Abstract
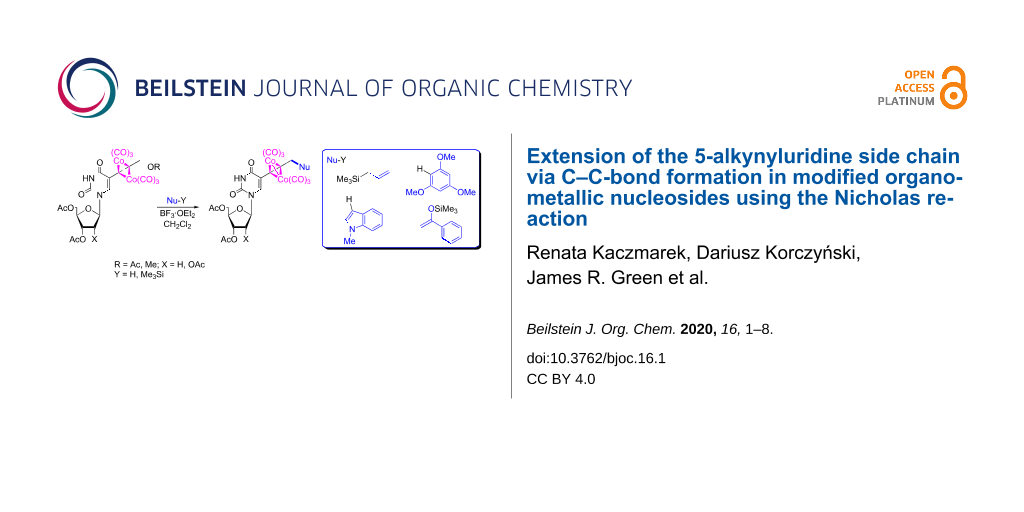
Dicobalt hexacarbonyl nucleoside complexes of propargyl ether or esters of 5-substituted uridines react with diverse C-nucleophiles. Synthetic outcomes confirmed that the Nicholas reaction can be carried out in a nucleoside presence, leading to a divergent synthesis of novel metallo-nucleosides enriched with alkene, arene, arylketo, and heterocyclic functions, in the deoxy and ribo series.

Graphical Abstract
Introduction
Nucleoside analogs are molecules of high pharmacological interest for the treatment of various conditions, especially cancer and viral diseases [1-5]. The substitution at C-5 of the uracil nucleobase provides a common framework for materials with potent biological properties [6-10]. Modification on this site of the nucleobase usually does not interfere with Watson–Crick base pairing. For example, C-5-modified pyrimidines are well tolerated by commercial polymerases [11,12]. Alkynyl modifications not only provide a biological impact but also create a synthetic handle for further functionalization/modification. Among others, alkynyl uridines undergo cycloisomerization to potent antiviral agents, furopyrimidines [13], related halofuropyrimidines [14], and can be converted into interstrand dimers [15].
In parallel, bioorganometallic chemistry provides new tools to influence biological interactions [16-24]. Cobalt possesses a diverse array of properties that can be manipulated to yield promising drug candidates [25]. The antiproliferative properties [26], as well as carbon monoxide-releasing properties [27,28] of dicobalt hexacarbonyl alkyne complexes have been noted, and their medicinal potential has been summarized [29-31].
Despite developments, the collection of metallo-nucleosides is limited. Hybridization of alkyl and aryl-substituted alkyne cobalt hexacarbonyls with 2'-deoxyuridines revealed pronounced in vitro activity against MCF-7 and MDA-MB-231 human breast cancer cells [32,33]. A recent investigation of hexacarbonyl dicobalt adducts of nucleosides containing derivatives of propargyl alcohol demonstrated their antiproliferative activities for the HeLa and K562 cell lines [31]. The formation of a reactive oxygen species in the presence of cobalt compounds was determined in K562 cells. The results indicate that the mechanism of action for most antiproliferative cobalt compounds may be related to the induction of oxidative stress [31]. Consequently, we aimed to develop methods that would synthetically extend the design of the metallo-nucleosides by introducing functionalized ligands in divergent synthesis. We decided to pursue the Nicholas reaction in the presence of the labile nucleoside unit, further modifying the already available material containing the propargyl alcohol derivative unit.
The chemistry of cationic propargyl dicobalt complexes, recognized as the Nicholas reaction, has become one of the most widely appreciated forms of metalorganic chemistry. These cations are generated most commonly from propargyl alcohol, -ether, or -acetate hexacarbonyl dicobalt complexes and a Lewis or Brønsted acid. A range of heteroatom nucleophiles have been incorporated into alkyne dicobalt complexes by this chemistry [34-40]. However, reactions with carbon-based nucleophiles provide an opportunity to access the structurally diverse products via formation of C–C bonds. Nucleophiles as diverse as electron-rich arenes or heteroarenes [41,42], alkenes [43], allylmetalloids [44-46], enol derivatives [47,48], and organometallics [49] are suitable for the Nicholas reaction. Allenic byproducts are rarely seen, and intramolecular versions of the reaction are also highly successful [50,51].
Although the Nicholas reaction has been employed to functionalize biomolecules, including amino acids [52,53], β-lactams [54], steroids [55], and carbohydrates [56-62], we are unaware of any examples of nucleoside functionalization by way of propargyl dicobalt cation chemistry. Nucleoside modifications are considerably challenging due to the presence of reactive functional groups. Since numerous uridine C-5 modifications play an important role in biochemistry, we considered exploration of pertinent methods development warranted, which at the same time may provide biologically active compounds.
Results and Discussion
Preparation of 5-alkynyluridines was carried out from acyl-protected 5-iodouridines (1a,b) [8,63] and the appropriate terminal alkyne in the presence of catalytic amounts of Pd(PPh3)4, copper(I) iodide, triethylamine, in DMF, and at room temperature – to avoid cycloisomerization to furopyrimidines (Scheme 1). The modified pyrimidine nucleoside scaffolds, propargyl acetate-substituted 2'-deoxyuridine (R = Ac, 2) and propargyl methyl ether-substituted uridine (R = Me, 3), were obtained in 87% and 61% yield, respectively. These specific combinations were not optimized since we presumed that acetate and methyl ether could be used interchangeably. Acetyl protection has been introduced to alcohol functions to prevent free hydroxy groups from competing with the C-nucleophiles in the Nicholas reaction. The structures of alkynyl nucleosides 2 and 3 were confirmed by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry [64,65].
![[1860-5397-16-1-i1]](/bjoc/content/inline/1860-5397-16-1-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of (2'-deoxy)-5-alkynyluridines 2 and 3, their dicobalt hexacarbonyl derivatives 4 and 5, and the subsequent Nicholas reaction.
Scheme 1: Preparation of (2'-deoxy)-5-alkynyluridines 2 and 3, their dicobalt hexacarbonyl derivatives 4 and 5...
The conversion of alkynyl nucleosides 2 and 3 into the corresponding dicobalt hexacarbonyl nucleosides complexes of 4 and 5 was accomplished at room temperature (Co2(CO)8, THF, 22 °C, 1 h) with 88–77% yield after silica gel column chromatography (Scheme 1). The structures of nucleosides 4 and 5 were confirmed by NMR and IR spectroscopy (for the synthesis of a related unprotected nucleoside, see [31]). The MS spectra of 4 and 5 exhibited appropriate high resolution molecular ions’ masses.
