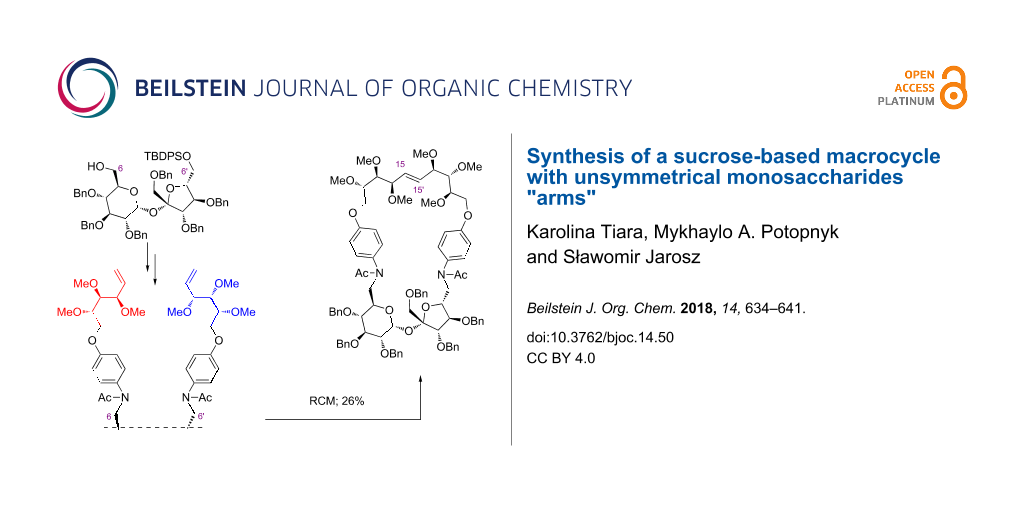
Abstract
An efficient methodology for the selective substitution of both terminal positions (C6 and C6’) in 1’,2,3,3’,4,4’-hexa-O-benzylsucrose with different unsaturated monosaccharide units is presented. Such a highly functionalized intermediate was cyclized under RCM conditions to afford a macrocyclic derivative containing a 31-membered ring in 26% yield.

Graphical Abstract
Introduction
Chiral macrocyclic compounds play an important role in supramolecular and biological systems [1,2]. Many of them serve as convenient receptors for cations [3], anions [4], ion pairs [5], neutral molecules [6] etc.
Binaphthols [7-9], amino acids [10], chiral diamines [11,12], carbohydrates [13], etc. are usually applied as building blocks for construction of such type of compounds.
We are engaged in the synthesis of such macrocyclic derivatives containing the most common natural disaccharide, sucrose [14,15]. Several different classes of macrocyclic derivatives, including: crown [16] and aza-crown [17,18] derivatives, macrocyclic dilactams [19,20], and ureas [21], were prepared in our laboratory.
Sucrose was also used by other groups as a precursor for the preparation of biodegradable polymers [22-24] and polymeric nanoparticles [25]. On the other hand, sucrose derivatives demonstrate antimicrobial and antitumor activities [26,27].
Results and Discussion
Recently, we have prepared sucrose-based macrocyclic derivative 4 in which the terminal positions of this disaccharide (C6 and C6’) are connected via a long polyhydroxylated bridge [28]. In this model study, both terminal positions in 6,6’-diamino-1’,2,3,3’,4,4’-hexa-O-benzyl-6,6’-dideoxysucrose (2) were elongated with the same polyhydroxylated unit 1 providing diamide 3, which subsequently underwent cyclization under the chosen ring-closing metathesis (RCM) conditions [29,30] to give the 21-membered macrocycle 4 (Scheme 1).
![[1860-5397-14-50-i1]](/bjoc/content/inline/1860-5397-14-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of macrocyclic derivative 4.
Scheme 1: Synthesis of macrocyclic derivative 4.
The reduction of the amide functions should lead to amines, which might be used as starting materials for the preparation of, e.g., cryptands 6 (Figure 1). All attempts, however, to reduce 4 to diamine 5 were unsuccessful.
![[1860-5397-14-50-1]](/bjoc/content/figures/1860-5397-14-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Possible route to sucrose cryptands 6.
Figure 1: Possible route to sucrose cryptands 6.
We have decided, therefore, to elaborate another method leading to functionalized sucrose amines of type 9 (Figure 2) which will be obtained by a selective introduction of different fragments 8 (obtained from, e.g., glucose, mannose, etc.).
![[1860-5397-14-50-2]](/bjoc/content/figures/1860-5397-14-50-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Possible route to dienes of type 9.
Figure 2: Possible route to dienes of type 9.
At this stage we were focused on the elaboration of a methodology allowing to introduce different fragments at the sucrose terminals. We chose, therefore, derivatives of methylated hexitols which are easy to prepare and are more convenient than benzyls in the interpretation of the NMR spectra.
We faced, however, a serious problem in the synthesis of amines of type 8. Treatment of aldehyde 11 – generated in situ from iodide 10 according to Vasellas' procedure [31,32] – with benzylamine under the reductive amination conditions afforded an inseparable mixture of two products differing in the configuration at the C2 center (12a and 12b; Scheme 2); such a phenomenon – epimerization under these conditions – is known [33].
The alternative way to the desired amine 12a, based on the SN2 reaction of the activated alcohol 13 [34,35] with benzylamine, also failed (Scheme 2).
![[1860-5397-14-50-i2]](/bjoc/content/inline/1860-5397-14-50-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Unsuccessful attempts to amines 12a and 13b.
Scheme 2: Unsuccessful attempts to amines 12a and 13b.
The Mitsunobu approach to convert the hydroxy group into an amine function was also unsuccessful. Although 13 reacted with phthalimide gave the desired product 13a, the deprotection of the amine function with hydrazine caused also reduction of the C5–C6 double bond; two peaks at 189 and 191 Da were observed in the MS spectrum of crude post-reaction mixture (for 13b and 13c, respectively).
We reasoned, that the all these problems may be overcome by an elongation of alcohol 13 (derived from D-glucose) with a rigid fragment and we decided to introduce the phenyl ring. Treatment of alcohol 13 with para-nitrophenol under Mitsunobu conditions afforded the nitro compound 14 in 63% yield. Stereoisomeric alditol 15, obtained from D-mannose, was converted analogously to 16 (in 60% yield). Both nitro compounds 14 and 16 were reduced to the corresponding amines 17 and 18 with sodium dithionite (Scheme 3).
