Abstract
There is tremendous effort put in the pursuit for cheap and efficient catalysts for photocatalytic hydrogen evolution systems. Herein, we report an active catalyst that uses the earth-abundant element cobalt and water-dispersible sulfonated graphene. The photocatalytic hydrogen evolution activity of the catalyst was tested by using triethanolamine (TEOA) as electron donor and eosin Y (EY) as the photosensitizer under LED irradiation at 525 nm. Hydrogen was produced constantly even after 20 h, and the turnover number (TON) reached 148 (H2/Co) in 4 h with respect to the initial concentration of the added cobalt salts was shown to be 5.6 times larger than that without graphene.
Introduction
Photocatalytic hydrogen evolution from water-splitting is a long-standing goal for researchers since it can help to supply the growing worldwide energy demand not only environmentally friendly but also sustainably [1-4]. Platinum, the most efficient hydrogen evolution co-catalyst, is rare and expensive, which limits its availability [5]. Hence, developing photocatalytic systems that rely only on earth-abundant elements are desired for making hydrogen a competitive alternative energy source. In recent years, systems based on iron complexes, nickel complexes or molybdenum complexes have been reported as promising candidates for catalyzing the hydrogen evolution [6-15]. Cobalt-based catalysts are particularly attractive catalysts that are easily obtained, environmentally benign and rely on earth-abundant elements [16]. Molecular cobalt catalysts [17], such as polypyridyl complexes [18,19], oxime complexes [20], have been proven to be efficient in the photocatalytic production of hydrogen, and the turnover number (TON) has become higher upon introducing more appropriate ligands. Besides, cobalt-based heterogeneous structures are also of interest [21,22]. A hybrid Coh–CdTe artificial catalyst for photocatalytic hydrogen evolution [23], for example, was simply constructed in situ from earth-abundant cobalt salts and CdTe quantum dots.
As a new carbon material with large surface area and excellent electrical properties, graphene has raised much attention since 2004 [24-33]. Specifically, graphene has been involved in photocatalytic hydrogen production systems [34], such as TiO2-(N)RGO-Pt [35-38], g-C3N4-RGO-Pt [39], CdS-RGO-Pt [40-43], MoS2-NRGO [44,45], EY-RGO-Pt [46] and BiVO4-RGO-Ru/SrTiO3:Rh [47] (RGO: reduced graphene oxide; EY: eosin Y). Graphene enhances the catalytic efficiency of hydrogen evolution remarkably. By using transient photovoltage and photocurrent techniques [48-50], the function of graphene was examined. More recently, our group has demonstrated the efficient forward electron-transfer mediated by graphene in terms of the unique spectroscopic property of photosensitizer EY [51]. The result stimulated us to explore graphene-based hydrogen evolution systems with earth-abundant co-catalysts.
In the present work, we report a new water-soluble graphene–cobalt-based hydrogen evolution system, showing a 5.6 times higher efficiency than that of the same system without graphene. Herein, sulfonated-graphene (G-SO3), being water-soluble and partially reduced [52,53], serves as a great platform [41,51] to support the catalysts. With TEOA (triethanolamine) as an electron donor, EY as a photosensitizer, Co(TEOA)22+ is formed in situ and adsorbed at the surface or around the G-SO3 when cobalt salts and G-SO3 are introduced into the hydrogen evolution system. Upon irradiation by visible light (525 nm LEDs as light source) for 4 h, the system is able to produce hydrogen with a TON up to 148 with the initial concentration of cobalt salts added. And hydrogen constantly evolves even after 20 h irradiation.
Results and Discussion
Fourier transform infrared spectroscopy (FTIR) is employed to characterize GO and G-SO3. As shown in Figure 1, compared to GO, G-SO3 has typical absorptions at 1177, 1123 and 1037 cm−1, which are assigned to νS-O and νS-phenyl confirming the modification of sulfanilic acid on graphene sheets [53]. Meanwhile, peaks attributed to C=O in carboxylic acid and carbonyl moieties (νC=O at 1720 cm−1), C–OH (νC–OH at 1365 cm−1) decrease sharply, implying a partial reduction of GO [54].
![[2190-4286-5-128-1]](/bjnano/content/figures/2190-4286-5-128-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: FTIR (a) and XPS (b) spectra of GO, G-SO3 and G-SO3 after photocatalytic hydrogen evolution
Figure 1: FTIR (a) and XPS (b) spectra of GO, G-SO3 and G-SO3 after photocatalytic hydrogen evolution
X-ray photoelectron spectroscopy (XPS) measurements were performed to confirm the differences between GO and G-SO3. Five different peaks centered at 284.5, 285.9, 286.6, 287.7 and 288.9 eV appear in the C1s deconvolution spectrum of GO, corresponding to C=C/C–C in aromatic rings, C–OH (hydroxy), C–O–C (epoxy), C=O (carbonyl), and C(O)O (carboxyl) groups, respectively [55]. For G-SO3, the peak centered at 284.6 eV becomes narrower, suggesting the partial restoration of the π-electron network in G-SO3. Other oxygen-containing carbon peaks, decreased sharply, indicating GO is reduced efficiently.
Further, Raman spectra and X-ray diffraction (XRD) patterns of GO and G-SO3 are compared in Figure 2. GO and G-SO3 both show the characteristic D band and G band at 1350 cm−1 and 1597 cm−1, but the enhanced ID/IG ratio for G-SO3 indicates the functionalization and reduction of GO. As confirmed by the XRD patterns, after reduction and functionalization, the d-spacing becomes wider since the angle 2θ shifted to the left from 8.85° to 6.92°. The decreased intensity, meanwhile, manifests a more disordered structure in G-SO3. As a result, the obtained G-SO3 is both reduced and functionalized, which guarantees not only its high conductivity for electron transfer, but also its great dispersibility to act as a platform to anchor catalysts.
