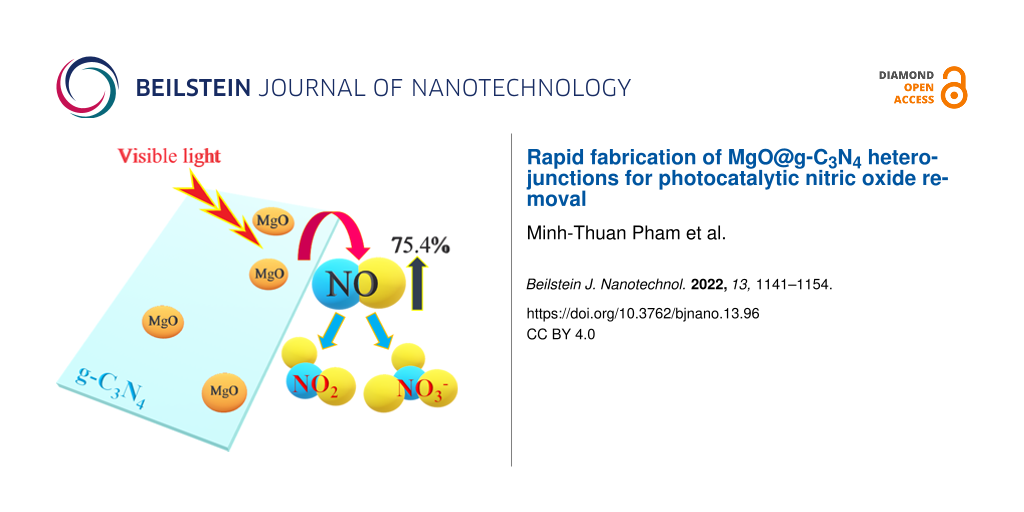
Abstract
Nitric oxide (NO) is an air pollutant impacting the environment, human health, and other biotas. Among the technologies to treat NO pollution, photocatalytic oxidation under visible light is considered an effective means. This study describes photocatalytic oxidation to degrade NO under visible light with the support of a photocatalyst. MgO@g-C3N4 heterojunction photocatalysts were synthesized by one-step pyrolysis of MgO and urea at 550 °C for two hours. The photocatalytic NO removal efficiency of the MgO@g-C3N4 heterojunctions was significantly improved and reached a maximum value of 75.4% under visible light irradiation. Differential reflectance spectroscopy (DRS) was used to determine the optical properties and bandgap energies of the material. The bandgap of the material decreases with increasing amounts of MgO. The photoluminescence spectra indicate that the recombination of electron–hole pairs is hindered by doping MgO onto g-C3N4. Also, NO conversion, DeNOx index, apparent quantum efficiency, trapping tests, and electron spin resonance measurements were carried out to understand the photocatalytic mechanism of the materials. The high reusability of the MgO@g-C3N4 heterojunction was shown by a five-cycle recycling test. This study provides a simple way to synthesize photocatalytic heterojunction materials with high reusability and the potential of heterojunction photocatalysts in the field of environmental remediation.
Introduction
The rapid development of industrialization has been continuously increasing the combustion of fossil fuels, which leads to a large extent of nitrogen oxide emissions. This particular type of air pollutant leads to environmental damage (e.g., smog and acid) and health problems (e.g., COPD and cardiovascular diseases) [1-3]. Presently, there are different approaches to mitigate NO pollution, including catalyst/non-catalyst [4], oxidation [5], bioprocesses [6], adsorption [7], absorption [8], and non-thermal plasma technologies [9]. Photocatalytic oxidation is considered a promising approach due to its ability to degrade various air pollutants with light under ambient conditions [10].
Due to its unique properties, such as high chemical stability and low synthesis cost, graphitic carbon nitride has attracted considerable attention in the realm of environmental remediation [11-13]. It is an organic semiconductor that effectively absorbs visible light due to its small bandgap below 2.7 eV. Because of this, it has been consistently regarded as a catalyst with excellent optical properties [14,15]. Unfortunately, its narrow bandgap leads to rapid recombination of electron–hole (e−–h+) pairs, and the valence band potential of g-C3N4 (+1.75 eV) is more negative than that of H2O/•OH (+2.40 eV), reducing the photocatalytic efficiency [16,17]. A well-known approach for overcoming this problem in order to achieve increased photocatalytic performance is to couple two semiconductors with optimal band alignment.
MgO is an alkaline metal oxide with wide bandgap (3.5–5 eV), high availability, non-toxicity, low cost, and native structural defects [18,19]. The large bandgap energy is the limitation of MgO, reducing the photocatalytic performance and applicability of MgO [20]. Various efforts have been made to enhance the absorption in the visible light region, including nonmetal and noble-metal doping, metal deposition, and formation of heterojunctions [21,22]. The construction of heterojunction structures has shown its effectiveness in improving photocatalytic performance by enhancing the separation of charge carriers and optimizing the redox potential by coupling two or more semiconductors [23,24], such as Bi2MoO6-based [25-29], BiOCl-based [30,31], g-C3N4-based [32-34], ZnO-based [35-37], TiO2-based [38,39], and MgO-based heterostructured photocatalysts [40]. Among these, the combination of MgO and g-C3N4 with a lower bandgap is an efficient process for improving the photocatalytic performance. Li and co-workers reported an improvement in the photocatalytic efficiency of MgO@g-C3N4 for the photoreduction of CO2 under visible light [33]. Similarly, MgO-modified g-C3N4 nanostructures enhanced the removal efficiency for NOx, NO, and NO2 [12]. However, these studies only focused on the synthesis of MgO from Mg(NO3)2·6H2O, increasing time and cost of the synthesis process. Commercial MgO as a precursor material for MgO@g-C3N4 heterojunctions has not been studied. Furthermore, there are no relevant reports on the fabrication of the MgO@g-C3N4 heterojunction via one-step pyrolysis nor on the photocatalytic pathway of the MgO@g-C3N4 heterojunction for photocatalytic NO removal under visible light.
In this study, a MgO@g-C3N4 heterojunction was synthesized via a one-step pyrolysis method using commercial MgO and urea and, subsequently, characterized. Charge transfer dynamics in the heterojunction and band structure were investigated to understand the effect of the heterojunction on the photocatalytic activity. Finally, the photocatalytic pathway of the MgO@g-C3N4 heterojunction was studied via trapping test, electron spin resonance (ESR) measurements, and other methods. This work might be helpful for the development of MgO@g-C3N4 heterojunction materials.
Experimental
Synthesis of MgO/g-C3N4
For the synthesis of g-C3N4, 30 g of urea was ground manually for 30 min and then placed in a 100 mL crucible. Then, the sample was annealed at 550 °C for two hours and let to cool to room temperature.
For preparation of the MgO@g-C3N4 heterojunction material, both MgO and urea were mixed, ground, and placed in a 100 mL crucible, followed by annealing at 550 °C for two hours (Figure 1). Then, the samples were let to cool to room temperature naturally. Different mass ratios of MgO and g-C3N4 were prepared, that is, 1%, 3% and 5%, named as x-MgO@g-C3N4 (x = 1, 3, and 5%).
![[2190-4286-13-96-1]](/bjnano/content/figures/2190-4286-13-96-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic diagram of pyrolysis synthesis process for MgO@g-C3N4 heterojunctions.
Figure 1: Schematic diagram of pyrolysis synthesis process for MgO@g-C3N4 heterojunctions.
Characterization
A variety of analytical techniques have been employed to evaluate the morphology and the physical, chemical, and optical properties of the materials. Scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM) were used to assess the morphology of the materials. The crystal phase of the materials was determined by X-ray diffraction (XRD) with a measurement range of 10°–80°. Fourier-transform infrared spectroscopy (FTIR) was used to determine the chemical bond composition of the materials. Differential reflectance spectroscopy (DRS) determined the change in the bandgap of the materials. The elements of the materials were identified by high-resolution X-ray photoelectron spectroscopy (HR-XPS). The photoluminescence (PL) spectra of the materials was carried out in the form of fluorescence analysis with an excitation wavelength range of 200–900 nm. Finally, the photocatalytic mechanism was determined by trapping tests and ESR measurements.
Photocatalytic performance
The photocatalytic activity of as-prepared MgO@g-C3N4 was evaluated by monitoring NO degradation. The photocatalytic NO removal experiments were performed using a 4.5 L reaction chamber and a Xenon lamp (300 W) as the visible light source. The initial NO concentration was 500 ppb, the flow rate was 1.5 L·min−1, and the dosage of catalysts was 0.2 g for all experiments. Before each catalytic experiment, 0.2 g of the sample was dispersed in 10 mL of DI water, evaporated at 80 °C, and placed in the dark to achieve adsorption–desorption equilibrium. Finally, the sample was illuminated by a Xenon lamp (300 W) for 30 min.
Trapping experiments were performed to evaluate the photocatalytic process mechanism for NO degradation. Three trapping agents were used representing different active species, namely isopropyl alcohol (IPA) for the hydroxyl radical (•OH), potassium dichromate (K2Cr2O7) for electrons (e−), and potassium iodide (KI) for holes (h+). The photocatalytic NO degradation experiments were performed under the previously described conditions.