![[1860-5397-14-50-i3]](/bjoc/content/inline/1860-5397-14-50-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Syntheses of "elongated" amines 17 and 18.
Scheme 3: Syntheses of "elongated" amines 17 and 18.
The synthesis of the macrocycle with different arms at both terminal positions was initiated from 6’-O-tert-butyldiphenylsilyl-1’,2,3,3’,4,4’-hexa-O-benzylsucrose (19) readily available by a selective silylation of 1’,2,3,3’,4,4’-hexa-O-benzylsucrose (7) [36].
Aldehyde 20 [37] – obtained by Swern oxidation [38] of alcohol 19 – was reacted with amine 17 to afford the desired amine isolated as acetate 21 in 85% total yield. Removal of the TBDPS protecting group from the C6’-position gave alcohol 22 in 97% yield. Under the same "Swern oxidation–reductive amination–acetylation" conditions, alcohol 22 was converted into aldehyde 23, which reacted further with amine 18, furnishing diolefin 24 in 64% total yield. Cyclization of precursor 24 induced by the Hoveyda–Grubbs catalyst (II gen.) afforded the target macrocycle 25 in 26% yield (Scheme 4). The E-configuration of the newly created C=C-bond in the final product was proven by 1H NMR analysis (J15-15’ = 15.9 Hz).
![[1860-5397-14-50-i4]](/bjoc/content/inline/1860-5397-14-50-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we proposed an efficient method of the synthesis of a 31-membered macrocycle with sucrose scaffold. The proposed methodology allows for the regioselective introduction of various polyhydroxylated unsaturated fragments (derived from different sugars) at either terminal position of sucrose which undergo an efficient cyclization under the RCM conditions. Although, for practical reasons, the method was elaborated for the methylated derivatives of hextitol pendants it might be, eventually, applied also for synthons with other protecting groups.
Experimental
General
The NMR spectra were recorded with a Varian VNMRS 600 MHz spectrometer for solutions in CDCl3 at 25 °C. The 13C NMR data for compound 25 were recorded with a Varian VNMRS 500 MHz spectrometer. The structures were assigned, whenever necessary, with the help of 2D correlation experiments (COSY, HSQC, HMBC). Chemical shifts were reported with reference to TMS. Optical rotations were measured with a Jasco P 1020 polarimeter (sodium light) in chloroform at room temperature. Mass spectra were recorded with a Synapt G2-S HDMS (Waters Inc) mass spectrometer equipped with an electrospray ion source and a q-TOF type mass analyzer. The instrument was controlled and recorded data were processed using the MassLynx V4.1 software package (Waters Inc). Thin-layer chromatography (TLC) was performed on silica gel plates coated with fluorescent indicator. Column chromatography was performed on silica gel (Merck, 230–400 mesh). Organic solutions were dried over anhydrous MgSO4.
Procedure for the synthesis of nitro compounds 14 and 16
To a solution of alcohol 13 (310 mg, 1.63 mmol) in dry THF (12 mL) and toluene (4 mL), Ph3P (1.28 g, 4.89 mmol) and para-nitrophenol (340 mg, 2.44 mmol) were added. After stirring during 15 min, DEAD (384 μL, 2.44 mmol) was added dropwise. The reaction mixture was stirred for 3 h at room temperature and then partitioned between diethyl ether (20 mL) and 5% NaOH aqueous solution (30 mL). The layers were separated and the aqueous one extracted with diethyl ether (3 × 15 mL). The combined organic solutions were washed with water (15 mL) and brine (15 mL), dried, concentrated, and the resulting residue was purified by flash chromatography (hexanes–ethyl acetate, 70:30) to afford pure product 15 (318 mg, 1.02 mmol, 63%) as a white solid. TLC [hexanes–AcOEt (2:1)]: Rf = 0.3; [α]D22 +28.5; 1H NMR δ 8.18 (d, J = 9.3 Hz, 2H, ArH), 6.99 (d, J = 9.3 Hz, 2H, ArH), 6.03 (ddd, J5,4 = 6.0 Hz, J5,6 = 10.7 Hz, J5.6 = 17.3 Hz, 1H, H-5), 5.39 (m, 2H, H-6, H-6’), 4.95 (dd, J4,5 = 6.2 Hz, J4,3 = 7.3 Hz, 1H, H-4), 3.63–3.54 (m, 3H, 2×H-1, H-2), 3.53 (dd, J3,2 = 2.5 Hz, 1H, H-3), 3.49 (s, 3H, OMe), 3.42 (s, 3H, OMe), 3.29 (s, 3H, OMe) ppm; 13C NMR δ 162.73 (C-Ar), 141.57 (C-Ar), 133.98 (C-5), 125.82 (2 × C-Ar), 119.07 (C-6), 115.51 (2 × C-Ar), 82.30 (C-3), 78.61 (C-2), 77.54 (C-4), 70.74 (C-1), 61.20 (OMe), 59.09 (OMe), 59.08 (OMe) ppm; HRMS (ESI) [M + Na]+: calcd for C15H21NO6Na, 334.1257; found, 334.1256; anal. calcd for C15H21NO6 (311.33): C, 57.87; H, 6.80; N, 4.50; found: C, 57.65; H, 6.79; N, 4.57.