![[2190-4286-5-128-2]](/bjnano/content/figures/2190-4286-5-128-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Raman (a) and XRD (b) spectra of GO (black), G-SO3 (red) and G-SO3 after photocatalytic hydrogen evolution (purple).
Figure 2: Raman (a) and XRD (b) spectra of GO (black), G-SO3 (red) and G-SO3 after photocatalytic hydrogen ev...
The photocatalytic hydrogen evolution was evaluated under irradiation at 525 nm by using TEOA as a sacrificial donor and EY as a photosensitizer, while cobalt salts and G-SO3 were added to serve as a catalyst in the reaction system (Figure 3). It is proposed that Co2+ forms a Co(TEOA)22+ complex in the presence of TEOA [56]. No significant amounts of hydrogen were detected in the absence of either irradiation or the photosensitizer EY, indicating that hydrogen was produced through the photochemical reaction. Evidently, Co(TEOA)22+ complexes can function as catalysts to reduce protons to hydrogen, similar to the observations of Sun and coworkers [57]. When G-SO3 was introduced, the amount of hydrogen obviously increased. Because our previous work [51] has demonstrated that G-SO3 acts as an electron mediator of EY and platinum nanoparticles co-catalyst, we consider that in the current study the electron transfer process from the EY radical anion (EY•−) to G-SO3 or in situ formed-Co(TEOA)22+ would be facilitated. Similar to the storage phenomenon observed in carbon nanotubes, a small fraction of the electrons may get stored in graphene sheets, thus making graphene an electron reservoir to continuously provide electrons to the catalytic center [58-60]. The positive synergetic effect consequently enhances the photocatalytic activity for hydrogen evolution of the system. To examine any counter anion effects, we further used four different kinds of cobalt salts in our photocatalytic hydrogen evolution system: cobalt chloride, cobalt nitrate, cobalt perchlorate and cobalt acetate. The amounts of evolved hydrogen in each system did not differ much, indicating that the catalytic behavior is independent of the anions used. The results also manifest the formation of Co(TEOA)22+ catalysts in the systems.
![[2190-4286-5-128-3]](/bjnano/content/figures/2190-4286-5-128-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Photocatalytic hydrogen evolution with different graphene (a) and cobalt salts (b) at pH 10.86 in H2O after 4 h; sample concentration: Co2+ (2.0 × 10−4 mol/L), graphene (0.04 mg/mL), EY (4.0 × 10−4 mol/L) and TEOA (0.2 mol/L).
Figure 3: Photocatalytic hydrogen evolution with different graphene (a) and cobalt salts (b) at pH 10.86 in H2...
The pH value of the solution greatly influences the hydrogen evolution process of the system. The system performed well over a wide range (pH 8–12), reaching a maximal turnover at pH 10.86 (Supporting Information File 1, Figure S1). However, when pH value was below 7.2, there was no detectable hydrogen produced from the system. This pH-dependency is due to a number of factors: in acidic medium, the protonation of TEOA inevitably results in a poor electron-donating ability and less Co(TEOA)22+ catalyst is formed. In a basic solution, the graphene dispersion was more stable and the light absorption of EY is stronger but the concentration of protons is too low.
To optimize the hydrogen evolution system, four sets of experiments were carried out: varying the concentration of CoSO4, G-SO3, EY and TEOA used while keeping a constant concentration of the other three components at pH 10.86. The results of these experiments are shown in Figure 4. With the addition of G-SO3, even at low concentrations, the amount of hydrogen evolution showed a remarkable increase and reached a maximum of 3.31 mL, which is 5.6 times larger than that of the system without G-SO3. Further increasing the concentration of G-SO3 resulted in a decrease in the amount of hydrogen generated. This phenomenon happened in many other reported works, which can be explained by the light shielding effect of graphene [61-63]. Varying the concentration of CoSO4, a similar tendency was observed. The concentration of EY also exercises a great influence on catalytic performance of the system. The amount of hydrogen evolution increases with the concentration of EY linearly when the concentration of EY is below 0.4 mM. After a further increase of the EY concentration to 0.8 mM or 1.6 mM, however, the amount of hydrogen still increases but at a relatively slower rate. This is because self-quenching and shield-effects inevitably decrease the ability of EY to act as the photosensitizer [64]. As for the electron donor TEOA, the highest hydrogen evolution efficiency was obtained at a concentration of 0.2 M. Figure S2 in Supporting Information File 1 shows the kinetic curve of the photocatalytic hydrogen evolution under the optimized conditions at pH 10.86 (the concentration of CoSO4, G-SO3, EY and TEOA are 2.0 × 10−4 mol/L, 0.04 mg/mL, 4.0 × 10−4 mol/L and 0.2 mol/L, respectively). The total amount of hydrogen evolved under LED irradiation at 525 nm was about 3.31 mL (148 μmol) and the TON reached 148 with respect to the initial concentration of cobalt. More hydrogen was produced from the system after prolonged irradiation times but at a slower rate. The reason for the decreased rate at longer irradiation times is attributed to the decomposition of EY. As described in our previous work [51], EY decomposes to fluorescein, which has a lower absorption but a higher stability. To confirm the result in the current study, we carried out control experiments that used fluorescein as photosensitizer for hydrogen evolution under the identical condition. As shown in Supporting Information File 1, Figure S2, the rate of hydrogen evolution is the same as that of the EY system after 1 h of irradiation.
