2 (9.25 g, 80.0 mmol, 2.00 equiv) was diluted with toluene (20 mL). This solution was transferred slowly, dropwise, over 20 min to the vigorously stirring reaction mixture at 60 °C. The reaction mixture was stirred at 60 °C for 4 h, at which time the slurry was cooled to ambient temperature, concentrated on a rotary evaporator under reduced pressure (40 °C, 40 mmHg), and dried under vacuum (0.15 mmHg) to give a white powder of amide chloride hydrochloric salt 11 (10.80 g, 98% yield). This material was used in the following step without purification. 1H NMR (500 MHz, DMSO-d6) δ 8.70 (ddd, J = 4.8, 2.0, 1.0 Hz, 1H), 8.66 (br d, J = 9.9 Hz, -NH), 8.10 (dt, J = 8.0, 1.0 Hz, 1H), 8.06 (td, J = 7.5, 1.4 Hz, 1H), 7.66 (ddd, J = 7.4, 4.8, 1.4 Hz, 1H), 4.08 (td, J = 9.9, 3.7 Hz, 1H), 3.97–3.90 (m, 2H), 0.93 (s, 9H); 13C NMR (125 MHz, DMSO-d6) δ 163.6, 149.0, 147.8, 138.1, 126.5, 122.0, 59.0, 44.9, 35.0, 26.3; IR (neat film, NaCl): 3368, 2963, 1680, 1520, 1465, 1434, 1369, 1285, 1239, 1087, 998 cm−1; HRMS (MultiMode ESI/APCI) m/z: [M + H]+ calcd for C12H18ClN2O, 241.1108; found, 241.1092; [α]25D +39.4 (c 0.96, MeOH, >99% ee).
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-187-i5.svg?max-width=637&scale=1.18182)
(S)-4-(tert-butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole (1): A 500 mL 3-neck round bottom flask was charged with a stir bar, amide chloride hydrochloric acid salt 11 (10.26 g, 37.0 mmol, 1.00 equiv) and MeOH (100 mL). To the clear solution was added powdered NaOMe (9.99 g, 185.0 mmol, 5.00 equiv), and the resulting mixture was heated to 55 °C in an oil bath. The slurry was stirred for 3 h until the free amide chloride was fully consumed, according to TLC analysis (4:1 hexanes/acetone). After removing the oil bath, toluene (100 mL) was added and the mixture was concentrated on a rotary evaporator (40 °C, 60 mmHg) to remove MeOH. The residual mixture was extracted with H2O (100 mL) and the aqueous phase was back extracted with toluene (40 mL × 2). The combined organic extracts were dried over Na2SO4 (10 g), filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography using American International Chemical ZEOprep® 60 ECO 40-63 micron silica gel (4:1 hexanes/acetone) to yield (S)-t-BuPyOx (1) as a white solid (5.44 g, 72% yield). Rf 0.44 with 3:2 hexanes/acetone; mp 70.2–71.0 °C; 1H NMR (500 MHz, CDCl3) δ 8.71 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H), 8.08 (dt, J = 7.9, 1.1 Hz, 1H), 7.77 (dt, J = 7.7, 1.7 Hz, 1H), 7.37 (ddd, J = 7.0, 4.5, 1.0 Hz, 1H), 4.45 (dd, J = 10.2, 8.7 Hz, 1H), 4.31 (t, J = 8.5 Hz, 1H), 4.12 (dd, J = 10.2, 8.5 Hz, 1H), 0.98 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 162.4, 149.6, 147.0, 136.5, 125.4, 124.0, 76.5, 69.3, 34.0, 26.0; IR (neat film, NaCl): 2981, 2960, 2863, 1641, 1587, 1466, 1442, 1358, 1273, 1097, 1038, 968 cm−1; HRMS (MultiMode ESI/APCI) m/z: [M + H]+ calcd for C12H17ON2, 205.1335; found, 205.1327; [α]25D −90.5 (c 1.15, CHCl3, >99% ee).
Supporting Information
Materials and methods, auxiliary experimental details, and relevant NMR spectra are provided.