The photocatalytic NO degradation efficiency (η), the yield of NO2 conversion (γ), the apparent quantum efficiency (AQE, φ), and the DeNOx index (αDeNOX αDeNOx) were calculated by using Equations 1–4: [41-43]:
![[2190-4286-13-96-i1]](/bjnano/content/inline/2190-4286-13-96-i1.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i2]](/bjnano/content/inline/2190-4286-13-96-i2.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i3]](/bjnano/content/inline/2190-4286-13-96-i3.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i4]](/bjnano/content/inline/2190-4286-13-96-i4.svg?max-width=590&scale=1.18182)
where CNO is the concentration of NO (ppb), CNO2 is the concentration of NO2 (ppb), the index “i” represents the initial concentration, and the index “f” represents the final concentration. NA is the Avogadro constant (mol−1), Vt is the flow rate of NO (L·min−1), and M is the molecular weight of NO (g·mol−1). The photon flux in the photocatalytic experiment is 2.72·1019 cm−2·min−1, the irradiation area for the 12 cm diameter petri dish is 113.1 cm2.
In addition, the bandgap energy of materials was calculated by using the Tauc and the Kubelka–Munk equation as described in Equations 5–7 [43]:
![[2190-4286-13-96-i5]](/bjnano/content/inline/2190-4286-13-96-i5.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i6]](/bjnano/content/inline/2190-4286-13-96-i6.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i7]](/bjnano/content/inline/2190-4286-13-96-i7.svg?max-width=590&scale=1.18182)
where E is the photon energy (eV), h is Planck’s constant (4.132·10−15 eV·s), ν is the photon frequency (s−1), c is the velocity of light (nm·s−1), λ is the wavelength (nm), α is the absorption coefficient, B is a constant, and Eg is the bandgap energy (eV), R is the reflectance value.
Results and Discussion
Photocatalytic performance
The photocatalytic NO removal efficiency of the materials is shown in Figure 2a. The efficiency gradually increased during the first 5 min of the photocatalytic reaction and remains stable until the end of the photocatalytic reaction. The photocatalytic NO removal efficiency values are 0.6%, 62.8%, 16.8%, 68.4%, 75.4%, and 72.1% for the blank sample, g-C3N4, MgO, 1% MgO@g-C3N4, 3% MgO@g-C3N4, and 5% MgO@g-C3N4, respectively. The photocatalytic NO removal efficiency is increased by combining MgO with g-C3N4. The results indicate that MgO@g-C3N4 heterojunction structures have been successfully synthesized with high photocatalytic NO degradation efficiency under visible light by one-step pyrolysis (see Table 1 for a comparison of the photocatalytic NO removal efficiency values). Also, the AQE has been calculated according to Equation 3. The AQE values (10−4%) of g-C3N4, MgO, 1% MgO@g-C3N4, 3% MgO@g-C3N4, and 5% MgO@g-C3N4 are 5.5, 1.4, 5.4, 6.7, and 6.2, respectively. The AQE results show (Figure 2b) that photons are most efficient in 3% MgO@g-C3N4. The heterojunction structure has enhanced the photocatalytic activities of the materials [44]. In addition, the photocatalytic reusability of 3% MgO@g-C3N4 was shown by a five-cycle recycling test under identical experimental conditions (Figure 2c). The photocatalytic NO degradation efficiency in the recycling test is 75.4%, 73.8%, 71.1%, 69.4%, and 68.3% after five cycles, respectively. The photocatalytic NO degradation efficiency decreased by 7% after five cycles. The FTIR spectra and the XRD patterns of the 3% MgO@g-C3N4 before and after the recycling test are shown in Figure S1a and Figure S1b of Supporting Information File 1, respectively. The results indicate the high reusability of 3% MgO@g-C3N4 [45].
![[2190-4286-13-96-2]](/bjnano/content/figures/2190-4286-13-96-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Photocatalytic NO degradation efficiency, (b) apparent quantum efficiency of the materials, and (c) photocatalytic recycling test of 3% MgO@g-C3N4.
Figure 2: (a) Photocatalytic NO degradation efficiency, (b) apparent quantum efficiency of the materials, and...
Table 1: Comparison of photocatalytic NO removal of current photocatalyst systems under visible light.
| Material |
Initial NO
concentration (ppb) |
Light source | Irradiation time (min) | Dosage (g) | Flowrate (L·min−1) | NO removal efficiency (%) | NO2 generation | Ref. |
|---|---|---|---|---|---|---|---|---|
|
BiOIO3/
g-C3N4 |
600 | xenon 300 W | 30 | 0.1 | 1.2 | 57 | 80 (ppb) | [72] |
|
SnO2/
g-C3N4 |
600 | tungsten halogen 150 W | 30 | 0.4 | N/A | 32 | 6% | [73] |
|
Ti3C2@TiO2/
g-C3N4 |
430 | xenon- 300 W | 30 | N/A | N/A | 29 | 18.7 (ppb) | [74] |
|
rGO/
Fe-doped g-C3N4 |
1000 | metal halide 250 W | 30 | N/A | N/A | 93.4 | N/A | [75] |
| FAPbBr3/g-C3N4 | 600 | xenon | 60 | 0.1 | 1.2 | 58 | 0.3 (ppb) | [76] |
| g-C3N4/SnO2 | 500 | xenon- 300 W | 30 | 0.2 | 0.6 | 35 | 2% | [77] |
| TiO2@g-C3N4 | 500 | xenon- 300 W | 30 | 0.2 | 0.5 | 90.2 | 5.3% | [44] |
|
g-C3N4@BiOCl/
Bi12O17Cl2 |
500 | tungsten halogen 100 W | 30 | 0.2 | 1 | 46.8 | N/A | [78] |
| MoS2/g-C3N4 | 600 | tungsten halogen 150 W | 30 | 0.2 | N/A | 51.7 | N/A | [79] |
The conversion rates of NO to NO2 and by-products have been calculated (Figure 3a). The conversion rates of NO to NO2 of g-C3N4, MgO, 1% MgO@g-C3N4, 3% MgO@g-C3N4, and 5% MgO@g-C3N4 are 34.6%, 9.6%, 34.9%, 21.9%, and 25.8%, respectively. Besides, the rates of converting NO to by-products of g-C3N4, MgO, 1% MgO@g-C3N4, 3% MgO@g-C3N4, and 5% MgO@g-C3N4 are 28.2%, 7.3%, 34.4%, 53.5%, and 46.3%, respectively. In this study, the by-products are defined as any nitrogen species (e.g., N2O5, N2O, and NO3−) except NO2, which are unstable and can be absorbed by plants [46]. MgO generates the lowest amount of NO2 and by-products due to the lowest photocatalytic NO removal efficiency (16.8%). 3% MgO@g-C3N4 has the lowest NO2 (21.9%) and highest by-product (53.5%) generation. In addition, the NO2 generation of g-C3N4 is almost equal to that of 1% MgO@g-C3N4. This can be explained by the low amount of MgO (only 1%). 3% MgO@g-C3N4 shows the highest photocatalytic NO removal efficiency with the lowest NO2 generation, which indicates a possible future application of 3% MgO@g-C3N4. Also, the DeNOx index values have been calculated according to Equation 4 [47]. The values of g-C3N4, MgO, 1% MgO@g-C3N4, 3% MgO@g-C3N4, and 5% MgO@g-C3N4 are −201.6%, −58.2%, −160.8%, 47.4%, and −25.8%, respectively.
![[2190-4286-13-96-3]](/bjnano/content/figures/2190-4286-13-96-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) NO conversion and (b) DeNOx index of the materials.
Figure 3: (a) NO conversion and (b) DeNOx index of the materials.
XRD and FTIR analyses
XRD patterns of the synthesized materials are shown in Figure 4a. There are two distinct diffraction peaks at 2θ = 13° and 27.4°, which were assigned to the (100) and (002) planes of g-C3N4, respectively [48]. Diffraction peaks of the pure MgO sample are detected at 36.9°, 42.9°, 62.5°, 74.8°, and 78.7°, which were attributed to the (111), (200), (220), (311), and (222) planes, respectively [AMCS: 000501] [49,50]. All MgO@g-C3N4 samples show the characteristic peaks of g-C3N4. No impurities are detected in the MgO@g-C3N4 samples. There are no reflections of MgO in all MgO@g-C3N4 samples, due to the low amount of MgO in the MgO@g-C3N4 samples. This agrees with previous studies, in which characteristic peaks of MgO were only detected when MgO amounts higher than 5% had been added [33,51].
![[2190-4286-13-96-4]](/bjnano/content/figures/2190-4286-13-96-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: (a) XRD patterns and (b) FTIR spectra of the materials.
Figure 4: (a) XRD patterns and (b) FTIR spectra of the materials.
Figure 4b shows the FTIR spectra of g-C3N4, MgO, and MgO@g-C3N4. For pure g-C3N4, the broad peak in the range of 3000–3600 cm−1 was attributed to the stretching vibrations of N–H and O–H bonds, indicating the existence of amino groups and adsorbed water molecules in the material [32,52]. The characteristic peaks at 1240, 1320, 1407, and 1465 cm−1 were associated with the stretching vibrations of aromatic C–N bonds, and the typical peaks at 1562 and 1642 cm−1 characterize the presence of C=O bonds [53,54]. In addition, the characteristic peak at 810 cm−1 matches with the typical breathing mode of triazine. After adding MgO, the distinct peaks of all MgO@g-C3N4 heterojunctions are similar to that of pure g-C3N4, indicating that the crystal structure of g-C3N4 remains unchanged. In addition, the small peak at 419 cm−1 proves the presence of MgO in MgO@g-C3N4 [55].