![[2190-4286-5-128-4]](/bjnano/content/figures/2190-4286-5-128-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Photocatalytic hydrogen evolution as a function of the CoSO4 (a), G-SO3 (b), EY (c) and TEOA (d) concentration at pH 10.86 in H2O after 4 h; other sample concentration: CoSO4 (2.0 × 10−4 mol/L), G-SO3 (0.04 mg/mL), EY (4.0 × 10−4 mol/L) and TEOA (0.2 mol/L).
Figure 4: Photocatalytic hydrogen evolution as a function of the CoSO4 (a), G-SO3 (b), EY (c) and TEOA (d) co...
It is worth noting that after irradiation, a black magnetic precipitate was observed and adsorbed on the magnetron in both cases with or without G-SO3. When rinsed with acetone more than three times, the precipitation was visualized by TEM (transmission electron microscopy). As shown in Figure 5, in the absence of G-SO3 nanoparticles aggregated in size of about hundreds nanometers. Each particle is composed of lots of small nanoparticles of several nanometers in diameter. The lattice fringes in the HRTEM (high resolution TEM) images suggest a well-defined crystal structure. The lattice spacing of about 0.191 and 0.203 nm can be assigned to the (101) and (002) planes of metallic cobalt Co, space group P63/mmc (JCPDS card 05-0727). When G-SO3 was added, the TEM images exhibited much difference. Firstly, nanoparticles were formed but dispersed on G-SO3 sheets instead. Secondly, the sizes of the nanoparticles were smaller. The HRTEM image also showed the lattice fringes, and the lattice spacing (0.191 and 0.203 nm) is consistent with those observed in the system without G-SO3. This phenomenon indicated that G-SO3 provides a platform to support cobalt catalysts, and at the same time G-SO3 avoids the aggregation of the catalyst to some extent. These results are consistent with the better performance and the higher hydrogen evolution from the system with G-SO3.
![[2190-4286-5-128-5]](/bjnano/content/figures/2190-4286-5-128-5.jpg?scale=2.4&max-width=1024&background=FFFFFF)
Figure 5: The TEM images nanoparticles after irradiation with (a–c) or without (d–f) G-SO3.
Figure 5: The TEM images nanoparticles after irradiation with (a–c) or without (d–f) G-SO3.
As mentioned above, the TEM results showed that cobalt metal nanoparticles may form during the process in both cases. XPS and ICP-MS (inductively coupled plasma mass spectrometry) were used to further investigate the magnetic precipitates obtained after the hydrogen evolution reaction. XPS spectra of the precipitates with or without G-SO3 showed the same peak pattern and location in the range from 776 to 810 eV, corresponding to the Co 2p orbital (Supporting Information File 1, Figure S3). The cobalt lines in the spectra, however, were assigned to cobalt(II) [65], not to cobalt(0). This is different from the TEM results and in contrast to the grown cobalt metal nanoparticles on graphene [66]. ICP-MS measurements were carried out by using the precipitates obtained from the system, which gave a cobalt content of 11.1% (with graphene) and 45.0% (without graphene), respectively. Either of these results was much higher than that calculated from XPS (3.8% and 20.8%). In consideration of the fact that XPS probes only a few nanometers below the surface, the discrepancy was tentatively interpreted to be because of Co2+ complexes around the cobalt metal particles, which hinder the effective detection of Co metal in XPS but allows its measurement with ICP-MS. In addition, FTIR spectra of G-SO3 (Figure 1a, purple line) showed a typical C–H stretching vibration at 2918 cm−1 after photocatalytic hydrogen evolution, which apparently comes from the catalytic CoII(TEOA)2 species on the surface of G-SO3.
Cyclic voltammetry (CV) spectra were used to investigate the hydrogen evolution system (Figure 6). And the results showed that Co(TEOA)22+ complex was active for electrocatalytic hydrogen evolution in 0.2 M K2SO4 and 0.4 M TEOA aqueous solution. The CoII(TEOA)2/CoI(TEOA)2 reduction band peaked at about −1.1 V (vs SCE), and is followed by a rapid rise in current at −1.25 V (vs SCE). This increase of current, accompanied by the evolution of bubbles, can be attributed to the catalytic generation of hydrogen from the aqueous solution [67]. In order to verify that Co(TEOA)22+ is responsible for the catalysis, control experiments were performed at room temperature. When the 0.2 M K2SO4 aqueous solution or 0.2 M K2SO4 and 0.4 M TEOA aqueous solution were studied, no catalytic current appeared until the potential was over −1.5 V (vs SCE). When G-SO3 was added, no new peak emerged, but the catalytic current intensity increased by about 20%. The observation implied that in the presence of G-SO3, electron transfer processes become faster, which results in a higher activity toward electrocatalytic hydrogen evolution. Analogously, G-SO3 is important for enhancing the performance of photocatalytic hydrogen evolution. For photocatalytic hydrogen evolution systems, the photosensitizer EY is often reduced by TEOA to form EY•− radical anions. Since the oxidation potential of EY•− (−1.05 V vs NHE) [68] is more negative than that of CoII(TEOA)2/CoI(TEOA)2 couple, an electron transfer from EY•− to cobalt-center is thermodynamically feasible and initiates the whole hydrogen evolution process.