The solutions of uridine complexes 4 and 5 in dichloromethane were subjected to Nicholas reactions with a variety of diverse nucleophiles in the presence of BF3·OEt2. Representatives of the major classes of C-based nucleophiles in Nicholas reaction chemistry were selected, including electron-rich arenes, π-excessive heterocycles, enol derivatives, and allylmetalloids. Specifically, the reactivity of 1,3,5-trimethoxybenzene, N-methylindole, acetophenone trimethylsilyl enol ether, and allyltrimethylsilane was investigated (Table 1). The Nicholas reaction products 6 and 7 (Figure 1) were obtained successfully in moderate to good yields (Table 1). The reactions progressed quite slowly and required an excess amount of the Lewis acid (4–5 equiv) to proceed at a preparatively reasonable rate (Table 1, entries 1–4). These observations can be attributed to the substantial number of potentially competing Lewis basic sites in 4 and 5. The use of tin(IV) chloride (stannic chloride) provided generally a slightly faster reaction but with slightly lower yields, except in the case of the 5/allyltrimethylsilane/7a combination (Table 1, entry 5). Reactions were more successful when the amount of nucleophile present was in slight excess relative to that of the Lewis acid, whereas limited amounts of nucleophile resulted in greater amounts of decomposition. Slightly more decomposition products were observed by TLC in reactions with ribo nucleoside 5 (Table 1, entries 4–7) than with 2'-deoxy derivative 4 (Table 1, entries 1–3), leading to higher yields for nucleosides 6a–c relative to 7a,b,d (Table 1).
Table 1: Preparation of modified uridine dicobalt hexacarbonyl derivatives 6 and 7 via the Nicholas reaction (BF3·OEt2, CH2Cl2, 0 °C to rt).
| entry | nucleoside | nucleophile | product | yield [%] |
| 1 | 4 | allyltrimethylsilane | 6a | 55 |
| 2 | 4 | 1,3,5-trimethoxybenzene | 6b | 89 |
| 3 | 4 | acetophenone trimethylsilyl enol ether | 6c | 49 |
| 4 | 5 | allyltrimethylsilane | 7a | 38 |
| 5 | 5 | allyltrimethylsilane | 7a | 37 (46)a,b |
| 6 | 5 | 1,3,5-trimethoxybenzene | 7b | 47 |
| 7 | 5 | N-methylindole | 7d | 40 |
aUsing SnCl4. bYield in parentheses is based on recovered starting material (brsm).
![[1860-5397-16-1-1]](/bjoc/content/figures/1860-5397-16-1-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of nucleosides 6 and 7, products of the Nicholas reaction.
Figure 1: Structures of nucleosides 6 and 7, products of the Nicholas reaction.
The reaction products were characterized by the disappearance of the formally diastereotopic propargylic methylene 1H NMR (CDCl3) spectral resonances (ca. 5.5 ppm in 4 and 4.8 ppm in 5) and their reappearance upfield in the reaction products (i.e., 3.17 ppm in 6a and 4.27 ppm in 6b). In the 13C NMR spectra, the slightly broadened resonance of the metal carbonyl carbons at 199–200 ppm (199.4 ppm in both 6a and 6b) were characteristic of the product alkyne–Co2(CO)6 complexes. In the IR spectra, the series of intense metal–CO stretching bands between 2000–2100 cm−1 (e.g., 6a, 2089, 2049, 2017 cm−1; 6b, 2088, 2048, 2018 cm−1) dominated even the organic carbonyl bands.
Conclusion
The Nicholas reaction, in which site of reactivity is well-defined and commonly free from formation of allenic byproducts, has been introduced into the repertoire of nucleosides modifications. The reaction of dicobalt hexacarbonyl propargylic alcohol uridine derivatives has been validated with diverse C-nucleophiles. By this means, alkene, arene, arylketo, and heterocyclic functions can be introduced onto metallo-nucleosides, preserving the dicobalt hexacarbonyl unit. This methodology allows for access in a divergent fashion to a variety of modified nucleosides with potential biological activity, and was shown to be viable for both 2'-deoxy- and regular uridines.
Experimental
General and instrumentation. All NMR measurements were carried out on Bruker Avance III spectrometers operating for 1H NMR at 500 MHz, 600 MHz or 300 MHz and for 13C NMR at 125 MHz or 150 MHz, at 22 °C. Mass spectra were recorded on an Agilent 6520 Q-TOF LCMS (HRMS). FTIR spectra were recorded on ATI Mattson Infinity Series AR60, Thermo Scientific Nicolet 6700 ATR, or Bruker Alpha-P ATR spectrometers. All reactions were carried out under a nitrogen atmosphere and all products were stored in a freezer at −10 °C.
3',5'-Di-O-acetyl-2'-deoxy-5-[3-(acetoxy)prop-1-yn-1-yl]uridine (2). A round-bottom flask was charged with 3',5'-di-O-acetyl-2'-deoxy-5-iodouridine (1a, 0.500 g, 1.14 mmol), Pd(PPh3)4 (0.066 g, 0.057 mmol), CuI (0.011 g, 0.057 mmol), DMF (10 mL), Et3N (396 µL, 2.85 mmol), and propargyl acetate (283 µL, 2.85 mmol). The reaction mixture was stirred at room temperature for 22 h. The solvent was removed by oil pump vacuum, and the residue was purified using silica gel column chromatography (230–400 mesh, eluent: 0→2% methanol in chloroform). The product was dried by oil pump vacuum for 2 h to give 2 as a white foam (0.405 g, 0.992 mmol, 87%). 1H NMR (500 MHz, DMSO-d6) δ 11.75 (s, 1H, N-H), 8.01 (s, 1H, H-6), 6.12 (t, J = 6.9 Hz, 1H, H-1'), 5.19–5.15 (m, 1H, H-3'), 4.87 (s, 2H, CH2), 4.27–4.23 (m, 2H, H-4', H-5'), 4.21–4.17 (m, 1H, H-5"), 2.52–2.47 (m, 1H, H-2'), 2.35–2.28 (m, 1H, H-2"), 2.07 (s, 3H, CH3), 2.05 (s, 6H, 2CH3); 13C NMR (125 MHz, DMSO-d6) δ 170.10, 170.03, 169.71, 161.30, 149.33, 144.40, 97.85, 87.05, 84.90, 81.48, 78.52, 73.66, 63.53, 52.20, 36.17, 20.76, 20.54, 20.46; IR (cm–1, KBr) 3442 m, 3389 m, 2987 m, 2823 m, 1701 s, 1627 s, 1467 m, 1288 m, 1052 m; TOF–ESI+–MS (m/z): [M + Na]+ calcd for C18H20N2NaO9, 431.1061; found, 431.1064.
2',3',5'-Tri-O-acetyl-5-(3-methoxyprop-1-yn-1-yl)uridine (3). A round-bottom flask was charged with 2',3',5'-tri-O-acetyl-5-iodouridine (1b, 0.500g, 1.01 mmol), Pd(PPh3)4 (0.058 g, 0.050 mmol), CuI (0.010 g, 0.050 mmol), DMF (10 mL), Et3N (351 µL, 2.52 mmol), and methyl propargyl ether (212 µL, 2.52 mmol). The reaction mixture was stirred at room temperature for 22 h. The solvent was removed by oil pump vacuum, and the residue was purified using silica gel column chromatography (230–400 mesh, eluent: 0→2% methanol in chloroform). The product was dried by oil pump vacuum for 2 h to give 3 as a white foam (0.270 g, 0.616 mmol, 61%). 1H NMR (500 MHz, CDCl3) δ 8.78 (s, 1H, N-H), 7.79 (s, 1H, H-6), 6.08–6.06 (m, 1H, H-1'), 5.35–5.30 (m, 2H, H-3', H-4'), 4.40–4.37 (m, 1H, H-2'), 4.37–4.35 (m, 2H, H-5', H-5"), 4.28 (s, 2H, CH2), 3.40 (s, 3H, OCH3), 2.21 (s, 3H, CH3), 2.12 (s, 3H, CH3), 2.11 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 170.13, 169.65, 169.55, 160.68, 142.06, 100.66, 90.35, 87.46, 80.19, 75.44, 73.20, 70.01, 62.91, 60.30, 57.88, 51.08, 20.83, 20.54, 20.45; IR (cm–1, KBr) 3208 br w, 3082 br w, 2938 br w, 2823 br w, 1743 s, 1692 vs, 1628 m, 1453 m, 1214 vs, 1092 s; TOF–ESI+–MS (m/z): [M + Na]+ calcd for C19H22N2NaO10, 461.1167; found, 461.1171.