SEM and TEM analyses
The morphology of g-C3N4, MgO, and 3%MgO@g-C3N4 has been determined through SEM and TEM analyses. The typical bulk structure of g-C3N4 is shown in Figure 5e,f. The difference between the morphologies of MgO and g-C3N4 is difficult to observe by SEM (Figure 5c,d). Figure 5a and Figure 5b show that the morphology of 3%MgO@ g-C3N4 is similar to that of pure g-C3N4. The results indicate that the morphology of 3% MgO@g-C3N4 is identical to that of g-C3N4 because the added amount of MgO is very low, as determined by EDS mapping. The EDS mapping images of 3% MgO@g-C3N4 are shown in Figure 6 and Figure S2 (Supporting Information File 1). The weight percentages of C, N, Mg, and O are 37, 52, 9, and 2 wt %, respectively. The weight fractions of Mg and O are the lowest, indicating that the amount of MgO in the MgO@g-C3N4 sample is too low. The shape of g-C3N4 is easy to observe in Figure 7c,d. However, the shape of MgO is complicated to determine by TEM and HR-TEM (Figure 7a,b). These results prove the presence of MgO in the compound.
![[2190-4286-13-96-5]](/bjnano/content/figures/2190-4286-13-96-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: SEM images of (a, b) 3%MgO@g-C3N4, (c, d) MgO, and (e, f) g-C3N4.
Figure 5: SEM images of (a, b) 3%MgO@g-C3N4, (c, d) MgO, and (e, f) g-C3N4.
![[2190-4286-13-96-6]](/bjnano/content/figures/2190-4286-13-96-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (a) Elemental composition and (b–e) EDS mappings of 3% MgO@g-C3N4.
Figure 6: (a) Elemental composition and (b–e) EDS mappings of 3% MgO@g-C3N4.
![[2190-4286-13-96-7]](/bjnano/content/figures/2190-4286-13-96-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: TEM and HR−TEM images of (a, b) MgO@g-C3N4 and (c, d) g-C3N4.
Figure 7: TEM and HR−TEM images of (a, b) MgO@g-C3N4 and (c, d) g-C3N4.
Chemical state analysis
XPS and HR-XPS have been employed to determine the chemical states of the materials. XPS survey scans of g-C3N4 MgO and 3% MgO@ g-C3N4 are shown in Figure 8a. The peaks at 87 and 530 eV were assigned to the Mg 2s and O 1s levels of MgO, respectively. The peaks at 287 and 397 eV were assigned to the C 1s and N 1s levels of g-C3N4, respectively [12,56,57]. In the MgO sample, the peaks of Mg 2p, Mg KLL, O loss, and O KLL levels are observed at 46, 304, 555, and 978 eV, respectively [58,59]. The HR-XPS of the C 1s level are shown in Figure 8b. The peak at 283 eV was assigned to the C–C coordination in MgO, and the peak at 287 eV was assigned to N–C=N bonds of g-C3N4. The latter peak only appears in g-C3N4 and 3% MgO@g-C3N4. However, the former peak of MgO and g-C3N4 occurs only in 3% MgO@g-C3N4. The C 1s peaks of the materials do not change during the pyrolysis. The HR-XPS of the N 1s level of the materials is shown in Figure 8c. The peaks at 397 and 399 eV correspond to the C–N=C bonds and the N–(C)3 structures of g-C3N4, respectively [60]. Figure 8d shows the peak of Mg–O bonds in MgO at 398 eV [61]. The Mg 2s peaks of MgO are shown in Figure 8e and Figure 8f. The peaks at 87 eV (Figure 8e) and 88 eV (Figure 8f) confirm the metallic state of Mg [62]. The peaks of the O 1s and Mg 2s levels in 3% MgO@g-C3N4 are too weak.
![[2190-4286-13-96-8]](/bjnano/content/figures/2190-4286-13-96-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: The XPS survey (a), HR−XPS C1s (b), N 1s (c), O 1s (d), C 1s (c), and Mg 2s (e, f) of the materials.
Figure 8: The XPS survey (a), HR−XPS C1s (b), N 1s (c), O 1s (d), C 1s (c), and Mg 2s (e, f) of the materials....
Optical properties
DRS spectra have been measured to understand the optical absorption characteristics of the materials (Figure 9). The g-C3N4 sample significantly absorbs light at a peak around 440 nm, corresponding to the direct and the indirect bandgap of 2.83 and 2.68 eV, respectively (Figure 9b,d). MgO with a wide bandgap shows absorption below 400 nm in both Kubelka–Munk and Tauc plots (Figure 9a,c). After adding MgO to g-C3N4, the absorption of the MgO@ g-C3N4 samples slightly shifts to the visible light region. The optical direct and indirect bandgap energy is slightly reduced, corresponding to the increase in MgO fraction. The indirect bandgap energies of 1% MgO@g-C3N4, 3% MgO@g-C3N4, and 5% MgO@g-C3N4 are 2.79, 2.73, and 2.69 eV, respectively. The trend of the direct bandgap is the same. The bandgap of the materials reduces with increasing amounts of added MgO. The bandgap reduction can be attributed to Mg−N bonds in the MgO@g-C3N4 materials, which promote charge transport, thus, increasing the photocatalytic efficiency [33,51]. Generally, smaller bandgaps lead to better light absorption, indicating the high photocatalytic activity of MgO@g-C3N4 under visible light and also the existence of MgO in the MgO@g-C3N4 heterojunction materials. However, with a smaller bandgap, the recombination of e−–h+ pairs will be faster, which decreases the photocatalytic activity of the materials [63]. As shown in Figure 9a and Figure 9c, the materials mostly absorb in the UV range (200–400 nm), with a sudden decrease in the visible range. The absorbance of 3% MgO@g-C3N4 is more substantial than the absorbance of 1% MgO@g-C3N4 and 5% MgO@g-C3N4 in the UV and visible ranges. These results indicate that the higher photocatalytic NO removal efficiency of the 3% MgO@g-C3N4 strongly depends on the optical properties.
![[2190-4286-13-96-9]](/bjnano/content/figures/2190-4286-13-96-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: (a) DRS reflectance spectra, (b) direct bandgap, (c) DRS absorbance spectra, and (d) indirect bandgap of the materials.
Figure 9: (a) DRS reflectance spectra, (b) direct bandgap, (c) DRS absorbance spectra, and (d) indirect bandg...
Photoluminescence
Fluorescence spectra of MgO and 3% MgO@g-C3N4 are shown in Figure 10a and Figure 10b, respectively. MgO shows strong fluorescence at 270 nm with an excitation wavelength (270 nm) in the UV range. MgO also shows another emission wavelength at 380 nm with an excitation wavelength (770 nm) in the visible range, which could be caused by the native structural defects in MgO [18]. 3% MgO@g-C3N4 shows intense fluorescence at 420 nm via excitation at 850 nm, due to the recombination of charge carriers. The photogenerated electrons from the valance band (VB) of g-C3N4 migrate to the conduction band (CB). The excited electrons in the CB of g-C3N4 can then return to energy bands between the CB and VB of g-C3N4 to produce an emission with an energy of about 1.5 eV. The energy band in g-C3N4 can be attributed to transitions between C atoms and N atoms [64-66]. Also, Liang and co-workers reported that the recombination of the e−–h+ pairs could be inhibited by doping MgO into g-C3N4 [32]. When MgO is added, the defect concentration increases and Mg and O vacancies are generated in MgO@g-C3N4. These defects work as the electron traps, which enhance the capacity to separate photogenerated e−−h pairs in MgO@g-C3N4. The photogenerated e−–h+ pairs in the defects also contribute to the photocatalytic reaction [67,68].
![[2190-4286-13-96-10]](/bjnano/content/figures/2190-4286-13-96-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: 3D fluorescence scan of (a) MgO and (b) 3% MgO@g-C3N4.
Figure 10: 3D fluorescence scan of (a) MgO and (b) 3% MgO@g-C3N4.
Photocatalytic mechanism
Trapping experiments were carried out to evaluate the involvement of electrons, holes, and reactive oxygen species. The used trapping agents were KI (h+), K2Cr2O7 (e−), and IPA (•OH). Figure 11a shows the reduction in efficiency when different scavengers are present. The photocatalytic NO degradation efficiency decreases significantly from 75.4% to 36.4% in the presence of KI. K2Cr2O7 as electron scavenger also reduces the NO decomposition by about 1.3 times. The weak contribution of •OH radicals in the NO degradation is clearly shown, with a reduction in efficiency by only about 1%. Hence, electrons and holes are the main contributors to the photocatalytic NO degradation.
![[2190-4286-13-96-11]](/bjnano/content/figures/2190-4286-13-96-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: (a) Trapping test results of the materials and (b) detection of radicals over over 3% MgO@g-C3N4 by ESR.
Figure 11: (a) Trapping test results of the materials and (b) detection of radicals over over 3% MgO@g-C3N4 by...
Also, ESR was used to determine accurately the reaction mechanism of the material. Figure 11b shows that under visible light and using DMPO–H2O and DMPO–OH, the material generates •OH and •O2 radicals. In contrast, only •OH radicals are generated in the dark, but to a very low extent. Hence, the generation of •O2 radicals contributes significantly to the photocatalytic NO degradation efficiency.