![[2190-4286-5-128-6]](/bjnano/content/figures/2190-4286-5-128-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: CV spectra of the 4.0 × 10−3 mol/L CoSO4 + 0.2 mol/L K2SO4 solution (black), 4.0 × 10−3 mol/L CoSO4 + 0.4 mol/L TEOA + 0.2 mol/L K2SO4 solution (blue), 4.0 × 10−3 mol/L CoSO4 + 0.4 mol/L TEOA + 0.04 mg/mL G-SO3 solution + 0.2 mol/L K2SO4 (red), 0.04 mg/mL G-SO3 solution + 0.2 mol/L K2SO4 (purple) and 0.4 mol/L TEOA + 0.2 mol/L K2SO4 solution (green).
Figure 6: CV spectra of the 4.0 × 10−3 mol/L CoSO4 + 0.2 mol/L K2SO4 solution (black), 4.0 × 10−3 mol/L CoSO4...
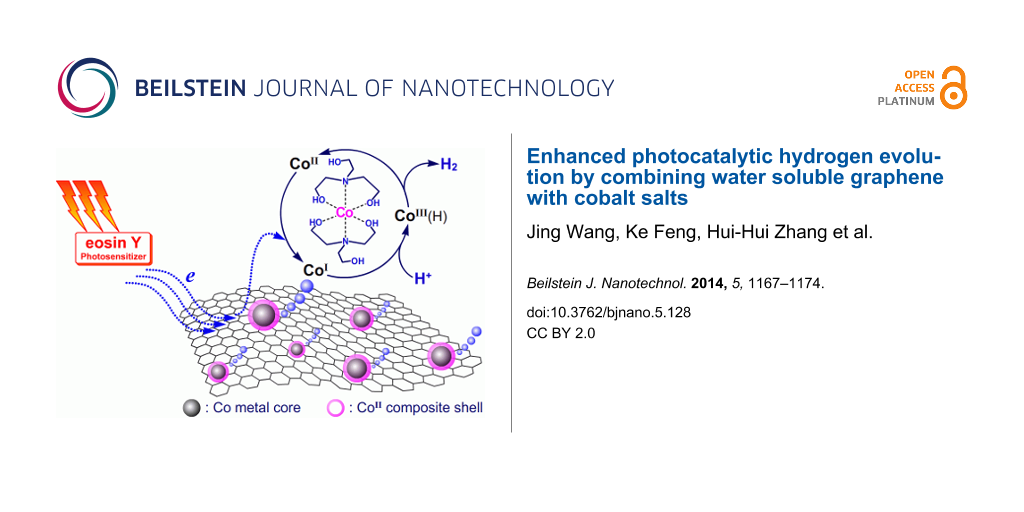
Taking into consideration all results of the TEM, XPS, ICP–MS and CV measurements, the photocatalytic process in this work can be described in Scheme 1. When all the components (TEOA, EY, G-SO3, CoSO4) were added into the reaction system, CoII(TEOA)2 complexes were formed in situ and are at well-adsorbed or surround the G-SO3. In fact, not all of the CoII(TEOA)2 complexes were on the surface of G-SO3, because ICP-MS measurements gave a cobalt content of 11.1%, which was much lower than the feeding ratio of 22.8%. Upon irradiation, the electrons of the EY•− radical anion generated from EY and TEOA, transfer to G-SO3 or directly to CoII(TEOA)2 to initiate the catalytic hydrogen evolution. Since graphene is an ideal electron acceptor and/or electron reservoir, an efficient multi-electron transfer toward the catalytic center CoII(TEOA)2 takes place. Regarding the reports about photocatalytic hydrogen evolution systems based on molecular cobalt complexes in the literature [20], it could be speculated that in the present work the reduction of CoII(TEOA)2 to CoI(TEOA)2 occurs firstly. CoI(TEOA)2, on the one hand, can be protonated to form CoIII(TEOA)2H hydride, which reacts with another hydride to eliminate hydrogen or further protonated to release hydrogen and CoIII(TEOA)2, which is subsequently reduced to CoII(TEOA)2 for the next catalytic circulation. On the other hand, the protonated CoIII(TEOA)2H can also be reduced further to yield CoII(TEOA)2H hydride, which experienced the above cycle for hydrogen evolution. Specifically, if the CoI(TEOA)2 species is not protonated at low concentrations of protons in the system, it can be reduced further to Co0(TEOA)2 [69]. Since there are ligands around Co0(TEOA)2, this Co0(TEOA)2 species can be protonated to form CoII(TEOA)2H that would either eliminate hydrogen as discussed above or release ligands to form metallic cobalt. The obtained metallic cobalt may function as nucleation center anchoring other cobalt-catalysts.
![[2190-4286-5-128-i1]](/bjnano/content/inline/2190-4286-5-128-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Schematic illustration of the photocatalytic hydrogen evolution process.
Scheme 1: Schematic illustration of the photocatalytic hydrogen evolution process.