General procedure for the synthesis of hexacarbonyl dicobalt 5-alkynyluridines (4 or 5): A round-bottom flask was charged under a nitrogen atmosphere with Co2(CO)8 (0.222 g, 0.650 mmol), alkynyl nucleoside 2 or 3 (0.500 mmol), and THF (10 mL). The mixture was stirred at room temperature (22 °C) for 1 h. The solvent was removed by rotary evaporation. Silica gel column chromatography (230–400 mesh, eluent: chloroform) gave reddish-brown compounds 4 or 5.
Hexacarbonyl dicobalt 3',5'-di-O-acetyl-2'-deoxy-5-[3-(acetoxy)prop-1-yn-1-yl]uridine (4). From alkynyl nucleoside 2 (0.204 g, 0.500 mmol); brown foam (0.305 g, 0.440 mmol, 88%); 1H NMR (600 MHz, CDCl3) δ 9.32 (s, 1H, NH), 7.83 (s, 1H, H-6), 6.26–6.22 (m, 1H, H-1'), 5.57–5.47 (m, 2H, CH2), 5.23–5.20 (m, 1H, H-3'), 4.41–4.37 (m, 1H, H-4'), 4.32–4.29 (m, 1H, H-5'), 4.28–4.24 (m, 1H, H-5"), 2.66–2.61 (m, 1H, H-2'), 2.17–2.11 (m, 7H, H-2", 2CH3), 2.07 (s, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ 198.71, 170.73, 170.29, 170.26, 160.23, 149.43, 138.26, 113.71, 94.82, 85.91, 82.58, 79.29, 74.03, 65.45, 63.65, 37.93, 20.90, 20.60, 20.54; IR (cm–1, KBr) 3356 br m, 3089 br w, 2960 br w, 2093 m, 2056 s, 2024 br s, 1736 vs, 1638 m, 1561 m, 1406 m, 1228 vs, 1024 s; TOF–ESI+–MS (m/z): [M + Na]+ calcd for C24H20Co2N2NaO15, 716.9420; found, 716.9426.
Hexacarbonyl dicobalt 2',3',5'-tri-O-acetyl-5-(3-methoxyprop-1-yn-1-yl)uridine (5). From alkynyl nucleoside 3 (0.219 g, 0.500 mmol); brown foam (0.279 g, 0.385 mmol, 77%); 1H NMR (500 MHz, CDCl3) δ 9.56 (s, 1H, NH), 7.74 (s, 1H, H-6), 6.10–6.00 (m, 1H, H-1'), 5.41–5.30 (m, 2H, H-3', H-4'), 4.79 (m, 2H, CH2), 4.41–4.27 (m, 3H, H-5', H-5", H-2'), 3.54 (s, 3H, OCH3), 2.15 (s, 3H, CH3), 2.12 (s, 3H, CH3), 2.10 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 198.97, 170.42, 169.66, 160.56, 150.01, 138.54, 114.36, 96.13, 87.92, 79.99, 79.19, 73.53, 72.44, 70.30, 63.34, 59.12, 20.69, 20.59, 20.44; IR (cm–1, KBr) 3234 br w, 2991 w, 2092 m, 2051 s, 2004 vs, 1746 m, 1688 m, 1447 m, 1214 br m, 1094 m, 750 vs; TOF–ESI+–MS (m/z): [M + Na]+ calcd for C25H22Co2N2NaO16, 746.9526; found, 746.9536.
Hexacarbonyl dicobalt 3',5'-di-O-acetyl-2'-deoxy-5-(hex-5-en-1-yn-1-yl)uridine (6a). To a solution of nucleoside complex 4 (0.0206 g, 29.7 μmol) in CH2Cl2 (5 mL) at 0 °C was added allyltrimethylsilane (25 μL, 0.16 mmol) and BF3·OEt2 (15 μL, 0.12 mmol). The solution was stirred over 12 h with gradual warming to room temperature, at which time starting material consumption was complete, as evidenced by TLC (1:1 petroleum ether/EtOAc). Then, NH4Cl (saturated aq, 0.25 mL) and NaHCO3 (saturated aq, 0.25 mL) were added, followed by MgSO4. The mixture was filtered through a plug of silica gel and washed with EtOAc. Concentration of the crude reaction product and purification by flash chromatography (2:1→3:2 petroleum ether/EtOAc) afforded 6a as a red-brown oil (0.0110 g, 16.3 μmol, 55%). 1H NMR (500 MHz, CDCl3) δ 8.85 (br s, 1H), 7.72 (s, 1H), 6.24 (dd, J = 8.0 Hz, 5.4 Hz, 1H), 5.93 (m, 1H), 5.22 (d, J = 6.3 Hz, 1H), 5.15 (d, J = 17.2 Hz, 1H), 5.05 (d, J = 10.1 Hz, 1H), 4.37 (dd, J = 11.6, 5.1 Hz, 1H), 4.30 (br s, 1H), 4.25 (dd, J = 11.6, 3.6 Hz, 1H), 3.17 (apparent t, J = 7.9 Hz, 2H), 2.61 (dd, J = 14.0, 4.6 Hz, 1H), 2.43 (dt, J = 8.1, 7.0 Hz, 2H), 2.13 (s, 3H), 2.12 (obscured, 1H), 2.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.4, 170.3, 159.9, 149.4, 137.2, 115.6, 114.6, 103.6, 85.7, 82.4, 81.8, 74.0, 63.7, 37.9, 35.6, 33.8, 20.9, 20.6; IR (neat, ATR) 3197, 3077, 2967, 2089, 2049, 2017, 1747, 1714, 1691, 1587 cm−1; ESI+–MS (m/z): [M]+ calcd for C25H22Co2N2O13, 698.9684; found, 698.9689.
Hexacarbonyl dicobalt 3',5'-di-O-acetyl-2'-deoxy-5-[3-(2,4,6-trimethoxyphenyl)prop-1-yn-1-yl]uridine (6b). To a solution of nucleoside complex 4 (0.0210 g, 30.3 μmol) in CH2Cl2 (5 mL) at 0 °C was added 1,3,5-trimethoxybenzene (0.0286 g, 0.170 mmol) and BF3·OEt2 (17 μL, 0.14 mmol). The solution was stirred for 0.5 h at 0 °C, followed by 1.5 h at room temperature. Then, NH4Cl (saturated aq, 0.25 mL) and NaHCO3 (saturated aq, 0.25 mL) were added, followed by a conventional extractive workup (CH2Cl2). Purification by preparative TLC (2:1 hexanes/EtOAc, 2 developments) afforded 6b (0.0218 g, 26.9 μmol, 89%) as a red-brown oil. 1H NMR (500 MHz, CDCl3) δ 8.95 (br s, 1H), 7.74 (s, 1H), 6.27 (dd, J = 8.8, 5.4 Hz, 1H), 6.14 (s, 2H), 5.22 (d, J = 6.5 Hz, 1H), 4.36 (dd, J = 11.6, 4.9 Hz, 1H), 4.29 (m, 1H), 4.27 (s, 2H), 4.25 (dd, J = 11.6, 3.7 Hz, 1H), 3.82 (s, 3H), 3.77 (s, 6H), 2.60 (ddd, J = 14.2, 5.3, 1.3 Hz, 1H), 2.15 (m, 1H), 2.13 (s, 3H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.4, 170.3, 160.3, 160.1, 158.9, 149.6, 136.2, 115.1, 108.7, 104.1, 90.0, 85.7, 82.3, 81.1, 74.1, 63.8, 55.3, 54.7, 37.7, 26.5, 20.9, 20.6; IR (neat, ATR) 3200, 2997, 2962, 2088, 2048, 2018, 1746, 1711, 1664, 1598 cm−1; ESI+–MS (m/z): [M]+ calcd for C31H28Co2N2O16, 825.0000; found, 825.0002.