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 803.3 KB | Download |
Acknowledgements
This publication is based on work supported by NIH-NIGMS (R01GM080269-01), and the authors additionally thank Amgen, Abbott, Boehringer Ingelheim, and Caltech for financial support. J.C.H. thanks the American Chemical Society Division of Organic Chemistry for a graduate research fellowship. We also thank Shionogi & Co., Ltd. for a research grant and fellowship to H.S.
References
-
Podhajsky, S. M.; Iwai, Y.; Cook-Sneathen, A.; Sigman, M. S. Tetrahedron 2011, 67, 4435–4441. doi:10.1016/j.tet.2011.02.027
Return to citation in text: [1] -
Aranda, C.; Cornejo, A.; Fraile, J. M.; García-Verdugo, E.; Gil, M. J.; Luis, S. V.; Mayoral, J. A.; Martínez-Merino, V.; Ochoa, Z. Green Chem. 2011, 13, 983–990. doi:10.1039/c0gc00775g
Return to citation in text: [1] -
Pathak, T. P.; Gligorich, K. M.; Welm, B. E.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 7870–7871. doi:10.1021/ja103472a
Return to citation in text: [1] -
Jiang, F.; Wu, Z.; Zhang, W. Tetrahedron Lett. 2010, 51, 5124–5126. doi:10.1016/j.tetlet.2010.07.084
Return to citation in text: [1] -
Jensen, K. H.; Pathak, T. P.; Zhang, Y.; Sigman, M. S. J. Am. Chem. Soc. 2009, 131, 17074–17075. doi:10.1021/ja909030c
Return to citation in text: [1] -
He, W.; Yip, K.-T.; Zhu, N.-Y.; Yang, D. Org. Lett. 2009, 11, 5626–5628. doi:10.1021/ol902348t
Return to citation in text: [1] -
Dai, H.; Lu, X. Tetrahedron Lett. 2009, 50, 3478–3481. doi:10.1016/j.tetlet.2009.03.005
Return to citation in text: [1] -
Linder, D.; Buron, F.; Constant, S.; Lacour, J. Eur. J. Org. Chem. 2008, 5778–5785. doi:10.1002/ejoc.200800854
Return to citation in text: [1] -
Schiffner, J. A.; Machotta, A. B.; Oestreich, M. Synlett 2008, 2271–2274. doi:10.1055/s-2008-1078271
Return to citation in text: [1] -
Koskinen, A. M. P.; Oila, M. J.; Tois, J. E. Lett. Org. Chem. 2008, 5, 11–16. doi:10.2174/157017808783330216
Return to citation in text: [1] -
Zhang, Y.; Sigman, M. S. J. Am. Chem. Soc. 2007, 129, 3076–3077. doi:10.1021/ja070263u
Return to citation in text: [1] -
Yoo, K. S.; Park, C. P.; Yoon, C. H.; Sakaguchi, S.; O’Neill, J.; Jung, K. W. Org. Lett. 2007, 9, 3933–3935. doi:10.1021/ol701584f
Return to citation in text: [1] -
Dhawan, R.; Dghaym, R. D.; St Cyr, D. J.; Arndtsen, B. A. Org. Lett. 2006, 8, 3927–3930. doi:10.1021/ol061308j
Return to citation in text: [1] -
Xu, W.; Kong, A.; Lu, X. J. Org. Chem. 2006, 71, 3854–3858. doi:10.1021/jo060288w
Return to citation in text: [1] -
Malkov, A. V.; Stewart Liddon, A. J. P.; Ramírez-López, P.; Bendová, L.; Haigh, D.; Kočovský, P. Angew. Chem., Int. Ed. 2006, 45, 1432–1435. doi:10.1002/anie.200503941
Return to citation in text: [1] -
Abrunhosa, I.; Delain-Bioton, L.; Gaumont, A.-C.; Gulea, M.; Masson, S. Tetrahedron 2004, 60, 9263–9272. doi:10.1016/j.tet.2004.07.048
Return to citation in text: [1] -
Brunner, H.; Kagan, H. B.; Kreutzer, G. Tetrahedron: Asymmetry 2003, 14, 2177–2187. doi:10.1016/S0957-4166(03)00433-6
Return to citation in text: [1] -