A photocatalysis mechanism of MgO@g-C3N4 is proposed taking into account the results of the DRS and ESR analyses and trapping tests. Because of the large bandgap of MgO, only g-C3N4 generates e−–h+ pairs under visible light (Equation 8). The holes can degrade NO directly by oxidizing NO into NO2 (Equations 9 and 10) [47,69]. Simultaneously, at the CB of g-C3N4, electrons are excited and react with O2 to produce •O2− radicals. In addition, these electrons also migrate across the CB of MgO, creating excess electrons in MgO and avoiding the recombination at g-C3N4. Then, the photogenerated electrons react with O2 to produce •O2− radicals (Equation 11). These •O2− radicals decompose NO to NO3 and prevent the formation of NO2 (Equation 12). Also, •O2− reacts with H2O to produce •HO2 radicals and OH− (Equation 13). Then, the •HO2 decomposes NO to form NO2 and •OH (Equation 14). Also, •HO2 also reacts with H2O and electrons to produce H2O2 (Equation 15). Finally, H2O2 generates two •OH radicals to decompose NO (Equations 16–19) [70,71].
![[2190-4286-13-96-i8]](/bjnano/content/inline/2190-4286-13-96-i8.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i9]](/bjnano/content/inline/2190-4286-13-96-i9.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i10]](/bjnano/content/inline/2190-4286-13-96-i10.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i11]](/bjnano/content/inline/2190-4286-13-96-i11.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i12]](/bjnano/content/inline/2190-4286-13-96-i12.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i13]](/bjnano/content/inline/2190-4286-13-96-i13.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i14]](/bjnano/content/inline/2190-4286-13-96-i14.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i15]](/bjnano/content/inline/2190-4286-13-96-i15.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i16]](/bjnano/content/inline/2190-4286-13-96-i16.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i17]](/bjnano/content/inline/2190-4286-13-96-i17.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i18]](/bjnano/content/inline/2190-4286-13-96-i18.svg?max-width=590&scale=1.18182)
![[2190-4286-13-96-i19]](/bjnano/content/inline/2190-4286-13-96-i19.svg?max-width=590&scale=1.18182)
Conclusion
MgO@g-C3N4 heterojunction materials were effectively synthesized by one-step pyrolysis of commercial MgO and urea. The photocatalytic efficiencies of the synthesized materials were increased dramatically by mixing MgO and g-C3N4. 3% MgO@g-C3N4 possessed the highest photocatalytic NO removal efficiency, reaching 75.4%. In addition, the photocatalytic NO removal efficiency of the MgO@g-C3N4 heterojunction materials was decreased when the amount of MgO exceeded 3%. An enhanced apparent quantum efficiency of (6.7 × 10−4)% as well as an extended lifetime of photogenerated electrons based on the heterojunction structure were obtained by combining MgO and g-C3N4. Moreover, the conversion rates of NO to NO2 and by-products of the 3% MgO@g-C3N4 were the lowest (21.9%) and the highest (53.5%), respectively. Also, 3% MgO@ g-C3N4 showed high reusability after a five-cycle recycling test, when the photocatalytic NO removal efficiency decreased only by 7.1%. The results indicated that 3% MgO@g-C3N4 could be applied in the future as an excellent photocatalyst with high removal efficiency and low generation of toxic products. FTIR, XPS, and EDS measurements were carried out to confirm the presence of MgO in the MgO@g-C3N4 heterojunctions. Although MgO was difficult to determine in the MgO@g-C3N4 heterojunctions, the addition of MgO affected the optical properties of the MgO@g-C3N4 heterojunctions. The DRS result showed that the bandgap of the MgO@g-C3N4 heterojunctions decreased by adding larger amounts of MgO. The light absorption of 3% MgO@g-C3N4 was higher than that of 1% MgO@g-C3N4 and 5% MgO@g-C3N4 in the visible and UV range, which increased the photocatalytic performance under visible light irradiation. The PL results confirmed the presence of vacancies in the MgO@g-C3N4 heterojunctions. MgO@g-C3N4 is promising for large-scale fabrication via this simple and fast method. This study provides a new way to synthesize a MgO@g-C3N4 heterojunction materials and an understanding of the photocatalytic mechanism of the MgO@g-C3N4 heterojunction applied in the removal of NO.
Supporting Information
| Supporting Information File 1: Additional figures. | ||
| Format: PDF | Size: 430.8 KB | Download |
Acknowledgements
This work was supported by Department of Civil Engineering Chung Yuan Christian University, Taoyuan City, 32023, Taiwan; Department of Environmental Engineering, Chung Yuan Christian University, Taoyuan City, 32023, Taiwan; Center for Environmental Risk Management, Chung Yuan Christian University, No.200, Taoyuan City, 32023, Taiwan; Faculty of Environment and Natural Resources, Ho Chi Minh City University of Technology (HCMUT), VNU-HCM, Vietnam.
References
-
Van Viet, P.; Hoang The Vinh, T.; Thi Ngoc Dung, N.; Minh Thi, C. Chem. Phys. Lett. 2021, 775, 138642. doi:10.1016/j.cplett.2021.138642
Return to citation in text: [1] -
Zhou, S.; Young, C. J.; VandenBoer, T. C.; Kahan, T. F. Environ. Sci.: Processes Impacts 2019, 21, 1374–1383. doi:10.1039/c9em00129h
Return to citation in text: [1] -
Pham, M.-T.; Hussain, A.; Bui, D.-P.; Nguyen, T.-M. T.; You, S.-J.; Wang, Y.-F. Environ. Technol. Innovation 2021, 23, 101755. doi:10.1016/j.eti.2021.101755
Return to citation in text: [1] -
Nguyen, V.-H.; Nguyen, B.-S.; Huang, C.-W.; Le, T.-T.; Nguyen, C. C.; Nhi Le, T. T.; Heo, D.; Ly, Q. V.; Trinh, Q. T.; Shokouhimehr, M.; Xia, C.; Lam, S. S.; Vo, D.-V. N.; Kim, S. Y.; Le, Q. V. J. Cleaner Prod. 2020, 270, 121912. doi:10.1016/j.jclepro.2020.121912
Return to citation in text: [1] -
Asghar, U.; Rafiq, S.; Anwar, A.; Iqbal, T.; Ahmed, A.; Jamil, F.; Khurram, M. S.; Akbar, M. M.; Farooq, A.; Shah, N. S.; Park, Y.-K. J. Environ. Chem. Eng. 2021, 9, 106064. doi:10.1016/j.jece.2021.106064
Return to citation in text: [1] -
Qie, F.; Zhu, J.; Rong, J.; Zong, B. Bioresour. Technol. 2019, 292, 122037. doi:10.1016/j.biortech.2019.122037
Return to citation in text: [1] -
Wang, S.; Xu, S.; Gao, S.; Xiao, P.; Jiang, M.; Zhao, H.; Huang, B.; Liu, L.; Niu, H.; Wang, J.; Guo, D. Sci. Rep. 2021, 11, 11003. doi:10.1038/s41598-021-90532-9