Hexacarbonyl dicobalt 3',5'-di-O-acetyl-2'-deoxy-5-(5-oxo-5-phenylhex-1-yn-1-yl)uridine (6c). To a solution of nucleoside complex 4 (0.0212 g, 30.6 μmol) in CH2Cl2 (5 mL) at 0 °C was added acetophenone trimethylsilyl enol ether (trimethyl(1-phenylvinyloxy)silane, 0.039 g, 0.20 mmol) and BF3·OEt2 (16 μL, 0.13 mmol). The solution was stirred over 12 h with gradual warming to room temperature. Then, NH4Cl (saturated saturated aq, 5 drops) and NaHCO3 (saturated aq, 5 drops) were added, followed by a conventional extractive workup (CH2Cl2). Purification by preparative TLC (3:2 hexanes/EtOAc, 2 developments) afforded 6c as brown oil (0.0108 g, 15.0 μmol, 49%). 1H NMR (500 MHz, CDCl3) δ 8.68 (s, 1H), 8.00 (d, J = 7.9 Hz, 2H), 7.83 (s, 1H), 7.58 (t, J = 7.4 Hz, 1H), 7.48 (apparent t, J = 7.7 Hz, 2H), 6.26 (dd, J = 8.7, 5.4 Hz, 1H), 5.23 (d, J = 6.4 Hz, 1H), 4.39 (dd, J = 11.7, 5.4 Hz, 1H), 4.31 (m, 1H), 4.26 (dd, J = 11.7, 3.8 Hz, 1H), 3.52 (m, 2H), 3.42 (t, J = 7.1 Hz, 2H), 2.71 (dd, J = 13.7, 4.8 Hz, 1H), 2.19 (m, 1H), 2.13 (s, 3H), 2.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.2, 198.3, 170.29, 170.26, 159.9, 149.3, 137.5, 136.6, 133.2, 128.7, 128.0, 114.1, 102.4, 85.6, 82.5, 81.8, 74.0, 63.7, 40.2, 37.8, 28.9, 20.9, 20.6; IR (neat, ATR) 3208, 2956, 2926, 2089, 2050, 2018, 1746, 1715, 1688, 1597 cm−1; ESI+–MS (m/z): [M + Na]+ calcd for C30H25Co2N2NaO14, 776.9789; found, 776.9788.
Hexacarbonyl dicobalt 2',3',5'-tri-O-acetyl-5-(hex-5-en-1-yn-1-yl)uridine (7a). To a solution of nucleoside complex 5 (20.6 mg, 28.4 μmol) and allyltrimethylsilane (100 μL, 0.629 mmol) at 0 °C was added SnCl4 (90 μL, 1.0 M, 0.090 mmol). The solution was stirred for 1 h at 0 °C, followed by 2 h at rt. Then, NH4Cl (saturated aq, 5 drops) and NaHCO3 (saturated aq, 5 drops) were added, and a conventional extractive workup was performed (CH2Cl2). Preparative TLC (3:2 petroleum ether/EtOAc) afforded, in order of elution, 7a (7.8 mg, 11 μmol, 37% yield, 46% brsm) and recovered 5 (3.7 mg, 5.1 μmol, 18% recovery). 7a: 1H NMR (300 MHz, CDCl3) δ 8.60 (s, 1H), 7.55 (s, 1H), 6.00 (d, J = 5.4 Hz, 1H), 5.94 (m, 1H), 5.30–5.40 (m, 2H), 5.14 (d, J = 17.1 Hz, 1H), 5.06 (d, J = 10.2 Hz, 1H), 4.23–4.43 (m, 3H), 3.17 (apparent t, J = 7.9 Hz, 2H), 2.43 (m, 2H), 2.16 (s, 3H), 2.13 (s, 3H), 2.10 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.2, 170.3, 169.5, 159.6, 149.4, 137.9, 137.2, 115.7, 115.2, 103.6, 88.0, 80.3, 80.1, 72.4, 70.3, 63.3, 35.6, 33.8, 20.6, 20.5, 20.4; IR (neat, ATR) 3219, 2956, 2924, 2089, 2049, 2014, 1749, 1718, 1692 cm−1; ESI+–MS (m/z): [M + Na]+ calcd for C27H24Co2N2NaO15, 756.9738; found, 756.9742.
Hexacarbonyl dicobalt 2',3',5'-tri-O-acetyl-5-[3-(2,4,6-trimethoxyphenyl)prop-1-yn-1-yl)]uridine (7b). To a solution of nucleoside complex 5 (20.4 mg, 28.2 μmol) and 1,3,5-trimethoxybenzene (23.6 mg, 140 μmol) in CH2Cl2 (5 mL) at 0 °C was added BF3·OEt2 (11 μL, 87 μmol). The solution was stirred for 45 min at 0 °C, followed by 1 h at rt. Then, NH4Cl (saturated aq, 5 drops) and NaHCO3 (saturated aq, 5 drops) were added, and a conventional extractive workup was performed (CH2Cl2). Preparative TLC (3:2 hexanes/EtOAc) gave 7b as viscous brown oil (11.3 mg, 13.2 μmol, 47%). 1H NMR (300 MHz, CDCl3) δ 8.64 (s, 1H), 7.56 (s, 1H), 6.14 (s, 2H), 5.92 (d, J = 5.7 Hz, 1H), 5.32–5.42 (m, 2H), 4.20–4.45 (m, 5H), 3.82 (s, 3H), 3.78 (s, 6H), 2.16 (s, 3H), 2.12 (s, 3H), 2.11 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.3, 170.3, 169.5, 160.4, 159.9, 158.9, 149.6, 136.7, 115.6, 108.7, 103.9, 90.0, 87.9, 80.5, 80.1, 72.4, 70.3, 63.3, 55.3, 54.8, 25.6, 20.6, 20.5, 20.3; IR (neat, ATR) 3211, 2956, 2924, 2087, 2047, 2010, 1748, 1716, 1693, 1597 cm−1; ESI+–MS (m/z): [M + Na]+ calcd for C33H30Co2N2NaO18, 883.0055; found, 883.0077.