Nitro compound 16 was obtained as a white solid in 60% yield (295 mg, 0.95 mmol), using the same procedure, from alcohol 15 (300 mg, 1.58 mmol), Ph3P (1.24 g, 4.73 mmol), para-nitrophenol (329 mg, 2.37 mmol), and DEAD (371 μL, 2.37 mmol). TLC [hexanes–AcOEt (2:1)]: Rf = 0.3. [α]D22 −18.9; 1H NMR δ 8.21 (d, J = 10 Hz, 2H, ArH), 7.03 (d, J = 10 Hz, 2H, ArH), 5.92 (ddd, J5,4 = 7.8 Hz, J5,6 = 10.4 Hz, J5.6’ = 17.3 Hz, 1H, H-5), 5.41–5.34 (m, 2H, 2 × H-6), 4.39 (dd, J1,1 = 10.3 Hz, J1,2 = 2.3 Hz, 1H, H-1), 4.19 (dd, J1,1 = 10.3 Hz, J1,2 = 4.7 Hz, 1H, H-1), 3.86 (m, 1H, H-4), 3.75 (m, 1H, H-2), 3.49 (s, 3H, OMe), 3.43 (m, 4H, OMe, H-3), 3.34 (s, 3H, OMe) ppm; 13C NMR δ 163.82 (C-Ar), 141.61 (C-Ar), 135.54 (C-5), 125.89 (2 × C-Ar), 118.71 (C-6), 114.58 (2 × C-Ar), 82.59 (C-3), 81.81 (C-4), 78.81 (C-2), 67.56 (C-1), 61.25, 58.45, 56.72 (3 × OMe) ppm; HRMS (ESI) [M + Na]+: calcd for C15H21NO6Na 334.1257; found: 334.1256; anal. calcd for C15H21NO6 (311.33): C, 57.87; H, 6.80; N, 4.50; found: C, 57.69; H, 6.81; N, 4.50.
Synthesis of amino compounds 17 and 18
To a solution of nitro compound 14 or 16 (232 mg, 0.74 mmol) in aq ethanol (14 mL, 1:1 v/v), K2CO3 (304 mg, 2.22 mmol) and Na2S2O4 (322 mg, 1.85 mmol) were added, and the mixture was stirred for 30 min at rt. Ethyl acetate (15 mL) was added, the layers were separated, and the aqueous one extracted with ethyl acetate (3 × 8 mL). The combined organic solutions were dried, concentrated, and the crude product 17 or 18 was used in the next step without further purification.
Synthesis of compound 21
A solution of amine 17 (203 mg, 0.72 mmol) in DCM (10 mL) was added to a solution of aldehyde 20 (202 mg, 0.18 mmol; prepared from alcohol 19 as described in our previous paper [37]) in DCM (10 mL) containing acetic acid (41 μL, 0.72 mmol) and MgSO4 (≈200 mg), and the mixture was stirred for 1 h at rt. Then, NaBH3CN (17 mg, 0.72 mmol) was added and stirring was continued overnight. Water (20 mL), 0.1 M solution of NH3 (2 mL), and DCM (15 mL) were added, the layers were separated, and the aqueous one extracted with DCM (3 × 10 mL). The combined organic solutions were washed with water (10 mL) and brine (10 mL), dried and concentrated. The residue was dissolved in 1,4-dioxane (10 mL) to which DMAP (4 mg, 0.04 mmol), Et3N (126 μL, 0.90 mmol), and acetic anhydride (51 μL, 0.54 mmol) were added and the mixture was stirred overnight at 100 °C. After cooling to rt, water (15 mL) and DCM (15 mL) were added, the layers were separated, and the aqueous one was extracted with DCM (3 × 10 mL). Combined organic solutions were washed with water (10 mL) and brine (10 mL), dried, concentrated, and the crude productwas purified by flash chromatography (hexanes–ethyl acetate, 80:20 to 50:50) to afford 21 (219 mg, 0.15 mmol, 85%) as a colorless oil. TLC [hexanes–AcOEt (2:1)]: Rf = 0.2; [α]D22 +13.6; 1H NMR δ 7.66 (m, 4H, ArH), 7.33–7.15 (m, 36H, ArH), 7.05 (d, J9,8 = 8.7 Hz, 2H, 2 × H-9), 6.77 (d, J8,9 = 8.7 Hz, 2H, 2 × H-8), 5.92 (ddd, J15,14 = 6.0 Hz, J15,16 = 10.6 Hz, J15,16 = 17.2 Hz, 1H, H-15), 5.76 (d, J1,2 = 3.3 Hz, 1H, H-1), 5.30 (d, J16,15 = 17.2 Hz, 1H, H-16), 5.21 (d, J16,15 = 10.6 Hz, 1H, H-16), 4.79 (d, J = 11.1 Hz, 1H, benzylic H), 4.77–4.71 (m, H-14, 3H, 2 × benzylic H), 4.63 (d, J = 11.8 Hz, 1H, benzylic H), 4.62 (d, J = 11.4 Hz, 1H, benzylic H), 4.56 (d, J = 11.9 Hz, 1H, benzylic H), 4.50 (d, J = 11.8 Hz, 1H, benzylic H), 4.46 (d, J = 10.8 Hz, 1H, benzylic H), 4.43 (d, J = 11.8 Hz, 1H, benzylic H), 4.42–4.37 (m, H-3’, 4H, 3 × benzylic H), 4.23 (dd, J4’,5’ = 5.9 Hz, J4’,3’ = 6.2 Hz, 1H, H-4’), 4.19 (m, 1H, H-5), 4.07 (dd, J5’,6’ = 11.7 Hz, J5’,4’ = 5.9 Hz, 1H, H-5’), 3.96 (m, 3H, 2 × H-6’, H-6), 3.83 (dd, J1’,1’ = 10.3 Hz, 1H, H-1’), 3.81 (m, 1H, H-3), 3.66 (dd, J5,6 = 6.7 Hz, J6,6 = 14.0 Hz, 1H, H-6), 3.61–3.51 (m, 4H, 2 × H-11, H-12, H-1’), 3.45 (s, 3H, OMe), 3.43 (dd, J = 2.25 Hz, J = 7.2 Hz, 1H, H-13), 3.39 (s, 3H, OMe), 3.38 (m, 1H, H-2), 3.22 (s, 3H, OMe), 3.19 (m, 1H, H-4), 1.66 (s, 3H, OAc), 1.05 (s, 9H, t-Bu) ppm; 13C NMR δ 170.84 (C=O), 156.83 (C-10), 138.73, 138.68, 138.42, 138.36, 138.24, 137.88 (6Cquat, 6 × Ph), 137.49 (C-7), 135.57 (2C-Ph), 135.51 (2C-Ph), 134.97 (C-15), 133.44, 133.28 (2Cquat, 2 × Ph), 129.63 (2C-Ph), 129.58 (2C-Ph), 129.24 (2 × C-9), 128.36–127.29 (m, 32C-Ar), 118.42 (C-16), 116.32 (2 × C-8), 105.17 (C-2’), 90.18 (C-1), 83.94 (C-3’), 83.59 (C-4’), 82.57 (C-13), 81.97 (C-5’), 81.47 (C-3), 80.01 (C-2), 79.85 (C-4), 78.66 (C-12), 77.21 (C-14), 75.41, 73.90, 73.41, 72.98, 72.35, 71.98 (6 × OBn), 71.02 (C-11), 70.37 (C-1’), 69.51 (C-5), 65.46 (C-6’), 61.10, 59.05, 59.05 (3 × OMe), 50.50 (C-6), 26.93 [3C, SiC(CH3)3], 22.90 (CH3CO2), 19.27 (Cquat-t-Bu) ppm; HRMS (ESI) [M + Na]+ calcd for C87H99O15SiNa, 1448.6669; found, 1448.6682; anal. calcd for C87H99NO15Si (1426.83): C, 73.24; H, 6.99; N, 0.98; found: C, 73.23; H, 7.10; N, 1.08.