Conclusion
In summary, we introduce a new water-soluble graphene-cobalt-based hydrogen evolution system. With TEOA as the electron donor, EY as the photosensitizer, Co(TEOA)22+ formed in situ from cobalt salts and TEOA on the surface of G-SO3 or around it as the initial catalyst, the effective hydrogen evolution system is established. By using 525 nm LEDs as the light source, this system shows a 5.6 times higher efficiency than that of the same system without G-SO3, and the hydrogen can continually evolve even after 20 h. With TEM, ICP-MS, and XPS measurements the magnetic precipitation after irradiation is confirmed to be Co metal surrounded by Co2+ species. CV results indicate the redox potential for the CoII(TEOA)2/CoI(TEOA)2, manifesting the feasible electron transfer process thermodynamically. The effects of the pH value, as well as the concentration of G-SO3, CoSO4 and TEOA were investigated in detail not only to optimize the catalytic activity for hydrogen evolution but also to understand the reaction mechanism. The enhanced activity of the photocatalytic system makes it attractive to design and synthesize new catalysts by using graphene and earth-abundant metal salts for the photocatalytic H2 production.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 596.7 KB | Download |
References
-
McKone, J. R.; Lewis, N. S.; Gray, H. B. Chem. Mater. 2013, 26, 407–414. doi:10.1021/cm4021518
Return to citation in text: [1] -
Armaroli, N.; Balzani, V. Angew. Chem., Int. Ed. 2007, 46, 52–66. doi:10.1002/anie.200602373
Return to citation in text: [1] -
Cook, T. R.; Dogutan, D. K.; Reece, S. Y.; Surendranath, Y.; Teets, T. S.; Nocera, D. G. Chem. Rev. 2010, 110, 6474–6502. doi:10.1021/cr100246c
Return to citation in text: [1] -
Armaroli, N.; Balzani, V. ChemSusChem 2011, 4, 21–36. doi:10.1002/cssc.201000182
Return to citation in text: [1] -
Harinipriya, S.; Sangaranarayanan, M. V. Langmuir 2002, 18, 5572–5578. doi:10.1021/la025548t
Return to citation in text: [1] -
Li, Z.-J.; Wang, J.-J.; Li, X.-B.; Fan, X.-B.; Meng, Q.-Y.; Feng, K.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Adv. Mater. 2013, 25, 6613–6618. doi:10.1002/adma.201370283
Return to citation in text: [1] -
Han, Z.; Qiu, F.; Eisenberg, R.; Holland, P. L.; Krauss, T. D. Science 2012, 338, 1321–1324. doi:10.1126/science.1227775
Return to citation in text: [1] -
Nippe, M.; Khnayzer, R. S.; Panetier, J. A.; Zee, D. Z.; Olaiya, B. S.; Head-Gordon, M.; Chang, C. J.; Castellano, F. N.; Long, J. R. Chem. Sci. 2013, 4, 3934–3945. doi:10.1039/c3sc51660a
Return to citation in text: [1] -
Wen, F.; Li, C. Acc. Chem. Res. 2013, 46, 2355–2364. doi:10.1021/ar300224u
Return to citation in text: [1] -
Ran, J.; Zhang, J.; Yu, J.; Jaroniec, M.; Qiao, S. Z. Chem. Soc. Rev. 2014, in press. doi:10.1039/c3cs60425j
Return to citation in text: [1] -
Kong, D.; Cha, J. J.; Wang, H.; Lee, H. R.; Cui, Y. Energy Environ. Sci. 2013, 6, 3553–3558. doi:10.1039/c3ee42413h
Return to citation in text: [1] -
Du, P.; Eisenberg, R. Energy Environ. Sci. 2012, 5, 6012–6021. doi:10.1039/C2EE03250C
Return to citation in text: [1] -
Thoi, V. S.; Sun, Y.; Long, J. R.; Chang, C. J. Chem. Soc. Rev. 2013, 42, 2388–2400. doi:10.1039/c2cs35272a
Return to citation in text: [1] -
Wang, M.; Sun, L. ChemSusChem 2010, 3, 551–554. doi:10.1002/cssc.201000062
Return to citation in text: [1] -
Wang, F.; Wang, W.-G.; Wang, H.-Y.; Si, G.; Tung, C.-H.; Wu, L.-Z. ACS Catal. 2012, 2, 407–416. doi:10.1021/cs200458b
Return to citation in text: [1] -
Losse, S.; Vos, J. G.; Rau, S. Coord. Chem. Rev. 2010, 254, 2492–2504. doi:10.1016/j.ccr.2010.06.004
Return to citation in text: [1] -
Natali, M.; Luisa, A.; Iengo, E.; Scandola, F. Chem. Commun. 2014, 50, 1842–1844. doi:10.1039/c3cc48882a
Return to citation in text: [1] -
Bachmann, C.; Guttentag, M.; Spingler, B.; Alberto, R. Inorg. Chem. 2013, 52, 6055–6061. doi:10.1021/ic4004017
Return to citation in text: [1] -