Return to citation in text: [1] -
Chen, R.; Zhang, T.; Guo, Y.; Wang, J.; Wei, J.; Yu, Q. Chem. Eng. J. 2021, 420, 127588. doi:10.1016/j.cej.2020.127588
Return to citation in text: [1] -
Talebizadeh, P.; Babaie, M.; Brown, R.; Rahimzadeh, H.; Ristovski, Z.; Arai, M. Renewable Sustainable Energy Rev. 2014, 40, 886–901. doi:10.1016/j.rser.2014.07.194
Return to citation in text: [1] -
He, F.; Jeon, W.; Choi, W. Nat. Commun. 2021, 12, 2528. doi:10.1038/s41467-021-22839-0
Return to citation in text: [1] -
Wang, S.; Li, C.; Wang, T.; Zhang, P.; Li, A.; Gong, J. J. Mater. Chem. A 2014, 2, 2885–2890. doi:10.1039/c3ta14576j
Return to citation in text: [1] -
Papailias, I.; Todorova, N.; Giannakopoulou, T.; Karapati, S.; Boukos, N.; Dimotikali, D.; Trapalis, C. Appl. Surf. Sci. 2018, 430, 225–233. doi:10.1016/j.apsusc.2017.08.084
Return to citation in text: [1] [2] [3] -
Van Pham, V.; Mai, D.-Q.; Bui, D.-P.; Van Man, T.; Zhu, B.; Zhang, L.; Sangkaworn, J.; Tantirungrotechai, J.; Reutrakul, V.; Cao, T. M. Environ. Pollut. 2021, 286, 117510. doi:10.1016/j.envpol.2021.117510
Return to citation in text: [1] -
Hoang The Vinh, T.; Minh Thi, C.; Van Viet, P. Mater. Lett. 2020, 281, 128637. doi:10.1016/j.matlet.2020.128637
Return to citation in text: [1] -
Zhao, G.-Q.; Zou, J.; Hu, J.; Long, X.; Jiao, F.-P. Sep. Purif. Technol. 2021, 279, 119769. doi:10.1016/j.seppur.2021.119769
Return to citation in text: [1] -
Van, K. N.; Huu, H. T.; Nguyen Thi, V. N.; Le Thi, T. L.; Truong, D. H.; Truong, T. T.; Dao, N. N.; Vo, V.; Tran, D. L.; Vasseghian, Y. Chemosphere 2022, 289, 133120. doi:10.1016/j.chemosphere.2021.133120
Return to citation in text: [1] -
Mousavi, M.; Habibi-Yangjeh, A.; Pouran, S. R. J. Mater. Sci.: Mater. Electron. 2018, 29, 1719–1747. doi:10.1007/s10854-017-8166-x
Return to citation in text: [1] -
Madona, J.; Sridevi, C. Inorg. Chem. Commun. 2022, 138, 109265. doi:10.1016/j.inoche.2022.109265
Return to citation in text: [1] [2] -
Huang, Z.; Zhao, X.; Xia, H.; Lu, F.; Hu, L.; Chu, P. K. J. Environ. Chem. Eng. 2021, 9, 105922. doi:10.1016/j.jece.2021.105922
Return to citation in text: [1] -
Sharmin, F.; Chandra Roy, D.; Basith, M. A. Int. J. Hydrogen Energy 2021, 46, 38232–38246. doi:10.1016/j.ijhydene.2021.09.072
Return to citation in text: [1] -
Qi, K.; Cheng, B.; Yu, J.; Ho, W. J. Alloys Compd. 2017, 727, 792–820. doi:10.1016/j.jallcom.2017.08.142
Return to citation in text: [1] -
Murillo-Sierra, J. C.; Hernández-Ramírez, A.; Hinojosa-Reyes, L.; Guzmán-Mar, J. L. Chem. Eng. J. Adv. 2021, 5, 100070. doi:10.1016/j.ceja.2020.100070
Return to citation in text: [1] -
Liu, D.; Kelly, T. L. Nat. Photonics 2014, 8, 133–138. doi:10.1038/nphoton.2013.342
Return to citation in text: [1] -
Low, J.; Yu, J.; Jaroniec, M.; Wageh, S.; Al-Ghamdi, A. A. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1601694. doi:10.1002/adma.201601694
Return to citation in text: [1] -
Li, S.; Wang, C.; Cai, M.; Yang, F.; Liu, Y.; Chen, J.; Zhang, P.; Li, X.; Chen, X. Chem. Eng. J. 2022, 428, 131158. doi:10.1016/j.cej.2021.131158
Return to citation in text: [1] -
Yu, H.; Jiang, L.; Wang, H.; Huang, B.; Yuan, X.; Huang, J.; Zhang, J.; Zeng, G. Small 2019, 1901008. doi:10.1002/smll.201901008
Return to citation in text: [1] -
Wang, C.; Cai, M.; Liu, Y.; Yang, F.; Zhang, H.; Liu, J.; Li, S. J. Colloid Interface Sci. 2022, 605, 727–740. doi:10.1016/j.jcis.2021.07.137
Return to citation in text: [1] -
Wang, C.; Li, S.; Cai, M.; Yan, R.; Dong, K.; Zhang, J.; Liu, Y. J. Colloid Interface Sci. 2022, 619, 307–321. doi:10.1016/j.jcis.2022.03.075
Return to citation in text: [1] -
Li, S.; Wang, C.; Liu, Y.; Cai, M.; Wang, Y.; Zhang, H.; Guo, Y.; Zhao, W.; Wang, Z.; Chen, X. Chem. Eng. J. 2022, 429, 132519. doi:10.1016/j.cej.2021.132519
Return to citation in text: [1] -
Li, S.; Cai, M.; Wang, C.; Liu, Y.; Li, N.; Zhang, P.; Li, X. J. Mater. Sci. Technol. 2022, 123, 177–190. doi:10.1016/j.jmst.2022.02.012
Return to citation in text: [1] -
Guo, M.; Zhou, Z.; Yan, S.; Zhou, P.; Miao, F.; Liang, S.; Wang, J.; Cui, X. Sci. Rep. 2020, 10, 18401. doi:10.1038/s41598-020-75003-x
Return to citation in text: [1] -
An, W.; Tian, L.; Hu, J.; Liu, L.; Cui, W.; Liang, Y. Appl. Surf. Sci. 2020, 534, 147518. doi:10.1016/j.apsusc.2020.147518
Return to citation in text: [1] [2] [3] -
Li, N.; Huang, M.; Zhou, J.; Liu, M.; Jing, D. Chin. J. Catal. 2021, 42, 781–794. doi:10.1016/s1872-2067(20)63690-7
Return to citation in text: [1] [2] [3] [4] -
Ge, L.; Peng, Z.; Wang, W.; Tan, F.; Wang, X.; Su, B.; Qiao, X.; Wong, P. K. J. Mater. Chem. A 2018, 6, 16421–16429. doi:10.1039/c8ta05488f
Return to citation in text: [1] -
Shelemanov, A. A.; Evstropiev, S. K.; Karavaeva, A. V.; Nikonorov, N. V.; Vasilyev, V. N.; Podruhin, Y. F.; Kiselev, V. M. Mater. Chem. Phys. 2022, 276, 125204. doi:10.1016/j.matchemphys.2021.125204
Return to citation in text: [1] -
Panchal, P.; Paul, D. R.; Sharma, A.; Hooda, D.; Yadav, R.; Meena, P.; Nehra, S. P. J. Photochem. Photobiol., A 2019, 385, 112049. doi:10.1016/j.jphotochem.2019.112049
Return to citation in text: [1] -
Dhiman, P.; Rana, G.; Kumar, A.; Sharma, G.; Vo, D.-V. N.; Naushad, M. Environ. Chem. Lett. 2022, 20, 1047–1081. doi:10.1007/s10311-021-01361-1
Return to citation in text: [1] -
Chen, J.; Xiong, J.; Song, Y.; Yu, Y.; Wu, L. Appl. Surf. Sci. 2018, 440, 1269–1276. doi:10.1016/j.apsusc.2018.01.228
Return to citation in text: [1] -
Huang, Y.-C.; Chang, S.-Y.; Jehng, J.-M. J. Phys. Chem. C 2017, 121, 19063–19068. doi:10.1021/acs.jpcc.7b05806
Return to citation in text: [1] -
Vaizogullar, A. I. Kinet. Catal. 2018, 59, 418–427. doi:10.1134/s0023158418040146
Return to citation in text: [1] -
Guerrand, H.; Pucheault, M.; Vaultier, M. Ionic Liquids. Green Process Engineering; CRC Press:: Boca Raton, FL, USA, 2015; pp 267–291.
Return to citation in text: [1] -
Ohtani, B. Adv. Inorg. Chem. 2011, 63, 395–430. doi:10.1016/b978-0-12-385904-4.00001-9
Return to citation in text: [1] -
Bui, D.-P.; Pham, M.-T.; Tran, H.-H.; Nguyen, T.-D.; Cao, T. M.; Pham, V. V. ACS Omega 2021, 6, 27379–27386. doi:10.1021/acsomega.1c04215
Return to citation in text: [1] [2] -
Pham, M.-T.; Luu, H. Q.; Nguyen, T.-M. T.; Tran, H.-H.; You, S.-J.; Wang, Y.-F. Aerosol Air Qual. Res. 2021, 21, 210276. doi:10.4209/aaqr.210276
Return to citation in text: [1] [2] -