Hexacarbonyl dicobalt 2',3',5'-tri-O-acetyl-5-[3-(1'-methylindol-3'-yl)prop-1-yn-1-yl)]uridine (7d). To a solution of nucleoside complex 5 (20.4 mg, 28.1 μmol) and N-methylindole (18.4 mg, 14.0 μmol) in CH2Cl2 (5 mL) at 0 °C was added BF3·OEt2 (14 μL, 0.11 mmol). The solution was stirred for 45 min at 0 °C, followed by rt for 45 min. Then, NH4Cl (saturated aq, 5 drops) and NaHCO3 (saturated aq, 5 drops) were added, and a conventional extractive workup was performed (CH2Cl2). Preparative TLC (3:2 hexanes/EtOAc) afforded 7d as brown oil (9.2 mg, 11 μmol, 40%). 1H NMR (300 MHz, CDCl3) δ 8.39 (br s, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.46 (s, 1H), 7.29 (d, obscured, 1H), 7.24 (apparent t, J = 7.0 Hz, 1H), 7.12 (apparent dt, J = 1.0, 7.4 Hz, 1H), 6.96 (s, 1H), 5.84 (d, J = 5.4 Hz, 1H), 5.28–5.38 (m, 2H), 4.52 (s, 2H), 4.23–4.40 (m, 3H), 3.79 (s, 3H), 2.15 (s, 3H), 2.11 (s, 3H), 2.09 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.1, 170.3, 169.53, 169.52, 158.9, 149.3, 138.1, 136.6, 127.7, 127.6, 121.6, 119.0, 118.7, 115.3, 113.8, 109.2, 106.6, 88.5, 81.3, 80.0, 72.4, 70.2, 63.2, 32.7, 29.6, 20.6, 20.5, 20.4; IR (neat, ATR) 3204, 2954, 2924, 2089, 2050, 2019, 1750, 1720, 1692 cm−1; ESI+–MS (m/z): [M + H]+ calcd for C33H27Co2N3O15, 823.0184; found, 823.0184.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra for compounds 2, 3, 4, 5, 6a–c, and 7a,b,d. | ||
| Format: PDF | Size: 2.9 MB | Download |
Acknowledgements
We thank the National Institutes of Health (NIH, CA111329), the Statutory Funds of CMMS PAS, and the Natural Sciences and Engineering Research Council (NSERC Canada, RG-PIN-2016-04946) for support of this research. The NSF awards (CHE-0821487, CHE-1048719) and OU Research Excellence Fund are also acknowledged. We are also thankful to Dr. Hiroyuki Hayakawa (Yamasa Corporation, Biochemicals Division) for a generous supply of nucleosides.
References
-
Herdewijn, P. Modified Nucleosides in Biochemistry, Biotechnology and Medicine; Wiley-VCH: Weinheim, Germany, 2008. doi:10.1002/9783527623112
Return to citation in text: [1] -
Godefridus, J. P., Ed. Deoxynucleoside Analogs in Cancer Therapy; Humana Press: Totowa, USA, 2006.
Return to citation in text: [1] -
Merino, P., Ed. Chemical Synthesis of Nucleoside Analogues; John Wiley & Sons: Hoboken, NJ, USA, 2013. doi:10.1002/9781118498088
Return to citation in text: [1] -
Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010
Return to citation in text: [1] -
Bobrovnikova-Marjon, E.; Hurov, J. B. Annu. Rev. Med. 2014, 65, 157–170. doi:10.1146/annurev-med-092012-112344
Return to citation in text: [1] -
Kore, A. R.; Charles, I. Curr. Org. Chem. 2012, 16, 1996–2013. doi:10.2174/138527212803251622
Return to citation in text: [1] -
Kapdi, A. R.; Maiti, D.; Sanghvi, Y. S., Eds. Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides; Elsevier: Amsterdam, Netherlands, 2018. doi:10.1016/c2016-0-00656-3
Return to citation in text: [1] -
Hilko, D. H.; Bornaghi, L. F.; Poulsen, S.-A. J. Org. Chem. 2018, 83, 11944–11955. doi:10.1021/acs.joc.8b01834
See for a recent representative example of C-5 modified uridines.
Return to citation in text: [1] [2] -
Bag, S. S.; Gogoi, H. J. Org. Chem. 2018, 83, 7606–7621. doi:10.1021/acs.joc.7b03097
See for a recent representative example of C-5 modified uridines.
Return to citation in text: [1] -
Barthes, N. P. F.; Karpenko, I. A.; Dziuba, D.; Spadafora, M.; Auffret, J.; Demchenko, A. P.; Mély, Y.; Benhida, R.; Michel, B. Y.; Burger, A. RSC Adv. 2015, 5, 33536–33545. doi:10.1039/c5ra02709h
See for a ecent representative example of C-5 modified uridines.
Return to citation in text: [1] -
Hottin, A.; Marx, A. Acc. Chem. Res. 2016, 49, 418–427. doi:10.1021/acs.accounts.5b00544
Return to citation in text: [1] -
Mei, H.; Chaput, J. C. Chem. Commun. 2018, 54, 1237–1240. doi:10.1039/c7cc09130c
Return to citation in text: [1] -
Sniady, A.; Durham, A.; Morreale, M. S.; Marcinek, A.; Szafert, S.; Lis, T.; Brzezinska, K. R.; Iwasaki, T.; Ohshima, T.; Mashima, K.; Dembinski, R. J. Org. Chem. 2008, 73, 5881–5889. doi:10.1021/jo8007995
and references cited therein.
Return to citation in text: [1] -
Rao, M. S.; Esho, N.; Sergeant, C.; Dembinski, R. J. Org. Chem. 2003, 68, 6788–6790. doi:10.1021/jo0345648
Return to citation in text: [1] -
Sniady, A.; Sevilla, M. D.; Meneni, S.; Lis, T.; Szafert, S.; Khanduri, D.; Finke, J. M.; Dembinski, R. Chem. – Eur. J. 2009, 15, 7569–7577. doi:10.1002/chem.200900481
Return to citation in text: [1] -
Franz, K. J.; Metzler-Nolte, N., Eds. Metals in Medicine. Chem. Rev. 2019, 119, 727–1624. doi:10.1021/acs.chemrev.8b00685
Return to citation in text: [1] -
Jaouen, G.; Salmain, M., Eds. Bioorganometallic Chemistry: Applications in Drug Discovery, Biocatalysis, and Imaging; John Wiley & Sons: Hoboken, NJ, USA, 2015.
Return to citation in text: [1] -
Kraatz, H. B.; Metzler-Nolte, N., Eds. Concepts and Models in Bioinorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2006.
Return to citation in text: [1] -
Jaouen, G., Ed. Bioorganometallic Chemistry; Wiley-VCH: Weinheim, Germany, 2006.
Return to citation in text: [1] -
Gasser, G. Chimia 2015, 69, 442–446. doi:10.2533/chimia.2015.442
Return to citation in text: [1] -
Gasser, G.; Ott, I.; Metzler-Nolte, N. J. Med. Chem. 2011, 54, 3–25. doi:10.1021/jm100020w
Return to citation in text: [1] -
Timerbaev, A. R.; Hartinger, C. G.; Aleksenko, S. S.; Keppler, B. K. Chem. Rev. 2006, 106, 2224–2248. doi:10.1021/cr040704h
Return to citation in text: [1] -
Hartinger, C. G.; Metzler-Nolte, N.; Dyson, P. J. Organometallics 2012, 31, 5677–5685. doi:10.1021/om300373t
Return to citation in text: [1] -
Ott, I.; Gust, R. Arch. Pharm. (Weinheim, Ger.) 2007, 340, 117–126. doi:10.1002/ardp.200600151
Return to citation in text: [1] -
Heffern, M. C.; Yamamoto, N.; Holbrook, R. J.; Eckermann, A. L.; Meade, T. J. Curr. Opin. Chem. Biol. 2013, 17, 189–196. doi:10.1016/j.cbpa.2012.11.019
Return to citation in text: [1] -
Li, J.; Zhang, J.; Zhang, Q.; Wang, Y.; Bai, Z.; Zhao, Q.; He, D.; Wang, Z.; Zhang, J.; Chen, Y. Bioorg. Med. Chem. 2019, 27, 115071. doi:10.1016/j.bmc.2019.115071
See for a recent representative example.