Synthesis of alcohol 22
A solution of TBAF (113 mg, 0.43 mmol) in THF (5 mL) was added to a solution of compound 21 (205 mg, 0.14 mmol) in dry THF (10 mL), and the resulting mixture was stirred for 1 h at rt, and concentrated. The crude product was purified by flash chromatography (hexanes–ethyl acetate, 50:50 to 40:60) to afford 22 (166 mg, 0.14 mmol, 97%) as a colorless oil. TLC [hexanes–AcOEt (1:2)]: Rf = 0.3; [α]D22 +27.3; 1H NMR δ 7.32–7.21 (m, 30H, ArH), 7.10 (d, J9,8 = 8.7 Hz, 2H, 2 × H-9), 6.84 (d, J8,9 = 8.7 Hz, 2H, 2 × H-8), 5.98 (ddd, J15,14 = 6.0 Hz, J15,16 = 10.4 Hz, J15,16 = 17.3 Hz, 1H, H-15), 5.52 (d, J1,2 = 3.0 Hz, 1H, H-1), 5.36 (d, J16,15 = 17.3 Hz, 1H, H-16), 5.28 (d, J16,15 = 10.4 Hz, 1H, H-16), 4.81 (m, H-14, 3H, 2 × benzylic H), 4.70 (d, J = 11.6 Hz, 1H, benzylic H), 4.66–4.57 (m, 4H, 4 × benzylic H), 4.52–4.48 (m, 3H, 3 × benzylic H), 4.42–4.38 (m, 2H, H-3’, benzylic H), 4.35 (d, J = 11.9 Hz, 1H, benzylic H), 4.32–4.28 (m, 2H, H-5, H-4’), 4.02–3.95 (m, 3H, H-3, H-5’, H-6), 3.85 (dd, J6,6 = 14.1 Hz, J6,5 = 5.8 Hz, 1H, H-6), 3.77–3.72 (m, 2H, H-1’,H-6’), 3.68–3.48 (m, 5H, 2 × H11, H-12, H-1’, H-6’), 3.45 (m, 5H, H-2, H-13, OMe), 3.40 (s, 3H, OMe), 3.37 (br s, 1H, OH), 3.26 (m, 1H, H-4), 3.25 (s, 3H, OMe), 1.78 (s, 3H, OAc) ppm; 13C NMR δ 171.83 (C=O), 156.89 (C-10), 138.54, 138.48, 138.39, 138.16, 138.09, 137.72 (Cquat, 6 × Ph), 137.46 (C-7), 135.01 (C-15), 129.18 (2 ×C-9), 127.40–128.37 (m, 30C-Ar), 118.47 (C-16), 116.31 (2 × C-8), 104.61 (C-2’), 90.64 (C-1), 83.71 (C-3’), 82.57 (C-13), 82.01 (C-5’), 81.46 (C-3), 81.23 (C-4’), 79.71 (C-2), 79.50 (C-4), 78.68 (C-12), 77.26 (C-14), 75.43, 74.10, 73.36, 72.78, 72.67, 72.53 (6 × OBn), 71.04 (C-11), 70.76 (C-1’), 70.15 (C-5), 62.23 (C-6’), 61.13, 59.07, 59.07 (3 × OMe), 50.15 (C-6), 22.93 (CH3CO2) ppm; HRMS (ESI) [M + Na]+ calcd for C71H81NO15Na, 1210.5504; found, 1210.5544; anal. calcd for C71H81NO15 (1187.42): C, 71.76; H, 6.87; N, 1.18; found: C, 71.93; H, 7.01; N, 1.08.