Li, S.; Hu, S.; Jiang, W.; Zhang, J.; Xu, K.; Wang, Z. J. Colloid Interface Sci. 2019, 556, 335–344. doi:10.1016/j.jcis.2019.08.077
Return to citation in text: [1] -
Roy, S.; Madras, G. Curr. Org. Chem. 2015, 19, 2122–2131. doi:10.2174/1385272819666150603235429
Return to citation in text: [1] -
Pham, M.-T.; Tran, H.-H.; Nguyen, T.-M. T.; Bui, D.-P.; Huang, Y.; Cao, J.; You, S.-J.; Van Viet, P.; Nam, V. H.; Wang, Y.-F. Acta Mater. 2021, 215, 117068. doi:10.1016/j.actamat.2021.117068
Return to citation in text: [1] [2] -
Kumar, A.; Singh, S.; Khanuja, M. Mater. Chem. Phys. 2020, 243, 122402. doi:10.1016/j.matchemphys.2019.122402
Return to citation in text: [1] -
Xu, L.; Gao, S.; Chen, M.; Wu, Y.; Shinozaki, K. Mater. Chem. Phys. 2020, 253, 123368. doi:10.1016/j.matchemphys.2020.123368
Return to citation in text: [1] -
Mohammed, W. M.; Yanilkin, I. V.; Gumarov, A. I.; Kiiamov, A. G.; Yusupov, R. V.; Tagirov, L. R. Beilstein J. Nanotechnol. 2020, 11, 807–813. doi:10.3762/bjnano.11.65
Return to citation in text: [1] -
Mao, N.; Jiang, J.-X. Appl. Surf. Sci. 2019, 476, 144–150. doi:10.1016/j.apsusc.2019.01.049
Return to citation in text: [1] [2] -
Yuan, Y.; Zhang, L.; Xing, J.; Utama, M. I. B.; Lu, X.; Du, K.; Li, Y.; Hu, X.; Wang, S.; Genç, A.; Dunin-Borkowski, R.; Arbiol, J.; Xiong, Q. Nanoscale 2015, 7, 12343–12350. doi:10.1039/c5nr02905h
Return to citation in text: [1] -
Chen, Z.; Sun, P.; Fan, B.; Liu, Q.; Zhang, Z.; Fang, X. Appl. Catal., B 2015, 170-171, 10–16. doi:10.1016/j.apcatb.2015.01.024
Return to citation in text: [1] -
Bojdys, M. J.; Müller, J.-O.; Antonietti, M.; Thomas, A. Chem. – Eur. J. 2008, 14, 8177–8182. doi:10.1002/chem.200800190
Return to citation in text: [1] -
Dobrucka, R. Iran. J. Sci. Technol. 2018, 42, 547–555. doi:10.1007/s40995-016-0076-x
Return to citation in text: [1] -
Li, D.; Xiao, Y.; Pu, M.; Zan, J.; Zuo, S.; Xu, H.; Xia, D. Mater. Chem. Phys. 2019, 231, 225–232. doi:10.1016/j.matchemphys.2019.04.016
Return to citation in text: [1] -
Tan, L.; Xu, J.; Zhang, X.; Hang, Z.; Jia, Y.; Wang, S. Appl. Surf. Sci. 2015, 356, 447–453. doi:10.1016/j.apsusc.2015.08.078
Return to citation in text: [1] -
Cimino, A. Mater. Chem. Phys. 1985, 13, 221–241. doi:10.1016/0254-0584(85)90057-4
Return to citation in text: [1] -
Peng, Q.; Dai, Y.; Liu, K.; Luo, X.; He, D.; Tang, X.; Huang, G. J. Mater. Sci. 2020, 55, 11267–11283. doi:10.1007/s10853-020-04822-0
Return to citation in text: [1] -
Wang, P.; Guan, Z.; Li, Q.; Yang, J. J. Mater. Sci. 2018, 53, 774–786. doi:10.1007/s10853-017-1540-5
Return to citation in text: [1] -
Vesali-Kermani, E.; Habibi-Yangjeh, A.; Ghosh, S. J. Ind. Eng. Chem. (Amsterdam, Neth.) 2020, 84, 185–195. doi:10.1016/j.jiec.2019.12.033
Return to citation in text: [1] -
Gu, W.; Lee, J. T.; Nitta, N.; Yushin, G. Nanomater. Nanotechnol. 2014, 4, 30. doi:10.5772/59931
Return to citation in text: [1] -
Lenes, M.; Morana, M.; Brabec, C. J.; Blom, P. W. M. Adv. Funct. Mater. 2009, 19, 1106–1111. doi:10.1002/adfm.200801514
Return to citation in text: [1] -
Wei, F.; Liu, Y.; Zhao, H.; Ren, X.; Liu, J.; Hasan, T.; Chen, L.; Li, Y.; Su, B.-L. Nanoscale 2018, 10, 4515–4522. doi:10.1039/c7nr09660g
Return to citation in text: [1] -
Li, Y.; Gu, M.; Zhang, X.; Fan, J.; Lv, K.; Carabineiro, S. A. C.; Dong, F. Mater. Today 2020, 41, 270–303. doi:10.1016/j.mattod.2020.09.004
Return to citation in text: [1] -
Li, S.; Wang, C.; Liu, Y.; Xue, B.; Jiang, W.; Liu, Y.; Mo, L.; Chen, X. Chem. Eng. J. 2021, 415, 128991. doi:10.1016/j.cej.2021.128991
Return to citation in text: [1] -
Mageshwari, K.; Mali, S. S.; Sathyamoorthy, R.; Patil, P. S. Powder Technol. 2013, 249, 456–462. doi:10.1016/j.powtec.2013.09.016
Return to citation in text: [1] -
Karthik, K.; Dhanuskodi, S.; Gobinath, C.; Prabukumar, S.; Sivaramakrishnan, S. J. Photochem. Photobiol., B 2019, 190, 8–20. doi:10.1016/j.jphotobiol.2018.11.001
Return to citation in text: [1] -
Wang, J.; Yu, J.; Fu, Q.; Yang, H.; Tong, Q.; Hao, Z.; Ouyang, G. ACS Cent. Sci. 2021, 7, 355–364. doi:10.1021/acscentsci.0c01600
Return to citation in text: [1] -
Lasek, J.; Yu, Y.-H.; Wu, J. C. S. J. Photochem. Photobiol., C 2013, 14, 29–52. doi:10.1016/j.jphotochemrev.2012.08.002
Return to citation in text: [1] -
Nikokavoura, A.; Trapalis, C. Appl. Surf. Sci. 2018, 430, 18–52. doi:10.1016/j.apsusc.2017.08.192
Return to citation in text: [1] -
Wang, B.; Chen, D.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. J. Colloid Interface Sci. 2020, 576, 426–434. doi:10.1016/j.jcis.2020.05.037
Return to citation in text: [1] -
Zou, Y.; Xie, Y.; Yu, S.; Chen, L.; Cui, W.; Dong, F.; Zhou, Y. Appl. Surf. Sci. 2019, 496, 143630. doi:10.1016/j.apsusc.2019.143630
Return to citation in text: [1] -
Zhang, X.; Nie, J.; Rao, F.; Liu, H.; Wang, Y.; Qu, D.; Wu, W.; Zhong, P.; Zhu, G. Ceram. Int. 2021, 47, 31302–31310. doi:10.1016/j.ceramint.2021.08.003
Return to citation in text: [1] -
Yang, X.; Cao, X.; Tang, B.; Shan, B.; Deng, M.; Liu, Y. J. Photochem. Photobiol., A 2019, 375, 40–47. doi:10.1016/j.jphotochem.2019.02.011
Return to citation in text: [1] -
Xie, B.; Chen, D.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. Chem. Eng. J. 2022, 430, 132968. doi:10.1016/j.cej.2021.132968
Return to citation in text: [1] -
Van Viet, P.; Nguyen, H.-P.; Tran, H.-H.; Bui, D.-P.; Hai, L. V.; Pham, M.-T.; You, S.-J.; Thi, C. M. J. Sci.: Adv. Mater. Devices 2021, 6, 551–559. doi:10.1016/j.jsamd.2021.07.005
Return to citation in text: [1] -
Zhang, W.; Liang, Y. Front. Chem. (Lausanne, Switz.) 2019, 7, 664. doi:10.3389/fchem.2019.00664
Return to citation in text: [1] -
Wen, M. Q.; Xiong, T.; Zang, Z. G.; Wei, W.; Tang, X. S.; Dong, F. Opt. Express 2016, 24, 10205–10212. doi:10.1364/oe.24.010205
Return to citation in text: [1]
| 72. | Wang, B.; Chen, D.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. J. Colloid Interface Sci. 2020, 576, 426–434. doi:10.1016/j.jcis.2020.05.037 |
| 73. | Zou, Y.; Xie, Y.; Yu, S.; Chen, L.; Cui, W.; Dong, F.; Zhou, Y. Appl. Surf. Sci. 2019, 496, 143630. doi:10.1016/j.apsusc.2019.143630 |
| 74. | Zhang, X.; Nie, J.; Rao, F.; Liu, H.; Wang, Y.; Qu, D.; Wu, W.; Zhong, P.; Zhu, G. Ceram. Int. 2021, 47, 31302–31310. doi:10.1016/j.ceramint.2021.08.003 |
| 46. | Roy, S.; Madras, G. Curr. Org. Chem. 2015, 19, 2122–2131. doi:10.2174/1385272819666150603235429 |