Return to citation in text: [1] -
Berrino, E.; Milazzo, L.; Micheli, L.; Vullo, D.; Angeli, A.; Bozdag, M.; Nocentini, A.; Menicatti, M.; Bartolucci, G.; di Cesare Mannelli, L.; Ghelardini, C.; Supuran, C. T.; Carta, F. J. Med. Chem. 2019, 62, 7233–7249. doi:10.1021/acs.jmedchem.9b00845
Return to citation in text: [1] -
Atkin, A. J.; Williams, S.; Sawle, P.; Motterlini, R.; Lynam, J. M.; Fairlamb, I. J. S. Dalton Trans. 2009, 3653–3656. doi:10.1039/b904627p
Return to citation in text: [1] -
Ott, I.; Kircher, B.; Dembinski, R.; Gust, R. Expert Opin. Ther. Pat. 2008, 18, 327–337. doi:10.1517/13543776.18.3.327
Return to citation in text: [1] -
Munteanu, C. R.; Suntharalingam, K. Dalton Trans. 2015, 44, 13796–13808. doi:10.1039/c5dt02101d
Return to citation in text: [1] -
Kaczmarek, R.; Korczyński, D.; Królewska-Golińska, K.; Wheeler, K. A.; Chavez, F. A.; Mikus, A.; Dembinski, R. ChemistryOpen 2018, 7, 237–247. doi:10.1002/open.201700168
and references cited therein.
Return to citation in text: [1] [2] [3] [4] -
Sergeant, C. D.; Ott, I.; Sniady, A.; Meneni, S.; Gust, R.; Rheingold, A. L.; Dembinski, R. Org. Biomol. Chem. 2008, 6, 73–80. doi:10.1039/b713371e
Return to citation in text: [1] -
Meneni, S.; Ott, I.; Sergeant, C. D.; Sniady, A.; Gust, R.; Dembinski, R. Bioorg. Med. Chem. 2007, 15, 3082–3088. doi:10.1016/j.bmc.2007.01.048
See for anticancer activity of precursors 5-alkynyl 2'-deoxyuridines.
Return to citation in text: [1] -
Kann, N. Curr. Org. Chem. 2012, 16, 322–334. doi:10.2174/138527212799499949
Return to citation in text: [1] -
Shea, K. M. Nicholas Reaction. In Name Reactions for Homologations, Part 1; Li, J. J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp 284–298.
Return to citation in text: [1] -
Díaz, D. D.; Betancort, J. M.; Martín, V. S. Synlett 2007, 343–359. doi:10.1055/s-2007-967958
Return to citation in text: [1] -
Teobald, B. J. Tetrahedron 2002, 58, 4133–4170. doi:10.1016/s0040-4020(02)00315-0
Return to citation in text: [1] -
Green, J. R. Curr. Org. Chem. 2001, 5, 809–826. doi:10.2174/1385272013375247
Return to citation in text: [1] -
Caffyn, A. J. M.; Nicholas, K. M. Transition Metal Alkyne Complexes: Transition Metal-stabilized Propargyl Systems. In Comprehensive Organometallic Chemistry II; Abel, E. W.; Stone, F. G. A.; Wilkinson, G., Eds.; Pergamon: Oxford, U.K., 1995; Vol. 12, pp 685–702. doi:10.1016/b978-008046519-7.00124-6
Return to citation in text: [1] -
Green, J. R.; Nicholas, K. M. Org. React., in press.
Return to citation in text: [1] -
Roth, K.-D. Synlett 1993, 529–533. doi:10.1055/s-1993-22518
Return to citation in text: [1] -
Taj, R. A.; Green, J. R. J. Org. Chem. 2010, 75, 8258–8270. doi:10.1021/jo102127q
Return to citation in text: [1] -
Krafft, M. E.; Cheung, Y. Y.; Wright, C.; Cali, R. J. Org. Chem. 1996, 61, 3912–3915. doi:10.1021/jo952160c
Return to citation in text: [1] -
O'Boyle, J. E.; Nicholas, K. M. Tetrahedron Lett. 1980, 21, 1595–1598. doi:10.1016/s0040-4039(00)77762-8
Return to citation in text: [1] -
Takano, S.; Sugihara, T.; Ogasawara, K. Synlett 1992, 70–72. doi:10.1055/s-1992-21270
Return to citation in text: [1] -
Green, J. R. Chem. Commun. 1998, 1751–1752. doi:10.1039/a803316a
Return to citation in text: [1] -
Nicholas, K. M.; Mulvaney, M.; Bayer, M. J. Am. Chem. Soc. 1980, 102, 2508–2510. doi:10.1021/ja00527a086
Return to citation in text: [1] -
Tanino, K.; Shimizu, T.; Miyama, M.; Kuwajima, I. J. Am. Chem. Soc. 2000, 122, 6116–6117. doi:10.1021/ja001003e
Return to citation in text: [1] -
St Onge, B.; Green, J. R. Synlett 2017, 28, 2923–2927. doi:10.1055/s-0036-1588528
Return to citation in text: [1] -
Green, J. R. Eur. J. Org. Chem. 2008, 6053–6062. doi:10.1002/ejoc.200800836
Return to citation in text: [1] -
Isobe, M.; Hamajima, A. Nat. Prod. Rep. 2010, 27, 1204–1226. doi:10.1039/b919467n
Return to citation in text: [1] -
Hernández, J. N.; Ramírez, M. A.; Rodríguez, M. L.; Martín, V. S. Org. Lett. 2008, 10, 2349–2352. doi:10.1021/ol800544a
Return to citation in text: [1] -
Miyazaki, A.; Asanuma, M.; Dodo, K.; Egami, H.; Sodeoka, M. Chem. – Eur. J. 2014, 20, 8116–8128. doi:10.1002/chem.201400056
Return to citation in text: [1] -
Prasad, J. S.; Liebeskind, L. S. Tetrahedron Lett. 1987, 28, 1857–1860. doi:10.1016/s0040-4039(00)95993-8
Return to citation in text: [1] -
Gruselle, M.; Cordier, C.; Salmain, M.; El Amouri, H.; Guerin, C.; Vaissermann, J.; Jaouen, G. Organometallics 1990, 9, 2993–2997. doi:10.1021/om00161a031
See for a representative example.
Return to citation in text: [1] -
Tanaka, S.; Isobe, M. Tetrahedron 1994, 50, 5633–5644. doi:10.1016/s0040-4020(01)85634-9
Return to citation in text: [1] -
Jiang, Y.; Isobe, M. Tetrahedron 1996, 52, 2877–2892. doi:10.1016/0040-4020(96)00008-7
Return to citation in text: [1] -
Mukai, C.; Itoh, T.; Hanaoka, M. Tetrahedron Lett. 1997, 38, 4595–4598. doi:10.1016/s0040-4039(97)00983-0
Return to citation in text: [1] -
Hosokawa, S.; Isobe, M. J. Org. Chem. 1999, 64, 37–48. doi:10.1021/jo980088n
Return to citation in text: [1] -
Gómez, A. M.; Uriel, C.; Valverde, S.; López, J. C. Org. Lett. 2006, 8, 3187–3190. doi:10.1021/ol060929+
Return to citation in text: [1] -
Lobo, F.; Gómez, A. M.; Miranda, S.; López, J. C. Chem. – Eur. J. 2014, 20, 10492–10502. doi:10.1002/chem.201402149
Return to citation in text: [1] -
Bag, S. S.; Das, S. K. Tetrahedron 2019, 75, 3024–3037. doi:10.1016/j.tet.2019.04.038
Return to citation in text: [1] -
Esho, N.; Davies, B.; Lee, J.; Dembinski, R. Chem. Commun. 2002, 332–333. doi:10.1039/b109501c
Return to citation in text: [1] -
Yamamoto, Y.; Seko, T.; Nakamura, H.; Nemoto, H. Heteroat. Chem. 1992, 3, 239–244. doi:10.1002/hc.520030308
To our knowledge, compounds 2 and 3 have not yet been reported. See for a related protected compound.