Guttentag, M.; Rodenberg, A.; Bachmann, C.; Senn, A.; Hamm, P.; Alberto, R. Dalton Trans. 2013, 42, 334–337. doi:10.1039/c2dt31699d
Return to citation in text: [1] -
Dempsey, J. L.; Brunschwig, B. S.; Winkler, J. R.; Gray, H. B. Acc. Chem. Res. 2009, 42, 1995–2004. doi:10.1021/ar900253e
Return to citation in text: [1] [2] -
Huang, J.; Mulfort, K. L.; Du, P.; Chen, L. X. J. Am. Chem. Soc. 2012, 134, 16472–16475. doi:10.1021/ja3062584
Return to citation in text: [1] -
Cao, S.-W.; Liu, X.-F.; Yuan, Y.-P.; Zhang, Z.-Y.; Fang, J.; Loo, S. C. J.; Barber, J.; Sum, T. C.; Xue, C. Phys. Chem. Chem. Phys. 2013, 15, 18363–18366. doi:10.1039/c3cp53350f
Return to citation in text: [1] -
Li, Z.-J.; Li, X.-B.; Wang, J.-J.; Yu, S.; Li, C.-B.; Tung, C.-H.; Wu, L.-Z. Energy Environ. Sci. 2013, 6, 465–469. doi:10.1039/C2EE23898E
Return to citation in text: [1] -
Chen, D.; Zhang, H.; Liu, Y.; Li, J. Energy Environ. Sci. 2013, 6, 1362–1387. doi:10.1039/c3ee23586f
Return to citation in text: [1] -
Li, D.; Kaner, R. B. Science 2008, 320, 1170–1171. doi:10.1126/science.1158180
Return to citation in text: [1] -
Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Science 2004, 306, 666–669. doi:10.1126/science.1102896
Return to citation in text: [1] -
Loh, K. P.; Bao, Q.; Ang, P. K.; Yang, J. J. Mater. Chem. 2010, 20, 2277–2289. doi:10.1039/b920539j
Return to citation in text: [1] -
Bolotin, K. I.; Sikes, K. J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H. L. Solid State Commun. 2008, 146, 351–355. doi:10.1016/j.ssc.2008.02.024
Return to citation in text: [1] -
Stoller, M. D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R. S. Nano Lett. 2008, 8, 3498–3502. doi:10.1021/nl802558y
Return to citation in text: [1] -
Park, S.; Ruoff, R. S. Nat. Nanotechnol. 2009, 4, 217–224. doi:10.1038/nnano.2009.58
Return to citation in text: [1] -
Huang, X.; Qi, X.; Boey, F.; Zhang, H. Chem. Soc. Rev. 2012, 41, 666–686. doi:10.1039/c1cs15078b
Return to citation in text: [1] -
Liu, Y.; Dong, X.; Chen, P. Chem. Soc. Rev. 2012, 41, 2283–2307. doi:10.1039/c1cs15270j
Return to citation in text: [1] -
Xu, C.; Xu, B.; Gu, Y.; Xiong, Z.; Sun, J.; Zhao, X. Energy Environ. Sci. 2013, 6, 1388–1414. doi:10.1039/c3ee23870a
Return to citation in text: [1] -
Xie, G.; Zhang, K.; Guo, B.; Liu, Q.; Fang, L.; Gong, J. R. Adv. Mater. 2013, 25, 3820–3839. doi:10.1002/adma.201301207
Return to citation in text: [1] -
Zhang, X.-Y.; Li, H.-P.; Cui, X.-L.; Lin, Y. J. Mater. Chem. 2010, 20, 2801–2806. doi:10.1039/b917240h
Return to citation in text: [1] -
Fan, W.; Lai, Q.; Zhang, Q.; Wang, Y. J. Phys. Chem. C 2011, 115, 10694–10701. doi:10.1021/jp2008804
Return to citation in text: [1] -
Pei, F.; Liu, Y.; Xu, S.; Lü, J.; Wang, C.; Cao, S. Int. J. Hydrogen Energy 2013, 38, 2670–2677. doi:10.1016/j.ijhydene.2012.12.045
Return to citation in text: [1] -
Jiang, B.; Tian, C.; Pan, Q.; Jiang, Z.; Wang, J.-Q.; Yan, W.; Fu, H. J. Phys. Chem. C 2011, 115, 23718–23725. doi:10.1021/jp207624x
Return to citation in text: [1] -
Xiang, Q.; Yu, J.; Jaroniec, M. J. Phys. Chem. C 2011, 115, 7355–7363. doi:10.1021/jp200953k
Return to citation in text: [1] -
Fang, Z.; Wang, Y.; Song, J.; Sun, Y.; Zhou, J.; Xu, R.; Duan, H. Nanoscale 2013, 5, 9830–9838. doi:10.1039/c3nr03043a
Return to citation in text: [1] -
Lv, X. J.; Fu, W. F.; Chang, H. X.; Zhang, H.; Cheng, J. S.; Zhang, G. J.; Song, Y.; Hu, C. Y.; Li, J. H. J. Mater. Chem. 2012, 22, 1539–1546. doi:10.1039/c1jm14502a
Return to citation in text: [1] [2] -
Khan, Z.; Chetia, T. R.; Vardhaman, A. K.; Barpuzary, D.; Sastri, C. V.; Qureshi, M. RSC Adv. 2012, 2, 12122–12128. doi:10.1039/c2ra21596a
Return to citation in text: [1] -
Li, Q.; Guo, B.; Yu, J.; Ran, J.; Zhang, B.; Yan, H.; Gong, J. R. J. Am. Chem. Soc. 2011, 133, 10878–10884. doi:10.1021/ja2025454
Return to citation in text: [1] -
Xiang, Q.; Yu, J.; Jaroniec, M. J. Am. Chem. Soc. 2012, 134, 6575–6578. doi:10.1021/ja302846n
Return to citation in text: [1] -