| 47. | Pham, M.-T.; Tran, H.-H.; Nguyen, T.-M. T.; Bui, D.-P.; Huang, Y.; Cao, J.; You, S.-J.; Van Viet, P.; Nam, V. H.; Wang, Y.-F. Acta Mater. 2021, 215, 117068. doi:10.1016/j.actamat.2021.117068 |
| 78. | Zhang, W.; Liang, Y. Front. Chem. (Lausanne, Switz.) 2019, 7, 664. doi:10.3389/fchem.2019.00664 |
| 79. | Wen, M. Q.; Xiong, T.; Zang, Z. G.; Wei, W.; Tang, X. S.; Dong, F. Opt. Express 2016, 24, 10205–10212. doi:10.1364/oe.24.010205 |
| 77. | Van Viet, P.; Nguyen, H.-P.; Tran, H.-H.; Bui, D.-P.; Hai, L. V.; Pham, M.-T.; You, S.-J.; Thi, C. M. J. Sci.: Adv. Mater. Devices 2021, 6, 551–559. doi:10.1016/j.jsamd.2021.07.005 |
| 44. | Pham, M.-T.; Luu, H. Q.; Nguyen, T.-M. T.; Tran, H.-H.; You, S.-J.; Wang, Y.-F. Aerosol Air Qual. Res. 2021, 21, 210276. doi:10.4209/aaqr.210276 |
| 75. | Yang, X.; Cao, X.; Tang, B.; Shan, B.; Deng, M.; Liu, Y. J. Photochem. Photobiol., A 2019, 375, 40–47. doi:10.1016/j.jphotochem.2019.02.011 |
| 76. | Xie, B.; Chen, D.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. Chem. Eng. J. 2022, 430, 132968. doi:10.1016/j.cej.2021.132968 |
| 48. | Kumar, A.; Singh, S.; Khanuja, M. Mater. Chem. Phys. 2020, 243, 122402. doi:10.1016/j.matchemphys.2019.122402 |
| 49. | Xu, L.; Gao, S.; Chen, M.; Wu, Y.; Shinozaki, K. Mater. Chem. Phys. 2020, 253, 123368. doi:10.1016/j.matchemphys.2020.123368 |
| 50. | Mohammed, W. M.; Yanilkin, I. V.; Gumarov, A. I.; Kiiamov, A. G.; Yusupov, R. V.; Tagirov, L. R. Beilstein J. Nanotechnol. 2020, 11, 807–813. doi:10.3762/bjnano.11.65 |
| 33. | Li, N.; Huang, M.; Zhou, J.; Liu, M.; Jing, D. Chin. J. Catal. 2021, 42, 781–794. doi:10.1016/s1872-2067(20)63690-7 |
| 51. | Mao, N.; Jiang, J.-X. Appl. Surf. Sci. 2019, 476, 144–150. doi:10.1016/j.apsusc.2019.01.049 |
| 61. | Vesali-Kermani, E.; Habibi-Yangjeh, A.; Ghosh, S. J. Ind. Eng. Chem. (Amsterdam, Neth.) 2020, 84, 185–195. doi:10.1016/j.jiec.2019.12.033 |
| 62. | Gu, W.; Lee, J. T.; Nitta, N.; Yushin, G. Nanomater. Nanotechnol. 2014, 4, 30. doi:10.5772/59931 |
| 58. | Cimino, A. Mater. Chem. Phys. 1985, 13, 221–241. doi:10.1016/0254-0584(85)90057-4 |
| 59. | Peng, Q.; Dai, Y.; Liu, K.; Luo, X.; He, D.; Tang, X.; Huang, G. J. Mater. Sci. 2020, 55, 11267–11283. doi:10.1007/s10853-020-04822-0 |
| 60. | Wang, P.; Guan, Z.; Li, Q.; Yang, J. J. Mater. Sci. 2018, 53, 774–786. doi:10.1007/s10853-017-1540-5 |
| 55. | Dobrucka, R. Iran. J. Sci. Technol. 2018, 42, 547–555. doi:10.1007/s40995-016-0076-x |
| 12. | Papailias, I.; Todorova, N.; Giannakopoulou, T.; Karapati, S.; Boukos, N.; Dimotikali, D.; Trapalis, C. Appl. Surf. Sci. 2018, 430, 225–233. doi:10.1016/j.apsusc.2017.08.084 |
| 56. | Li, D.; Xiao, Y.; Pu, M.; Zan, J.; Zuo, S.; Xu, H.; Xia, D. Mater. Chem. Phys. 2019, 231, 225–232. doi:10.1016/j.matchemphys.2019.04.016 |
| 57. | Tan, L.; Xu, J.; Zhang, X.; Hang, Z.; Jia, Y.; Wang, S. Appl. Surf. Sci. 2015, 356, 447–453. doi:10.1016/j.apsusc.2015.08.078 |
| 32. | An, W.; Tian, L.; Hu, J.; Liu, L.; Cui, W.; Liang, Y. Appl. Surf. Sci. 2020, 534, 147518. doi:10.1016/j.apsusc.2020.147518 |
| 52. | Yuan, Y.; Zhang, L.; Xing, J.; Utama, M. I. B.; Lu, X.; Du, K.; Li, Y.; Hu, X.; Wang, S.; Genç, A.; Dunin-Borkowski, R.; Arbiol, J.; Xiong, Q. Nanoscale 2015, 7, 12343–12350. doi:10.1039/c5nr02905h |
| 53. | Chen, Z.; Sun, P.; Fan, B.; Liu, Q.; Zhang, Z.; Fang, X. Appl. Catal., B 2015, 170-171, 10–16. doi:10.1016/j.apcatb.2015.01.024 |
| 54. | Bojdys, M. J.; Müller, J.-O.; Antonietti, M.; Thomas, A. Chem. – Eur. J. 2008, 14, 8177–8182. doi:10.1002/chem.200800190 |
| 63. | Lenes, M.; Morana, M.; Brabec, C. J.; Blom, P. W. M. Adv. Funct. Mater. 2009, 19, 1106–1111. doi:10.1002/adfm.200801514 |
| 18. | Madona, J.; Sridevi, C. Inorg. Chem. Commun. 2022, 138, 109265. doi:10.1016/j.inoche.2022.109265 |
| 33. | Li, N.; Huang, M.; Zhou, J.; Liu, M.; Jing, D. Chin. J. Catal. 2021, 42, 781–794. doi:10.1016/s1872-2067(20)63690-7 |
| 51. | Mao, N.; Jiang, J.-X. Appl. Surf. Sci. 2019, 476, 144–150. doi:10.1016/j.apsusc.2019.01.049 |
| 1. | Van Viet, P.; Hoang The Vinh, T.; Thi Ngoc Dung, N.; Minh Thi, C. Chem. Phys. Lett. 2021, 775, 138642. doi:10.1016/j.cplett.2021.138642 |
| 2. | Zhou, S.; Young, C. J.; VandenBoer, T. C.; Kahan, T. F. Environ. Sci.: Processes Impacts 2019, 21, 1374–1383. doi:10.1039/c9em00129h |
| 3. | Pham, M.-T.; Hussain, A.; Bui, D.-P.; Nguyen, T.-M. T.; You, S.-J.; Wang, Y.-F. Environ. Technol. Innovation 2021, 23, 101755. doi:10.1016/j.eti.2021.101755 |
| 7. | Wang, S.; Xu, S.; Gao, S.; Xiao, P.; Jiang, M.; Zhao, H.; Huang, B.; Liu, L.; Niu, H.; Wang, J.; Guo, D. Sci. Rep. 2021, 11, 11003. doi:10.1038/s41598-021-90532-9 |
| 23. | Liu, D.; Kelly, T. L. Nat. Photonics 2014, 8, 133–138. doi:10.1038/nphoton.2013.342 |
| 24. | Low, J.; Yu, J.; Jaroniec, M.; Wageh, S.; Al-Ghamdi, A. A. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1601694. doi:10.1002/adma.201601694 |
| 6. | Qie, F.; Zhu, J.; Rong, J.; Zong, B. Bioresour. Technol. 2019, 292, 122037. doi:10.1016/j.biortech.2019.122037 |
| 25. | Li, S.; Wang, C.; Cai, M.; Yang, F.; Liu, Y.; Chen, J.; Zhang, P.; Li, X.; Chen, X. Chem. Eng. J. 2022, 428, 131158. doi:10.1016/j.cej.2021.131158 |
| 26. | Yu, H.; Jiang, L.; Wang, H.; Huang, B.; Yuan, X.; Huang, J.; Zhang, J.; Zeng, G. Small 2019, 1901008. doi:10.1002/smll.201901008 |
| 27. | Wang, C.; Cai, M.; Liu, Y.; Yang, F.; Zhang, H.; Liu, J.; Li, S. J. Colloid Interface Sci. 2022, 605, 727–740. doi:10.1016/j.jcis.2021.07.137 |
| 28. | Wang, C.; Li, S.; Cai, M.; Yan, R.; Dong, K.; Zhang, J.; Liu, Y. J. Colloid Interface Sci. 2022, 619, 307–321. doi:10.1016/j.jcis.2022.03.075 |
| 29. | Li, S.; Wang, C.; Liu, Y.; Cai, M.; Wang, Y.; Zhang, H.; Guo, Y.; Zhao, W.; Wang, Z.; Chen, X. Chem. Eng. J. 2022, 429, 132519. doi:10.1016/j.cej.2021.132519 |
| 5. | Asghar, U.; Rafiq, S.; Anwar, A.; Iqbal, T.; Ahmed, A.; Jamil, F.; Khurram, M. S.; Akbar, M. M.; Farooq, A.; Shah, N. S.; Park, Y.-K. J. Environ. Chem. Eng. 2021, 9, 106064. doi:10.1016/j.jece.2021.106064 |
| 20. | Sharmin, F.; Chandra Roy, D.; Basith, M. A. Int. J. Hydrogen Energy 2021, 46, 38232–38246. doi:10.1016/j.ijhydene.2021.09.072 |
| 70. | Lasek, J.; Yu, Y.-H.; Wu, J. C. S. J. Photochem. Photobiol., C 2013, 14, 29–52. doi:10.1016/j.jphotochemrev.2012.08.002 |
| 71. | Nikokavoura, A.; Trapalis, C. Appl. Surf. Sci. 2018, 430, 18–52. doi:10.1016/j.apsusc.2017.08.192 |