Return to citation in text: [1] -
Tolstikov, V. V.; Stetsenko, D. A.; Potapov, V. K.; Sverdlov, E. D. Nucleosides Nucleotides 1997, 16, 215–225. doi:10.1080/07328319708001343
See for a related protected compound.
Return to citation in text: [1]
| 31. |
Kaczmarek, R.; Korczyński, D.; Królewska-Golińska, K.; Wheeler, K. A.; Chavez, F. A.; Mikus, A.; Dembinski, R. ChemistryOpen 2018, 7, 237–247. doi:10.1002/open.201700168
and references cited therein. |
| 1. | Herdewijn, P. Modified Nucleosides in Biochemistry, Biotechnology and Medicine; Wiley-VCH: Weinheim, Germany, 2008. doi:10.1002/9783527623112 |
| 2. | Godefridus, J. P., Ed. Deoxynucleoside Analogs in Cancer Therapy; Humana Press: Totowa, USA, 2006. |
| 3. | Merino, P., Ed. Chemical Synthesis of Nucleoside Analogues; John Wiley & Sons: Hoboken, NJ, USA, 2013. doi:10.1002/9781118498088 |
| 4. | Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010 |
| 5. | Bobrovnikova-Marjon, E.; Hurov, J. B. Annu. Rev. Med. 2014, 65, 157–170. doi:10.1146/annurev-med-092012-112344 |
| 14. | Rao, M. S.; Esho, N.; Sergeant, C.; Dembinski, R. J. Org. Chem. 2003, 68, 6788–6790. doi:10.1021/jo0345648 |
| 34. | Kann, N. Curr. Org. Chem. 2012, 16, 322–334. doi:10.2174/138527212799499949 |
| 35. | Shea, K. M. Nicholas Reaction. In Name Reactions for Homologations, Part 1; Li, J. J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp 284–298. |
| 36. | Díaz, D. D.; Betancort, J. M.; Martín, V. S. Synlett 2007, 343–359. doi:10.1055/s-2007-967958 |
| 37. | Teobald, B. J. Tetrahedron 2002, 58, 4133–4170. doi:10.1016/s0040-4020(02)00315-0 |
| 38. | Green, J. R. Curr. Org. Chem. 2001, 5, 809–826. doi:10.2174/1385272013375247 |
| 39. | Caffyn, A. J. M.; Nicholas, K. M. Transition Metal Alkyne Complexes: Transition Metal-stabilized Propargyl Systems. In Comprehensive Organometallic Chemistry II; Abel, E. W.; Stone, F. G. A.; Wilkinson, G., Eds.; Pergamon: Oxford, U.K., 1995; Vol. 12, pp 685–702. doi:10.1016/b978-008046519-7.00124-6 |
| 40. | Green, J. R.; Nicholas, K. M. Org. React., in press. |
| 13. |
Sniady, A.; Durham, A.; Morreale, M. S.; Marcinek, A.; Szafert, S.; Lis, T.; Brzezinska, K. R.; Iwasaki, T.; Ohshima, T.; Mashima, K.; Dembinski, R. J. Org. Chem. 2008, 73, 5881–5889. doi:10.1021/jo8007995
and references cited therein. |
| 41. | Roth, K.-D. Synlett 1993, 529–533. doi:10.1055/s-1993-22518 |
| 42. | Taj, R. A.; Green, J. R. J. Org. Chem. 2010, 75, 8258–8270. doi:10.1021/jo102127q |
| 11. | Hottin, A.; Marx, A. Acc. Chem. Res. 2016, 49, 418–427. doi:10.1021/acs.accounts.5b00544 |
| 12. | Mei, H.; Chaput, J. C. Chem. Commun. 2018, 54, 1237–1240. doi:10.1039/c7cc09130c |
| 31. |
Kaczmarek, R.; Korczyński, D.; Królewska-Golińska, K.; Wheeler, K. A.; Chavez, F. A.; Mikus, A.; Dembinski, R. ChemistryOpen 2018, 7, 237–247. doi:10.1002/open.201700168
and references cited therein. |
| 6. | Kore, A. R.; Charles, I. Curr. Org. Chem. 2012, 16, 1996–2013. doi:10.2174/138527212803251622 |
| 7. | Kapdi, A. R.; Maiti, D.; Sanghvi, Y. S., Eds. Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides; Elsevier: Amsterdam, Netherlands, 2018. doi:10.1016/c2016-0-00656-3 |
| 8. |
Hilko, D. H.; Bornaghi, L. F.; Poulsen, S.-A. J. Org. Chem. 2018, 83, 11944–11955. doi:10.1021/acs.joc.8b01834
See for a recent representative example of C-5 modified uridines. |
| 9. |
Bag, S. S.; Gogoi, H. J. Org. Chem. 2018, 83, 7606–7621. doi:10.1021/acs.joc.7b03097
See for a recent representative example of C-5 modified uridines. |
| 10. |
Barthes, N. P. F.; Karpenko, I. A.; Dziuba, D.; Spadafora, M.; Auffret, J.; Demchenko, A. P.; Mély, Y.; Benhida, R.; Michel, B. Y.; Burger, A. RSC Adv. 2015, 5, 33536–33545. doi:10.1039/c5ra02709h
See for a ecent representative example of C-5 modified uridines. |
| 31. |
Kaczmarek, R.; Korczyński, D.; Królewska-Golińska, K.; Wheeler, K. A.; Chavez, F. A.; Mikus, A.; Dembinski, R. ChemistryOpen 2018, 7, 237–247. doi:10.1002/open.201700168
and references cited therein. |
| 26. |
Li, J.; Zhang, J.; Zhang, Q.; Wang, Y.; Bai, Z.; Zhao, Q.; He, D.; Wang, Z.; Zhang, J.; Chen, Y. Bioorg. Med. Chem. 2019, 27, 115071. doi:10.1016/j.bmc.2019.115071
See for a recent representative example. |
| 29. | Ott, I.; Kircher, B.; Dembinski, R.; Gust, R. Expert Opin. Ther. Pat. 2008, 18, 327–337. doi:10.1517/13543776.18.3.327 |
| 30. | Munteanu, C. R.; Suntharalingam, K. Dalton Trans. 2015, 44, 13796–13808. doi:10.1039/c5dt02101d |
| 31. |
Kaczmarek, R.; Korczyński, D.; Królewska-Golińska, K.; Wheeler, K. A.; Chavez, F. A.; Mikus, A.; Dembinski, R. ChemistryOpen 2018, 7, 237–247. doi:10.1002/open.201700168
and references cited therein. |
| 25. | Heffern, M. C.; Yamamoto, N.; Holbrook, R. J.; Eckermann, A. L.; Meade, T. J. Curr. Opin. Chem. Biol. 2013, 17, 189–196. doi:10.1016/j.cbpa.2012.11.019 |
| 32. | Sergeant, C. D.; Ott, I.; Sniady, A.; Meneni, S.; Gust, R.; Rheingold, A. L.; Dembinski, R. Org. Biomol. Chem. 2008, 6, 73–80. doi:10.1039/b713371e |