Cornejo, A.; Fraile, J. M.; García, J. I.; Gil, M. J.; Herrerías, C. I.; Legarreta, G.; Martínez-Merino, V.; Mayoral, J. A. J. Mol. Catal. A: Chem. 2003, 196, 101–108. doi:10.1016/S1381-1169(02)00638-6
Return to citation in text: [1] -
Zhang, Q.; Lu, X.; Han, X. J. Org. Chem. 2001, 66, 7676–7684. doi:10.1021/jo0105181
Return to citation in text: [1] -
Zhang, Q.; Lu, X. J. Am. Chem. Soc. 2000, 122, 7604–7605. doi:10.1021/ja001379s
Return to citation in text: [1] -
Perch, N. S.; Pei, T.; Widenhoefer, R. A. J. Org. Chem. 2000, 65, 3836–3845. doi:10.1021/jo0003192
Return to citation in text: [1] -
Bremberg, U.; Rahm, F.; Moberg, C. Tetrahedron: Asymmetry 1998, 9, 3437–3443. doi:10.1016/S0957-4166(98)00346-2
Return to citation in text: [1] -
Brunner, H.; Obermann, U.; Wimmer, P. Organometallics 1989, 8, 821–826. doi:10.1021/om00105a039
Return to citation in text: [1] -
Kikushima, K.; Holder, J. C.; Gatti, M.; Stoltz, B. M. J. Am. Chem. Soc. 2011, 133, 6902–6905. doi:10.1021/ja200664x
Return to citation in text: [1] -
In fact, we have come to accept that the reaction requires a small amount of water to run efficiently. Typically, 5 equiv water are added to each reaction.
Return to citation in text: [1] -
Brunner, H.; Obermann, U. Chem. Ber. 1989, 122, 499–507. doi:10.1002/cber.19891220318
Return to citation in text: [1] -
This route, reported by our group in [24], is adapted from the synthesis reported in [26]. See Supporting Information File 1 for experimental details.
Return to citation in text: [1] -
Jensen, K. H.; Webb, J. D.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 17471–17482. doi:10.1021/ja108106h
Return to citation in text: [1] -
Degradation experiments demonstrate that ligand 1 is susceptible to hydrolysis. Exposure of t-BuPyOx to 3 N HCl results in complete hydrolysis to amide 4 as observed by 1H NMR.
Return to citation in text: [1] -
Attempts to isolate t-BuPyOx as a salt were successful with HBF4, however the resulting compound was very unstable to atmospheric moisture. See Supporting Information File 1.
Return to citation in text: [1]
| 1. | Podhajsky, S. M.; Iwai, Y.; Cook-Sneathen, A.; Sigman, M. S. Tetrahedron 2011, 67, 4435–4441. doi:10.1016/j.tet.2011.02.027 |
| 2. | Aranda, C.; Cornejo, A.; Fraile, J. M.; García-Verdugo, E.; Gil, M. J.; Luis, S. V.; Mayoral, J. A.; Martínez-Merino, V.; Ochoa, Z. Green Chem. 2011, 13, 983–990. doi:10.1039/c0gc00775g |
| 3. | Pathak, T. P.; Gligorich, K. M.; Welm, B. E.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 7870–7871. doi:10.1021/ja103472a |
| 4. | Jiang, F.; Wu, Z.; Zhang, W. Tetrahedron Lett. 2010, 51, 5124–5126. doi:10.1016/j.tetlet.2010.07.084 |
| 5. | Jensen, K. H.; Pathak, T. P.; Zhang, Y.; Sigman, M. S. J. Am. Chem. Soc. 2009, 131, 17074–17075. doi:10.1021/ja909030c |
| 6. | He, W.; Yip, K.-T.; Zhu, N.-Y.; Yang, D. Org. Lett. 2009, 11, 5626–5628. doi:10.1021/ol902348t |
| 7. | Dai, H.; Lu, X. Tetrahedron Lett. 2009, 50, 3478–3481. doi:10.1016/j.tetlet.2009.03.005 |
| 8. | Linder, D.; Buron, F.; Constant, S.; Lacour, J. Eur. J. Org. Chem. 2008, 5778–5785. doi:10.1002/ejoc.200800854 |
| 9. | Schiffner, J. A.; Machotta, A. B.; Oestreich, M. Synlett 2008, 2271–2274. doi:10.1055/s-2008-1078271 |