| 4. | Nguyen, V.-H.; Nguyen, B.-S.; Huang, C.-W.; Le, T.-T.; Nguyen, C. C.; Nhi Le, T. T.; Heo, D.; Ly, Q. V.; Trinh, Q. T.; Shokouhimehr, M.; Xia, C.; Lam, S. S.; Vo, D.-V. N.; Kim, S. Y.; Le, Q. V. J. Cleaner Prod. 2020, 270, 121912. doi:10.1016/j.jclepro.2020.121912 |
| 21. | Qi, K.; Cheng, B.; Yu, J.; Ho, W. J. Alloys Compd. 2017, 727, 792–820. doi:10.1016/j.jallcom.2017.08.142 |
| 22. | Murillo-Sierra, J. C.; Hernández-Ramírez, A.; Hinojosa-Reyes, L.; Guzmán-Mar, J. L. Chem. Eng. J. Adv. 2021, 5, 100070. doi:10.1016/j.ceja.2020.100070 |
| 11. | Wang, S.; Li, C.; Wang, T.; Zhang, P.; Li, A.; Gong, J. J. Mater. Chem. A 2014, 2, 2885–2890. doi:10.1039/c3ta14576j |
| 12. | Papailias, I.; Todorova, N.; Giannakopoulou, T.; Karapati, S.; Boukos, N.; Dimotikali, D.; Trapalis, C. Appl. Surf. Sci. 2018, 430, 225–233. doi:10.1016/j.apsusc.2017.08.084 |
| 13. | Van Pham, V.; Mai, D.-Q.; Bui, D.-P.; Van Man, T.; Zhu, B.; Zhang, L.; Sangkaworn, J.; Tantirungrotechai, J.; Reutrakul, V.; Cao, T. M. Environ. Pollut. 2021, 286, 117510. doi:10.1016/j.envpol.2021.117510 |
| 16. | Van, K. N.; Huu, H. T.; Nguyen Thi, V. N.; Le Thi, T. L.; Truong, D. H.; Truong, T. T.; Dao, N. N.; Vo, V.; Tran, D. L.; Vasseghian, Y. Chemosphere 2022, 289, 133120. doi:10.1016/j.chemosphere.2021.133120 |
| 17. | Mousavi, M.; Habibi-Yangjeh, A.; Pouran, S. R. J. Mater. Sci.: Mater. Electron. 2018, 29, 1719–1747. doi:10.1007/s10854-017-8166-x |
| 67. | Mageshwari, K.; Mali, S. S.; Sathyamoorthy, R.; Patil, P. S. Powder Technol. 2013, 249, 456–462. doi:10.1016/j.powtec.2013.09.016 |
| 68. | Karthik, K.; Dhanuskodi, S.; Gobinath, C.; Prabukumar, S.; Sivaramakrishnan, S. J. Photochem. Photobiol., B 2019, 190, 8–20. doi:10.1016/j.jphotobiol.2018.11.001 |
| 10. | He, F.; Jeon, W.; Choi, W. Nat. Commun. 2021, 12, 2528. doi:10.1038/s41467-021-22839-0 |
| 18. | Madona, J.; Sridevi, C. Inorg. Chem. Commun. 2022, 138, 109265. doi:10.1016/j.inoche.2022.109265 |
| 19. | Huang, Z.; Zhao, X.; Xia, H.; Lu, F.; Hu, L.; Chu, P. K. J. Environ. Chem. Eng. 2021, 9, 105922. doi:10.1016/j.jece.2021.105922 |
| 47. | Pham, M.-T.; Tran, H.-H.; Nguyen, T.-M. T.; Bui, D.-P.; Huang, Y.; Cao, J.; You, S.-J.; Van Viet, P.; Nam, V. H.; Wang, Y.-F. Acta Mater. 2021, 215, 117068. doi:10.1016/j.actamat.2021.117068 |
| 69. | Wang, J.; Yu, J.; Fu, Q.; Yang, H.; Tong, Q.; Hao, Z.; Ouyang, G. ACS Cent. Sci. 2021, 7, 355–364. doi:10.1021/acscentsci.0c01600 |
| 9. | Talebizadeh, P.; Babaie, M.; Brown, R.; Rahimzadeh, H.; Ristovski, Z.; Arai, M. Renewable Sustainable Energy Rev. 2014, 40, 886–901. doi:10.1016/j.rser.2014.07.194 |
| 64. | Wei, F.; Liu, Y.; Zhao, H.; Ren, X.; Liu, J.; Hasan, T.; Chen, L.; Li, Y.; Su, B.-L. Nanoscale 2018, 10, 4515–4522. doi:10.1039/c7nr09660g |
| 65. | Li, Y.; Gu, M.; Zhang, X.; Fan, J.; Lv, K.; Carabineiro, S. A. C.; Dong, F. Mater. Today 2020, 41, 270–303. doi:10.1016/j.mattod.2020.09.004 |
| 66. | Li, S.; Wang, C.; Liu, Y.; Xue, B.; Jiang, W.; Liu, Y.; Mo, L.; Chen, X. Chem. Eng. J. 2021, 415, 128991. doi:10.1016/j.cej.2021.128991 |
| 8. | Chen, R.; Zhang, T.; Guo, Y.; Wang, J.; Wei, J.; Yu, Q. Chem. Eng. J. 2021, 420, 127588. doi:10.1016/j.cej.2020.127588 |
| 14. | Hoang The Vinh, T.; Minh Thi, C.; Van Viet, P. Mater. Lett. 2020, 281, 128637. doi:10.1016/j.matlet.2020.128637 |
| 15. | Zhao, G.-Q.; Zou, J.; Hu, J.; Long, X.; Jiao, F.-P. Sep. Purif. Technol. 2021, 279, 119769. doi:10.1016/j.seppur.2021.119769 |
| 32. | An, W.; Tian, L.; Hu, J.; Liu, L.; Cui, W.; Liang, Y. Appl. Surf. Sci. 2020, 534, 147518. doi:10.1016/j.apsusc.2020.147518 |
| 35. | Shelemanov, A. A.; Evstropiev, S. K.; Karavaeva, A. V.; Nikonorov, N. V.; Vasilyev, V. N.; Podruhin, Y. F.; Kiselev, V. M. Mater. Chem. Phys. 2022, 276, 125204. doi:10.1016/j.matchemphys.2021.125204 |
| 36. | Panchal, P.; Paul, D. R.; Sharma, A.; Hooda, D.; Yadav, R.; Meena, P.; Nehra, S. P. J. Photochem. Photobiol., A 2019, 385, 112049. doi:10.1016/j.jphotochem.2019.112049 |
| 37. | Dhiman, P.; Rana, G.; Kumar, A.; Sharma, G.; Vo, D.-V. N.; Naushad, M. Environ. Chem. Lett. 2022, 20, 1047–1081. doi:10.1007/s10311-021-01361-1 |
| 30. | Li, S.; Cai, M.; Wang, C.; Liu, Y.; Li, N.; Zhang, P.; Li, X. J. Mater. Sci. Technol. 2022, 123, 177–190. doi:10.1016/j.jmst.2022.02.012 |
| 31. | Guo, M.; Zhou, Z.; Yan, S.; Zhou, P.; Miao, F.; Liang, S.; Wang, J.; Cui, X. Sci. Rep. 2020, 10, 18401. doi:10.1038/s41598-020-75003-x |
| 32. | An, W.; Tian, L.; Hu, J.; Liu, L.; Cui, W.; Liang, Y. Appl. Surf. Sci. 2020, 534, 147518. doi:10.1016/j.apsusc.2020.147518 |
| 33. | Li, N.; Huang, M.; Zhou, J.; Liu, M.; Jing, D. Chin. J. Catal. 2021, 42, 781–794. doi:10.1016/s1872-2067(20)63690-7 |
| 34. | Ge, L.; Peng, Z.; Wang, W.; Tan, F.; Wang, X.; Su, B.; Qiao, X.; Wong, P. K. J. Mater. Chem. A 2018, 6, 16421–16429. doi:10.1039/c8ta05488f |
| 44. | Pham, M.-T.; Luu, H. Q.; Nguyen, T.-M. T.; Tran, H.-H.; You, S.-J.; Wang, Y.-F. Aerosol Air Qual. Res. 2021, 21, 210276. doi:10.4209/aaqr.210276 |
| 45. | Li, S.; Hu, S.; Jiang, W.; Zhang, J.; Xu, K.; Wang, Z. J. Colloid Interface Sci. 2019, 556, 335–344. doi:10.1016/j.jcis.2019.08.077 |
| 41. | Guerrand, H.; Pucheault, M.; Vaultier, M. Ionic Liquids. Green Process Engineering; CRC Press:: Boca Raton, FL, USA, 2015; pp 267–291. |
| 42. | Ohtani, B. Adv. Inorg. Chem. 2011, 63, 395–430. doi:10.1016/b978-0-12-385904-4.00001-9 |
| 43. | Bui, D.-P.; Pham, M.-T.; Tran, H.-H.; Nguyen, T.-D.; Cao, T. M.; Pham, V. V. ACS Omega 2021, 6, 27379–27386. doi:10.1021/acsomega.1c04215 |
| 43. | Bui, D.-P.; Pham, M.-T.; Tran, H.-H.; Nguyen, T.-D.; Cao, T. M.; Pham, V. V. ACS Omega 2021, 6, 27379–27386. doi:10.1021/acsomega.1c04215 |
| 33. | Li, N.; Huang, M.; Zhou, J.; Liu, M.; Jing, D. Chin. J. Catal. 2021, 42, 781–794. doi:10.1016/s1872-2067(20)63690-7 |
| 12. | Papailias, I.; Todorova, N.; Giannakopoulou, T.; Karapati, S.; Boukos, N.; Dimotikali, D.; Trapalis, C. Appl. Surf. Sci. 2018, 430, 225–233. doi:10.1016/j.apsusc.2017.08.084 |
| 38. | Chen, J.; Xiong, J.; Song, Y.; Yu, Y.; Wu, L. Appl. Surf. Sci. 2018, 440, 1269–1276. doi:10.1016/j.apsusc.2018.01.228 |
| 39. | Huang, Y.-C.; Chang, S.-Y.; Jehng, J.-M. J. Phys. Chem. C 2017, 121, 19063–19068. doi:10.1021/acs.jpcc.7b05806 |
| 40. | Vaizogullar, A. I. Kinet. Catal. 2018, 59, 418–427. doi:10.1134/s0023158418040146 |
© 2022 Pham et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjnano/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.