| 33. |
Meneni, S.; Ott, I.; Sergeant, C. D.; Sniady, A.; Gust, R.; Dembinski, R. Bioorg. Med. Chem. 2007, 15, 3082–3088. doi:10.1016/j.bmc.2007.01.048
See for anticancer activity of precursors 5-alkynyl 2'-deoxyuridines. |
| 16. | Franz, K. J.; Metzler-Nolte, N., Eds. Metals in Medicine. Chem. Rev. 2019, 119, 727–1624. doi:10.1021/acs.chemrev.8b00685 |
| 17. | Jaouen, G.; Salmain, M., Eds. Bioorganometallic Chemistry: Applications in Drug Discovery, Biocatalysis, and Imaging; John Wiley & Sons: Hoboken, NJ, USA, 2015. |
| 18. | Kraatz, H. B.; Metzler-Nolte, N., Eds. Concepts and Models in Bioinorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2006. |
| 19. | Jaouen, G., Ed. Bioorganometallic Chemistry; Wiley-VCH: Weinheim, Germany, 2006. |
| 20. | Gasser, G. Chimia 2015, 69, 442–446. doi:10.2533/chimia.2015.442 |
| 21. | Gasser, G.; Ott, I.; Metzler-Nolte, N. J. Med. Chem. 2011, 54, 3–25. doi:10.1021/jm100020w |
| 22. | Timerbaev, A. R.; Hartinger, C. G.; Aleksenko, S. S.; Keppler, B. K. Chem. Rev. 2006, 106, 2224–2248. doi:10.1021/cr040704h |
| 23. | Hartinger, C. G.; Metzler-Nolte, N.; Dyson, P. J. Organometallics 2012, 31, 5677–5685. doi:10.1021/om300373t |
| 24. | Ott, I.; Gust, R. Arch. Pharm. (Weinheim, Ger.) 2007, 340, 117–126. doi:10.1002/ardp.200600151 |
| 15. | Sniady, A.; Sevilla, M. D.; Meneni, S.; Lis, T.; Szafert, S.; Khanduri, D.; Finke, J. M.; Dembinski, R. Chem. – Eur. J. 2009, 15, 7569–7577. doi:10.1002/chem.200900481 |
| 27. | Berrino, E.; Milazzo, L.; Micheli, L.; Vullo, D.; Angeli, A.; Bozdag, M.; Nocentini, A.; Menicatti, M.; Bartolucci, G.; di Cesare Mannelli, L.; Ghelardini, C.; Supuran, C. T.; Carta, F. J. Med. Chem. 2019, 62, 7233–7249. doi:10.1021/acs.jmedchem.9b00845 |
| 28. | Atkin, A. J.; Williams, S.; Sawle, P.; Motterlini, R.; Lynam, J. M.; Fairlamb, I. J. S. Dalton Trans. 2009, 3653–3656. doi:10.1039/b904627p |
| 47. | Nicholas, K. M.; Mulvaney, M.; Bayer, M. J. Am. Chem. Soc. 1980, 102, 2508–2510. doi:10.1021/ja00527a086 |
| 48. | Tanino, K.; Shimizu, T.; Miyama, M.; Kuwajima, I. J. Am. Chem. Soc. 2000, 122, 6116–6117. doi:10.1021/ja001003e |
| 43. | Krafft, M. E.; Cheung, Y. Y.; Wright, C.; Cali, R. J. Org. Chem. 1996, 61, 3912–3915. doi:10.1021/jo952160c |
| 44. | O'Boyle, J. E.; Nicholas, K. M. Tetrahedron Lett. 1980, 21, 1595–1598. doi:10.1016/s0040-4039(00)77762-8 |
| 45. | Takano, S.; Sugihara, T.; Ogasawara, K. Synlett 1992, 70–72. doi:10.1055/s-1992-21270 |
| 46. | Green, J. R. Chem. Commun. 1998, 1751–1752. doi:10.1039/a803316a |
| 8. |
Hilko, D. H.; Bornaghi, L. F.; Poulsen, S.-A. J. Org. Chem. 2018, 83, 11944–11955. doi:10.1021/acs.joc.8b01834
See for a recent representative example of C-5 modified uridines. |
| 63. | Esho, N.; Davies, B.; Lee, J.; Dembinski, R. Chem. Commun. 2002, 332–333. doi:10.1039/b109501c |
| 64. |
Yamamoto, Y.; Seko, T.; Nakamura, H.; Nemoto, H. Heteroat. Chem. 1992, 3, 239–244. doi:10.1002/hc.520030308
To our knowledge, compounds 2 and 3 have not yet been reported. See for a related protected compound. |
| 65. |
Tolstikov, V. V.; Stetsenko, D. A.; Potapov, V. K.; Sverdlov, E. D. Nucleosides Nucleotides 1997, 16, 215–225. doi:10.1080/07328319708001343
See for a related protected compound. |
| 55. |
Gruselle, M.; Cordier, C.; Salmain, M.; El Amouri, H.; Guerin, C.; Vaissermann, J.; Jaouen, G. Organometallics 1990, 9, 2993–2997. doi:10.1021/om00161a031
See for a representative example. |
| 56. | Tanaka, S.; Isobe, M. Tetrahedron 1994, 50, 5633–5644. doi:10.1016/s0040-4020(01)85634-9 |
| 57. | Jiang, Y.; Isobe, M. Tetrahedron 1996, 52, 2877–2892. doi:10.1016/0040-4020(96)00008-7 |
| 58. | Mukai, C.; Itoh, T.; Hanaoka, M. Tetrahedron Lett. 1997, 38, 4595–4598. doi:10.1016/s0040-4039(97)00983-0 |
| 59. | Hosokawa, S.; Isobe, M. J. Org. Chem. 1999, 64, 37–48. doi:10.1021/jo980088n |
| 60. | Gómez, A. M.; Uriel, C.; Valverde, S.; López, J. C. Org. Lett. 2006, 8, 3187–3190. doi:10.1021/ol060929+ |
| 61. | Lobo, F.; Gómez, A. M.; Miranda, S.; López, J. C. Chem. – Eur. J. 2014, 20, 10492–10502. doi:10.1002/chem.201402149 |
| 62. | Bag, S. S.; Das, S. K. Tetrahedron 2019, 75, 3024–3037. doi:10.1016/j.tet.2019.04.038 |
| 52. | Hernández, J. N.; Ramírez, M. A.; Rodríguez, M. L.; Martín, V. S. Org. Lett. 2008, 10, 2349–2352. doi:10.1021/ol800544a |
| 53. | Miyazaki, A.; Asanuma, M.; Dodo, K.; Egami, H.; Sodeoka, M. Chem. – Eur. J. 2014, 20, 8116–8128. doi:10.1002/chem.201400056 |
| 54. | Prasad, J. S.; Liebeskind, L. S. Tetrahedron Lett. 1987, 28, 1857–1860. doi:10.1016/s0040-4039(00)95993-8 |
| 49. | St Onge, B.; Green, J. R. Synlett 2017, 28, 2923–2927. doi:10.1055/s-0036-1588528 |
| 50. | Green, J. R. Eur. J. Org. Chem. 2008, 6053–6062. doi:10.1002/ejoc.200800836 |
| 51. | Isobe, M.; Hamajima, A. Nat. Prod. Rep. 2010, 27, 1204–1226. doi:10.1039/b919467n |
© 2020 Kaczmarek et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)