Synthesis of diolefin 24
To a cooled solution (−78 °C) of oxalyl chloride (39 μL, 0.46 mmol) in DCM (10 mL), a solution of DMSO (93 μL, 1.30 mmol) in DCM (3 mL) was added within 5 min. After 10 min alcohol 22 (155 mg, 0.13 mmol) in DCM (3 mL) was added dropwise and stirring was continued for 10 min at −78 °C. Then, Et3N (145 μL, 1.04 mmol) was added and the mixture was allowed to attain rt. Water (7 mL) was added, the organic layer was separated, dried, and concentrated. Crude aldehyde 23 was dissolved in DCM (10 mL) and this solution was added to a solution of amine 18 (147 mg, 0.52 mmol), acetic acid (30 μL, 0.52 mmol), and MgSO4 (≈200 mg) in DCM (10 mL), and the resulting mixture was stirred for 1 h at rt. Then, NaBH3CN (12 mg, 0.20 mmol) was added and the stirring was continued overnight. Water (20 mL), 0.1 M solution of NH3 (2 mL), and DCM (15 mL) were added, the layers were separated, and the aqueous one extracted with DCM (3 × 10 mL). Combined organic solutions were washed with water (10 mL) and brine (10 mL), dried, concentrated, and the residue was dissolved in 1,4-dioxane (10 mL). DMAP (3 mg, 0.03 mmol), Et3N (91 μL, 0.65 mmol), acetic anhydride (37 μL, 0.39 mmol) were added, and the mixture was stirred overnight at 100 °C. After cooling, water (15 mL) and DCM (15 mL) were added, the layers were separated, and the aqueous one was extracted with DCM (3 × 10 mL). Combined organic solutions were washed with water (10 mL) and brine (10 mL), dried, concentrated, and the product was isolated by flash chromatography (hexanes–ethyl acetate, 25:75) to afford 24 (120 mg, 0.08 mmol, 64%) as a colorless oil. TLC [hexanes–AcOEt (1:3)]: Rf = 0.25; [α]D22 +32.7; 1H NMR δ 7.25–7.06 (m, 30H, ArH), 7.06–6.95 (m, 4H, 2 × H-9, 2 × H-9’), 6.79–6.72 (m, 4H, 2 × H-8, 2 × H-8’), 5.91 (m, 1H, H-15), 5.82 (m, 1H, H-15’), 5.41 (d, J1,2 = 3.2 Hz, 1H, H-1), 5.31–5.16 (m, 4H, 2 × H-16, 2 × H-16’), 4.78 (J = 11.1 Hz, 1H, benzylic H), 4.75–4.69 (m, 2H, benzylic H, H-14), 4.57–4.45 (m, 4H, 4 × benzylic H), 4.43–4.36 (m, 3H, 2 × benzylic H, H-12’), 4.35–4.26 (m, 3H, 2 × benzylic H, H-4’), 4.18 (m, 1H, H-5), 4.12 (dd, J11’,11’ = 10.0 Hz, J11’,12’ = 2.1 Hz, 1H, H-11’), 4.06–4.02 (m, 2H, H-5’, H-3’), 3.94–3.82 (m, 4H, 2 × H-6, H-6’, H-11’), 3.79–3.70 (m, 3H, H-6’, H-14’, H-11,), 3.63–3.56 (3H, m, H-3, H-11, H-12), 3.54–3.43 (m, 4H, 2 × H-1’, H-13, H-13’), 3.40 (s, 3H, OMe), 3.37 (s, 3H, OMe), 3.33–3.28 (m, 7H, 2×OMe, H-2), 3.25 (s, 3H, OMe), 3.21 (m, 1H, H-4), 3.16 (s, 3H, OMe), 1.67 (s, 3H, OAc), 1.64 (s, 3H, OAc) ppm; 13C NMR δ 171.13, 170.77 (2 × C=O), 157.98, 156.79 (C-10, C-10’), 138.97, 138.79, 138.46, 138.32, 138.32, 137.99, 137.89, 136.04 (Cquat, 6 × Ph, C-7, C-7’), 135.74 (C-15’), 135.09 (C-15), 129.52, 129.44 (2 × C-9, 2 × C-9’), 128.31–127.25 (m, 30 × C-Ar), 118.48, 118.43 (C-16, C-16’), 116.23, 115.18 (2 × C-8, 2 × C-8’), 104.93 (C-2’), 89.27 (C-1), 84.05 (C-3’), 83.54 (C-4’), 82.56 (C-13), 81.81 (C-14’), 81.73 (C-12’), 80.13 (C-2), 79.75 (C-13’), 79.23 (C-4), 78.81 (C-3), 78.73 (C-12), 77.54 (C-14), 77.48 (C-5’), 75.22, 73.95, 73.26, 72.67, 72.27, 72.25 (6 × OBn), 71.16 (C-11), 71.04 (C-1’), 70.13 (C-5), 66.55 (C-11’), 61.20, 61.10, 59.05, 59.01, 58.24, 56.70 (6 × OMe), 52.08 (C-6’), 49.66 (C-6), 22.90, 22.75 (2 × CH3CO2) ppm; HRMS (ESI) [M + Na]+calcd for C88H104N2O19Na, 1515.7153; found, 1515.7131; anal. calcd for C88H104N2O19 (1493.80): C, 70.76; H, 7.02; N, 1.88; found: C, 70.62; H, 7.10; N, 1.90.
Synthesis of macrocyclic compound 25
To a solution of diene 24 (85.0 mg, 0.060 mmol) in degassed, anhydrous toluene (10 mL), Hoveyda–Grubbs catalyst 2nd generation (3.7 mg, 0.006 mmol) was added, and the mixture was stirred and heated at 95 °C for 48 h. The mixture was concentrated and the product was purified by flash chromatography (hexanes–ethyl acetate, 15:85) to give macrocycle 25 (22.8 mg, 0.016 mmol, 26%) as a white amorphous foam. TLC [hexanes–AcOEt (1:5)]: Rf = 0.2; [α]D22 +13.2; 1H NMR δ 7.40–7.02 (m, 36H, ArH), 6.60 (m, 2H, ArH), 5.97 (dd, J15’,15 = 15.9 Hz, J15’,14’ = 6.4 Hz, 1H, H-15’), 5.74–5.68 (m, 2H, H-1, H-15), 4.87 (m, 1H, H-14’), 4.70 (d, J = 12.0 Hz, 1H, benzylic H), 4.66 (m, 1H, H-5’), 4.59 (d, J =10.8 Hz, 1H, benzylic H), 4.58 (d, J =11.7 