Maitra, U.; Gupta, U.; De, M.; Datta, R.; Govindaraj, A.; Rao, C. N. R. Angew. Chem., Int. Ed. 2013, 52, 13057–13061. doi:10.1002/anie.201306918
Return to citation in text: [1] -
Min, S. X.; Lu, G. X. J. Phys. Chem. C 2011, 115, 13938–13945. doi:10.1021/jp203750z
Return to citation in text: [1] -
Iwase, A.; Ng, Y. H.; Ishiguro, Y.; Kudo, A.; Amal, R. J. Am. Chem. Soc. 2011, 133, 11054–11057. doi:10.1021/ja203296z
Return to citation in text: [1] -
Kaniyankandy, S.; Rawalekar, S.; Ghosh, H. N. J. Phys. Chem. C 2012, 116, 16271–16275. doi:10.1021/jp303712y
Return to citation in text: [1] -
Lightcap, I. V.; Kamat, P. V. J. Am. Chem. Soc. 2012, 134, 7109–7116. doi:10.1021/ja3012929
Return to citation in text: [1] -
Cao, A.; Liu, Z.; Chu, S.; Wu, M.; Ye, Z.; Cai, Z.; Chang, Y.; Wang, S.; Gong, Q.; Liu, Y. Adv. Mater. 2010, 22, 103–106. doi:10.1002/adma.200901920
Return to citation in text: [1] -
Zhang, H.-H.; Feng, K.; Chen, B.; Meng, Q.-Y.; Li, Z.-J.; Tung, C.-H.; Wu, L.-Z. Catal. Sci. Technol. 2013, 3, 1815–1821. doi:10.1039/c3cy00098b
Return to citation in text: [1] [2] [3] [4] -
Huang, W.; Ouyang, X.; Lee, L. J. ACS Nano 2012, 6, 10178–10185. doi:10.1021/nn303917p
Return to citation in text: [1] -
Si, Y.; Samulski, E. T. Nano Lett. 2008, 8, 1679–1682. doi:10.1021/nl080604h
Return to citation in text: [1] [2] -
Stankovich, S.; Dikin, D. A.; Piner, R. D.; Kohlhaas, K. A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S. T.; Ruoff, R. S. Carbon 2007, 45, 1558–1565. doi:10.1016/j.carbon.2007.02.034
Return to citation in text: [1] -
Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. J. Phys. Chem. C 2011, 115, 17009–17019. doi:10.1021/jp203741y
Return to citation in text: [1] -
Hughes, M. N.; Rutt, K. J. J. Chem. Soc. A 1968, 0, 2788–2790. doi:10.1039/j19680002788
Return to citation in text: [1] -
Dong, J. F.; Wang, M.; Li, X. Q.; Chen, L.; He, Y.; Sun, L. C. ChemSusChem 2012, 5, 2133–2138. doi:10.1002/cssc.201200490
Return to citation in text: [1] -
Kamat, P. V. J. Phys. Chem. Lett. 2011, 2, 242–251. doi:10.1021/jz101639v
Return to citation in text: [1] -
Williams, G.; Seger, B.; Kamat, P. V. ACS Nano 2008, 2, 1487–1491. doi:10.1021/nn800251f
Return to citation in text: [1] -
Kongkanand, A.; Kamat, P. V. ACS Nano 2007, 1, 13–21. doi:10.1021/nn700036f
Return to citation in text: [1] -
Zhu, M.; Li, Z.; Xiao, B.; Lu, Y.; Du, Y.; Yang, P.; Wang, X. ACS Appl. Mater. Interfaces 2013, 5, 1732–1740. doi:10.1021/am302912v
Return to citation in text: [1] -
Zhou, J.; Tian, G.; Chen, Y.; Meng, X.; Shi, Y.; Cao, X.; Pan, K.; Fu, H. Chem. Commun. 2013, 49, 2237–2239. doi:10.1039/c3cc38999e
Return to citation in text: [1] -
Mou, Z.; Yin, S.; Zhu, M.; Du, Y.; Wang, X.; Yang, P.; Zheng, J.; Lu, C. Phys. Chem. Chem. Phys. 2013, 15, 2793–2799. doi:10.1039/c2cp44270a
Return to citation in text: [1] -
Valdes-Aguilera, O.; Neckers, D. C. Acc. Chem. Res. 1989, 22, 171–177. doi:10.1021/ar00161a002
Return to citation in text: [1] -
Biesinger, M. C.; Payne, B. P.; Grosvenor, A. P.; Lau, L. W. M.; Gerson, A. R.; Smart, R. S. C. Appl. Surf. Sci. 2011, 257, 2717–2730. doi:10.1016/j.apsusc.2010.10.051
Return to citation in text: [1] -
Bai, S.; Shen, X.; Zhu, G.; Li, M.; Xi, H.; Chen, K. ACS Appl. Mater. Interfaces 2012, 4, 2378–2386. doi:10.1021/am300310d
Return to citation in text: [1] -
Sun, Y.; Bigi, J. P.; Piro, N. A.; Tang, M. L.; Long, J. R.; Chang, C. J. J. Am. Chem. Soc. 2011, 133, 9212–9215. doi:10.1021/ja202743r
Return to citation in text: [1] -
Sharma, G. D.; Balraju, P.; Kumar, M.; Roy, M. S. Mater. Sci. Eng., B 2009, 162, 32–39. doi:10.1016/j.mseb.2009.01.033
Return to citation in text: [1] -
Stubbert, B. D.; Peters, J. C.; Gray, H. B. J. Am. Chem. Soc. 2011, 133, 18070–18073. doi:10.1021/ja2078015
Return to citation in text: [1]
| 58. | Kamat, P. V. J. Phys. Chem. Lett. 2011, 2, 242–251. doi:10.1021/jz101639v |
| 59. | Williams, G.; Seger, B.; Kamat, P. V. ACS Nano 2008, 2, 1487–1491. doi:10.1021/nn800251f |