| 10. | Koskinen, A. M. P.; Oila, M. J.; Tois, J. E. Lett. Org. Chem. 2008, 5, 11–16. doi:10.2174/157017808783330216 |
| 11. | Zhang, Y.; Sigman, M. S. J. Am. Chem. Soc. 2007, 129, 3076–3077. doi:10.1021/ja070263u |
| 12. | Yoo, K. S.; Park, C. P.; Yoon, C. H.; Sakaguchi, S.; O’Neill, J.; Jung, K. W. Org. Lett. 2007, 9, 3933–3935. doi:10.1021/ol701584f |
| 13. | Dhawan, R.; Dghaym, R. D.; St Cyr, D. J.; Arndtsen, B. A. Org. Lett. 2006, 8, 3927–3930. doi:10.1021/ol061308j |
| 14. | Xu, W.; Kong, A.; Lu, X. J. Org. Chem. 2006, 71, 3854–3858. doi:10.1021/jo060288w |
| 15. | Malkov, A. V.; Stewart Liddon, A. J. P.; Ramírez-López, P.; Bendová, L.; Haigh, D.; Kočovský, P. Angew. Chem., Int. Ed. 2006, 45, 1432–1435. doi:10.1002/anie.200503941 |
| 16. | Abrunhosa, I.; Delain-Bioton, L.; Gaumont, A.-C.; Gulea, M.; Masson, S. Tetrahedron 2004, 60, 9263–9272. doi:10.1016/j.tet.2004.07.048 |
| 17. | Brunner, H.; Kagan, H. B.; Kreutzer, G. Tetrahedron: Asymmetry 2003, 14, 2177–2187. doi:10.1016/S0957-4166(03)00433-6 |
| 18. | Cornejo, A.; Fraile, J. M.; García, J. I.; Gil, M. J.; Herrerías, C. I.; Legarreta, G.; Martínez-Merino, V.; Mayoral, J. A. J. Mol. Catal. A: Chem. 2003, 196, 101–108. doi:10.1016/S1381-1169(02)00638-6 |
| 19. | Zhang, Q.; Lu, X.; Han, X. J. Org. Chem. 2001, 66, 7676–7684. doi:10.1021/jo0105181 |
| 20. | Zhang, Q.; Lu, X. J. Am. Chem. Soc. 2000, 122, 7604–7605. doi:10.1021/ja001379s |
| 21. | Perch, N. S.; Pei, T.; Widenhoefer, R. A. J. Org. Chem. 2000, 65, 3836–3845. doi:10.1021/jo0003192 |
| 22. | Bremberg, U.; Rahm, F.; Moberg, C. Tetrahedron: Asymmetry 1998, 9, 3437–3443. doi:10.1016/S0957-4166(98)00346-2 |
| 23. | Brunner, H.; Obermann, U.; Wimmer, P. Organometallics 1989, 8, 821–826. doi:10.1021/om00105a039 |
| 27. | This route, reported by our group in [24], is adapted from the synthesis reported in [26]. See Supporting Information File 1 for experimental details. |
| 26. | Brunner, H.; Obermann, U. Chem. Ber. 1989, 122, 499–507. doi:10.1002/cber.19891220318 |
| 25. | In fact, we have come to accept that the reaction requires a small amount of water to run efficiently. Typically, 5 equiv water are added to each reaction. |
| 24. | Kikushima, K.; Holder, J. C.; Gatti, M.; Stoltz, B. M. J. Am. Chem. Soc. 2011, 133, 6902–6905. doi:10.1021/ja200664x |
| 24. | Kikushima, K.; Holder, J. C.; Gatti, M.; Stoltz, B. M. J. Am. Chem. Soc. 2011, 133, 6902–6905. doi:10.1021/ja200664x |
| 30. | Attempts to isolate t-BuPyOx as a salt were successful with HBF4, however the resulting compound was very unstable to atmospheric moisture. See Supporting Information File 1. |
| 29. | Degradation experiments demonstrate that ligand 1 is susceptible to hydrolysis. Exposure of t-BuPyOx to 3 N HCl results in complete hydrolysis to amide 4 as observed by 1H NMR. |
| 28. | Jensen, K. H.; Webb, J. D.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 17471–17482. doi:10.1021/ja108106h |
| 26. | Brunner, H.; Obermann, U. Chem. Ber. 1989, 122, 499–507. doi:10.1002/cber.19891220318 |
© 2013 Shimizu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)