Hz, 1H, benzylic H), 4.54 (d, J = 10.8 Hz, 1H, benzylic H), 4.50 (d, J = 11.2 Hz, 1H, benzylic H), 4.48 (d, J = 11.7 Hz, 1H, benzylic H), 4.48 (d, J = 11.7 Hz, 1H, benzylic H), 4.46 (dd, J = 7.3 Hz, J = 6.0 Hz, 1H, H-4’), 4.41–4.33 (m, 4H, 3 × benzylic H, H-6’), 4.30 (d, J = 11.0 Hz, 1H, benzylic H), 4.15 (d, J = 10.8 Hz, 1H, benzylic H), 4.02–3.96 (m, 2H, H-6, H-3’), 3.95–3.90 (m, 2H, H-1’, H-14), 3.82 (m, 1H, H-5), 3.77 (m, 1H, H-11’), 3.72–3.61 (5H, H-12, H-12’, H-1’, H-11, H-11’), 3.58 (m, 1H, H-11), 3.55–3.51 (m, 2H, H-13, H-13’), 3.50 (s, 3H, OMe), 3.49 (s, 3H, OMe), 3.41 (s, 3H, OMe), 3.41 (s, 3H, OMe), 3.39 (s, 3H, OMe), 3.37 (s, 3H, OMe), 3.35 (m, 1H, H-6’), 3.23 (dd, J2,3 = 6.7 Hz, J2,1 = 4.0 Hz, 1H, H-2), 3.14 (dd, J3,4 = 9.5 Hz, 1H, H-3), 3.07 (m, 1H, H-6), 3.00 (dd, J4,5 = 9.0 Hz, 1H, H-4), 1.90 (s, 3H, OAc), 1.73 (s, 3H, OAc) ppm; 13C NMR (125 MHz) δ 171.21, 170.78 (2 × C=O), 157.56, 157.49 (C-10, C-10’), 138.50, 138.50, 138.39, 138.13, 138.00, 137.83, 137.53, 137.36 (Cquat, 6 × Ph, C-7, C-7’), 131.73 (C-15’), 130.50 (C-15), 129.46, 129.02 (2 × C-9, 2 × C-9’), 128.98–128.41 (m, 30 × C-Ar), 119.31, 115.03 (2 × C-8, 2 × C-8’), 105.77 (C-2’), 91.08 (C-1), 84.23 (C-3’), 84.23 (C-4’), 83.73 (C-13), 82.10 (C-13’), 81.79 (C-3), 81.27 (C-14), 79.68 (C-12’), 79.41 (C-5’), 79.41 (C-2), 78.95 (C-14’), 78.32 (C-12), 76.75 (C-4), 75.65, 73.72, 73.43, 73.28, 72.39, 72.23 (6 × OBn), 70.69 (C-11), 70.56 (C-5), 69.05 (C-1’), 67.27 (C-11’), 61.22, 59.54, 59.12, 59.01, 58.66, 57.07 (6 × OMe), 56.43 (C-6’), 49.57 (C-6), 23.06, 22.68 (2 × CH3CO2) ppm; HRMS (ESI) [M + Na]+ calcd for C86H100N2O19Na, 1487.6857; found, 1487.6971; anal. calcd for C86H100N2O19 (1465.74): C, 70.47; H, 6.88; N, 1.91; found: C, 70.62; H, 6.96; N, 1.84.
Supporting Information
| Supporting Information File 1: Copies of NMR spectra. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Zhang, X.; Yin, J.; Yoon, J. Chem. Rev. 2014, 114, 4918–4959. doi:10.1021/cr400568b
Return to citation in text: [1] -
Kolesnichenko, I. V.; Anslyn, E. V. Chem. Soc. Rev. 2017, 46, 2385–2390. doi:10.1039/C7CS00078B
Return to citation in text: [1] -
Amato, M. E.; Ballistreri, F. P.; Gentile, S.; Pappalardo, A.; Tomaselli, G. A.; Toscano, R. M. J. Org. Chem. 2010, 75, 1437–1443. doi:10.1021/jo902328y
Return to citation in text: [1] -
Akdeniz, A.; Minami, T.; Watanabe, S.; Yokoyama, M.; Ema, T.; Anzenbacher, P., Jr. Chem. Sci. 2016, 7, 2016–2022. doi:10.1039/C5SC04235F
Return to citation in text: [1] -
McConnell, A. J.; Beer, P. D. Angew. Chem., Int. Ed. 2012, 51, 5052–5061. doi:10.1002/anie.201107244
Return to citation in text: [1] -
Ema, T.; Ura, N.; Eguchi, K.; Ise, Y.; Sakai, T. Chem. Commun. 2011, 47, 6090–6092. doi:10.1039/C1CC11572C
Return to citation in text: [1] -
Caricato, M.; Leza, N. J.; Gargiulli, C.; Gattuso, G.; Dondi, D.; Pasini, D. Beilstein J. Org. Chem. 2012, 8, 967–976. doi:10.3762/bjoc.8.109
Return to citation in text: [1] -
Pu, L. Acc. Chem. Res. 2012, 45, 150–163. doi:10.1021/ar200048d
Return to citation in text: [1] -
Ema, T.; Yokoyama, M.; Watanabe, S.; Sasaki, S.; Ota, H.; Takaishi, K. Org. Lett. 2017, 19, 4070–4073. doi:10.1021/acs.orglett.7b01838
Return to citation in text: [1] -
Brea, R. J.; Reiriz, C.; Granja, J. R. Chem. Soc. Rev. 2010, 39, 1448–1456. doi:10.1039/B805753M
Return to citation in text: [1] -
Quinn, T. P.; Atwood, P. D.; Tanski, J. M.; Moore, T. F.; Folmer-Andersen, J. F. J. Org. Chem. 2011, 76, 10020–10030. doi:10.1021/jo2018203
Return to citation in text: [1] -
Šolomek, T.; Powers-Riggs, N. E.; Wu, Y.-L.; Young, R. M.; Krzyaniak, M. D.; Horwitz, N. E.; Wasielewski, M. R. J. Am. Chem. Soc. 2017, 139, 3348–3351. doi:10.1021/jacs.7b00233
Return to citation in text: [1] -
Potopnyk, M. A.; Jarosz, S. Adv. Carbohydr. Chem. Biochem. 2014, 71, 227–295. doi:10.1016/B978-0-12-800128-8.00003-0
Return to citation in text: [1] -
Jarosz, S.; Potopnyk, M. A.; Kowalski, M. Carbohydr. Chem. 2014, 40, 236–256. doi:10.1039/9781849739986-00236
Return to citation in text: [1] -
Potopnyk, M. A.; Jarosz, S. “Sweet” Sucrose Macrocycles via a “Click Chemistry” Route. In Click Chemistry in Glycoscience; Witczak, Z. J.; Bielski, R., Eds.; Wiley, 2013; pp 235–250. doi:10.1002/9781118526996.ch9
Return to citation in text: [1] -
Jarosz, S.; Listkowski, A.; Lewandowski, B.; Ciunik, Z.; Brzuszkiewicz, A. Tetrahedron 2005, 61, 8485–8492. doi:10.1016/j.tet.2005.06.046
Return to citation in text: [1] -
Potopnyk, M. A.; Lewandowski, B.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1474–1479. doi:10.1016/j.tetasy.2012.10.003
Return to citation in text: [1] -
Potopnyk, M. A.; Jarosz, S. Eur. J. Org. Chem. 2013, 5117–5126. doi:10.1002/ejoc.201300427
Return to citation in text: [1] -
Potopnyk, M. A.; Cmoch, P.; Jarosz, S. Org. Lett. 2012, 14, 4258–4261. doi:10.1021/ol301993d
Return to citation in text: [1] -
Potopnyk, M. A.; Jarosz, S. Monatsh. Chem. 2013, 144, 437–443. doi:10.1007/s00706-012-0894-2
Return to citation in text: [1] -
Łęczycka-Wilk, K.; Dąbrowa, K.; Cmoch, P.; Jarosz, S. Org. Lett. 2017, 19, 4596–4599. doi:10.1021/acs.orglett.7b02198
Return to citation in text: [1] -
Barros, M. T.; Petrova, K. T.; Singh, R. P. Eur. Polym. J. 2010, 46, 1151–1157. doi:10.1016/j.eurpolymj.2010.02.002
Return to citation in text: [1] -
Petrova, K. T.; Potewar, T. M.; Ascenso, O. S.; Barros, M. T. Carbohydr. Polym. 2014, 110, 38–46. doi:10.1016/j.carbpol.2014.03.050
Return to citation in text: [1] -
Petrova, K. T.; Correia-da-Silva, P.; Crucho, C. I. C.; Barros, M. T. Curr. Org. Chem. 2014, 18, 1788–1802. doi:10.2174/1385272819666140527231535
Return to citation in text: [1] -
Crucho, C. I. C.; Barros, M. T. J. Mater. Chem. B 2014, 2, 3946–3955. doi:10.1039/C3TB21632B
Return to citation in text: [1] -
Raposo, C. D.; Petrova, K. T.; Barros, M. T.; Calhelha, R. C.; Soković, M.; Ferreira, I. C. F. R. Med. Chem. 2016, 12, 22–29. doi:10.2174/1573406410666150807111029
Return to citation in text: [1] -
Petrova, K. T.; Barros, M. T.; Calhelha, R. C.; Soković, C.; Ferreira, I. C. F. R. Med. Chem. Res. 2018, 27, 980–988. doi:10.1007/s00044-017-2121-5
Return to citation in text: [1] -
Łęczycka, K.; Jarosz, S. Tetrahedron 2015, 71, 9216–9222. doi:10.1016/j.tet.2015.10.046
Return to citation in text: [1] -
Wakchaure, S.; Einsiedel, J.; Waibel, R.; Gmeiner, P. Synthesis 2012, 44, 2682–2694. doi:10.1055/s-0032-1316758
Return to citation in text: [1] -
Mao, Z.-Y.; Si, C.-M.; Liu, Y.-W.; Dong, H.-Q.; Wei, B.-G.; Lin, G.-Q. J. Org. Chem. 2017, 82, 10830–10845. doi:10.1021/acs.joc.7b01598
Return to citation in text: [1] -
Bernet, B.; Vasella, A. Helv. Chim. Acta 1979, 62, 1990–2016. doi:10.1002/hlca.19790620629
Return to citation in text: [1] -
Bernet, B.; Vasella, A. Helv. Chim. Acta 1979, 62, 2400–2410. doi:10.1002/hlca.19790620736
Return to citation in text: [1] -
Jensen, T.; Mikkelsen, M.; Lauritsen, A.; Andresen, T. L.; Gotfredsen, C. H.; Madsen, R. J. Org. Chem. 2009, 74, 8886–8889. doi:10.1021/jo9019495
Return to citation in text: [1] -
Fürstner, A.; Weidmann, H. J. Org. Chem. 1989, 54, 2307–2311. doi:10.1021/jo00271a012
Return to citation in text: [1] -
Fürstner, A.; Weidmann, H. J. Org. Chem. 1990, 55, 1363–1366. doi:10.1021/jo00291a053
Return to citation in text: [1] -
Gaweł, A.; Jarosz, S. J. Carbohydr. Chem. 2010, 29, 332–347. doi:10.1080/07328303.2010.524958
Return to citation in text: [1] -
Szyszka, Ł.; Osuch-Kwiatkowska, A.; Potopnyk, M. A.; Jarosz, S. Beilstein J. Org. Chem. 2017, 13, 2146–2152. doi:10.3762/bjoc.13.213
Return to citation in text: [1] [2] -
Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041
Return to citation in text: [1]
| 1. | Zhang, X.; Yin, J.; Yoon, J. Chem. Rev. 2014, 114, 4918–4959. doi:10.1021/cr400568b |
| 2. | Kolesnichenko, I. V.; Anslyn, E. V. Chem. Soc. Rev. 2017, 46, 2385–2390. doi:10.1039/C7CS00078B |
| 6. | Ema, T.; Ura, N.; Eguchi, K.; Ise, Y.; Sakai, T. Chem. Commun. 2011, 47, 6090–6092. doi:10.1039/C1CC11572C |
| 22. | Barros, M. T.; Petrova, K. T.; Singh, R. P. Eur. Polym. J. 2010, 46, 1151–1157. doi:10.1016/j.eurpolymj.2010.02.002 |
| 23. | Petrova, K. T.; Potewar, T. M.; Ascenso, O. S.; Barros, M. T. Carbohydr. Polym. 2014, 110, 38–46. doi:10.1016/j.carbpol.2014.03.050 |
| 24. | Petrova, K. T.; Correia-da-Silva, P.; Crucho, C. I. C.; Barros, M. T. Curr. Org. Chem. 2014, 18, 1788–1802. doi:10.2174/1385272819666140527231535 |
| 5. | McConnell, A. J.; Beer, P. D. Angew. Chem., Int. Ed. 2012, 51, 5052–5061. doi:10.1002/anie.201107244 |
| 25. | Crucho, C. I. C.; Barros, M. T. J. Mater. Chem. B 2014, 2, 3946–3955. doi:10.1039/C3TB21632B |
| 4. | Akdeniz, A.; Minami, T.; Watanabe, S.; Yokoyama, M.; Ema, T.; Anzenbacher, P., Jr. Chem. Sci. 2016, 7, 2016–2022. doi:10.1039/C5SC04235F |
| 19. | Potopnyk, M. A.; Cmoch, P.; Jarosz, S. Org. Lett. 2012, 14, 4258–4261. doi:10.1021/ol301993d |
| 20. | Potopnyk, M. A.; Jarosz, S. Monatsh. Chem. 2013, 144, 437–443. doi:10.1007/s00706-012-0894-2 |
| 3. | Amato, M. E.; Ballistreri, F. P.; Gentile, S.; Pappalardo, A.; Tomaselli, G. A.; Toscano, R. M. J. Org. Chem. 2010, 75, 1437–1443. doi:10.1021/jo902328y |
| 21. | Łęczycka-Wilk, K.; Dąbrowa, K.; Cmoch, P.; Jarosz, S. Org. Lett. 2017, 19, 4596–4599. doi:10.1021/acs.orglett.7b02198 |
| 13. | Potopnyk, M. A.; Jarosz, S. Adv. Carbohydr. Chem. Biochem. 2014, 71, 227–295. doi:10.1016/B978-0-12-800128-8.00003-0 |
| 16. | Jarosz, S.; Listkowski, A.; Lewandowski, B.; Ciunik, Z.; Brzuszkiewicz, A. Tetrahedron 2005, 61, 8485–8492. doi:10.1016/j.tet.2005.06.046 |
| 11. | Quinn, T. P.; Atwood, P. D.; Tanski, J. M.; Moore, T. F.; Folmer-Andersen, J. F. J. Org. Chem. 2011, 76, 10020–10030. doi:10.1021/jo2018203 |
| 12. | Šolomek, T.; Powers-Riggs, N. E.; Wu, Y.-L.; Young, R. M.; Krzyaniak, M. D.; Horwitz, N. E.; Wasielewski, M. R. J. Am. Chem. Soc. 2017, 139, 3348–3351. doi:10.1021/jacs.7b00233 |
| 17. | Potopnyk, M. A.; Lewandowski, B.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1474–1479. doi:10.1016/j.tetasy.2012.10.003 |
| 18. | Potopnyk, M. A.; Jarosz, S. Eur. J. Org. Chem. 2013, 5117–5126. doi:10.1002/ejoc.201300427 |
| 10. | Brea, R. J.; Reiriz, C.; Granja, J. R. Chem. Soc. Rev. 2010, 39, 1448–1456. doi:10.1039/B805753M |
| 7. | Caricato, M.; Leza, N. J.; Gargiulli, C.; Gattuso, G.; Dondi, D.; Pasini, D. Beilstein J. Org. Chem. 2012, 8, 967–976. doi:10.3762/bjoc.8.109 |
| 8. | Pu, L. Acc. Chem. Res. 2012, 45, 150–163. doi:10.1021/ar200048d |
| 9. | Ema, T.; Yokoyama, M.; Watanabe, S.; Sasaki, S.; Ota, H.; Takaishi, K. Org. Lett. 2017, 19, 4070–4073. doi:10.1021/acs.orglett.7b01838 |
| 14. | Jarosz, S.; Potopnyk, M. A.; Kowalski, M. Carbohydr. Chem. 2014, 40, 236–256. doi:10.1039/9781849739986-00236 |
| 15. | Potopnyk, M. A.; Jarosz, S. “Sweet” Sucrose Macrocycles via a “Click Chemistry” Route. In Click Chemistry in Glycoscience; Witczak, Z. J.; Bielski, R., Eds.; Wiley, 2013; pp 235–250. doi:10.1002/9781118526996.ch9 |
| 29. | Wakchaure, S.; Einsiedel, J.; Waibel, R.; Gmeiner, P. Synthesis 2012, 44, 2682–2694. doi:10.1055/s-0032-1316758 |
| 30. | Mao, Z.-Y.; Si, C.-M.; Liu, Y.-W.; Dong, H.-Q.; Wei, B.-G.; Lin, G.-Q. J. Org. Chem. 2017, 82, 10830–10845. doi:10.1021/acs.joc.7b01598 |
| 26. | Raposo, C. D.; Petrova, K. T.; Barros, M. T.; Calhelha, R. C.; Soković, M.; Ferreira, I. C. F. R. Med. Chem. 2016, 12, 22–29. doi:10.2174/1573406410666150807111029 |
| 27. | Petrova, K. T.; Barros, M. T.; Calhelha, R. C.; Soković, C.; Ferreira, I. C. F. R. Med. Chem. Res. 2018, 27, 980–988. doi:10.1007/s00044-017-2121-5 |
| 28. | Łęczycka, K.; Jarosz, S. Tetrahedron 2015, 71, 9216–9222. doi:10.1016/j.tet.2015.10.046 |
| 37. | Szyszka, Ł.; Osuch-Kwiatkowska, A.; Potopnyk, M. A.; Jarosz, S. Beilstein J. Org. Chem. 2017, 13, 2146–2152. doi:10.3762/bjoc.13.213 |
| 37. | Szyszka, Ł.; Osuch-Kwiatkowska, A.; Potopnyk, M. A.; Jarosz, S. Beilstein J. Org. Chem. 2017, 13, 2146–2152. doi:10.3762/bjoc.13.213 |
| 38. | Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041 |
| 34. | Fürstner, A.; Weidmann, H. J. Org. Chem. 1989, 54, 2307–2311. doi:10.1021/jo00271a012 |
| 35. | Fürstner, A.; Weidmann, H. J. Org. Chem. 1990, 55, 1363–1366. doi:10.1021/jo00291a053 |
| 36. | Gaweł, A.; Jarosz, S. J. Carbohydr. Chem. 2010, 29, 332–347. doi:10.1080/07328303.2010.524958 |
| 31. | Bernet, B.; Vasella, A. Helv. Chim. Acta 1979, 62, 1990–2016. doi:10.1002/hlca.19790620629 |
| 32. | Bernet, B.; Vasella, A. Helv. Chim. Acta 1979, 62, 2400–2410. doi:10.1002/hlca.19790620736 |
| 33. | Jensen, T.; Mikkelsen, M.; Lauritsen, A.; Andresen, T. L.; Gotfredsen, C. H.; Madsen, R. J. Org. Chem. 2009, 74, 8886–8889. doi:10.1021/jo9019495 |
© 2018 Tiara et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)