| 60. | Kongkanand, A.; Kamat, P. V. ACS Nano 2007, 1, 13–21. doi:10.1021/nn700036f |
| 61. | Zhu, M.; Li, Z.; Xiao, B.; Lu, Y.; Du, Y.; Yang, P.; Wang, X. ACS Appl. Mater. Interfaces 2013, 5, 1732–1740. doi:10.1021/am302912v |
| 62. | Zhou, J.; Tian, G.; Chen, Y.; Meng, X.; Shi, Y.; Cao, X.; Pan, K.; Fu, H. Chem. Commun. 2013, 49, 2237–2239. doi:10.1039/c3cc38999e |
| 63. | Mou, Z.; Yin, S.; Zhu, M.; Du, Y.; Wang, X.; Yang, P.; Zheng, J.; Lu, C. Phys. Chem. Chem. Phys. 2013, 15, 2793–2799. doi:10.1039/c2cp44270a |
| 64. | Valdes-Aguilera, O.; Neckers, D. C. Acc. Chem. Res. 1989, 22, 171–177. doi:10.1021/ar00161a002 |
| 1. | McKone, J. R.; Lewis, N. S.; Gray, H. B. Chem. Mater. 2013, 26, 407–414. doi:10.1021/cm4021518 |
| 2. | Armaroli, N.; Balzani, V. Angew. Chem., Int. Ed. 2007, 46, 52–66. doi:10.1002/anie.200602373 |
| 3. | Cook, T. R.; Dogutan, D. K.; Reece, S. Y.; Surendranath, Y.; Teets, T. S.; Nocera, D. G. Chem. Rev. 2010, 110, 6474–6502. doi:10.1021/cr100246c |
| 4. | Armaroli, N.; Balzani, V. ChemSusChem 2011, 4, 21–36. doi:10.1002/cssc.201000182 |
| 17. | Natali, M.; Luisa, A.; Iengo, E.; Scandola, F. Chem. Commun. 2014, 50, 1842–1844. doi:10.1039/c3cc48882a |
| 44. | Xiang, Q.; Yu, J.; Jaroniec, M. J. Am. Chem. Soc. 2012, 134, 6575–6578. doi:10.1021/ja302846n |
| 45. | Maitra, U.; Gupta, U.; De, M.; Datta, R.; Govindaraj, A.; Rao, C. N. R. Angew. Chem., Int. Ed. 2013, 52, 13057–13061. doi:10.1002/anie.201306918 |
| 69. | Stubbert, B. D.; Peters, J. C.; Gray, H. B. J. Am. Chem. Soc. 2011, 133, 18070–18073. doi:10.1021/ja2078015 |
| 16. | Losse, S.; Vos, J. G.; Rau, S. Coord. Chem. Rev. 2010, 254, 2492–2504. doi:10.1016/j.ccr.2010.06.004 |
| 46. | Min, S. X.; Lu, G. X. J. Phys. Chem. C 2011, 115, 13938–13945. doi:10.1021/jp203750z |
| 6. | Li, Z.-J.; Wang, J.-J.; Li, X.-B.; Fan, X.-B.; Meng, Q.-Y.; Feng, K.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Adv. Mater. 2013, 25, 6613–6618. doi:10.1002/adma.201370283 |
| 7. | Han, Z.; Qiu, F.; Eisenberg, R.; Holland, P. L.; Krauss, T. D. Science 2012, 338, 1321–1324. doi:10.1126/science.1227775 |
| 8. | Nippe, M.; Khnayzer, R. S.; Panetier, J. A.; Zee, D. Z.; Olaiya, B. S.; Head-Gordon, M.; Chang, C. J.; Castellano, F. N.; Long, J. R. Chem. Sci. 2013, 4, 3934–3945. doi:10.1039/c3sc51660a |
| 9. | Wen, F.; Li, C. Acc. Chem. Res. 2013, 46, 2355–2364. doi:10.1021/ar300224u |
| 10. | Ran, J.; Zhang, J.; Yu, J.; Jaroniec, M.; Qiao, S. Z. Chem. Soc. Rev. 2014, in press. doi:10.1039/c3cs60425j |
| 11. | Kong, D.; Cha, J. J.; Wang, H.; Lee, H. R.; Cui, Y. Energy Environ. Sci. 2013, 6, 3553–3558. doi:10.1039/c3ee42413h |
| 12. | Du, P.; Eisenberg, R. Energy Environ. Sci. 2012, 5, 6012–6021. doi:10.1039/C2EE03250C |
| 13. | Thoi, V. S.; Sun, Y.; Long, J. R.; Chang, C. J. Chem. Soc. Rev. 2013, 42, 2388–2400. doi:10.1039/c2cs35272a |
| 14. | Wang, M.; Sun, L. ChemSusChem 2010, 3, 551–554. doi:10.1002/cssc.201000062 |
| 15. | Wang, F.; Wang, W.-G.; Wang, H.-Y.; Si, G.; Tung, C.-H.; Wu, L.-Z. ACS Catal. 2012, 2, 407–416. doi:10.1021/cs200458b |
| 39. | Xiang, Q.; Yu, J.; Jaroniec, M. J. Phys. Chem. C 2011, 115, 7355–7363. doi:10.1021/jp200953k |
| 68. | Sharma, G. D.; Balraju, P.; Kumar, M.; Roy, M. S. Mater. Sci. Eng., B 2009, 162, 32–39. doi:10.1016/j.mseb.2009.01.033 |
| 5. | Harinipriya, S.; Sangaranarayanan, M. V. Langmuir 2002, 18, 5572–5578. doi:10.1021/la025548t |
| 40. | Fang, Z.; Wang, Y.; Song, J.; Sun, Y.; Zhou, J.; Xu, R.; Duan, H. Nanoscale 2013, 5, 9830–9838. doi:10.1039/c3nr03043a |
| 41. | Lv, X. J.; Fu, W. F.; Chang, H. X.; Zhang, H.; Cheng, J. S.; Zhang, G. J.; Song, Y.; Hu, C. Y.; Li, J. H. J. Mater. Chem. 2012, 22, 1539–1546. doi:10.1039/c1jm14502a |
| 42. | Khan, Z.; Chetia, T. R.; Vardhaman, A. K.; Barpuzary, D.; Sastri, C. V.; Qureshi, M. RSC Adv. 2012, 2, 12122–12128. doi:10.1039/c2ra21596a |
| 43. | Li, Q.; Guo, B.; Yu, J.; Ran, J.; Zhang, B.; Yan, H.; Gong, J. R. J. Am. Chem. Soc. 2011, 133, 10878–10884. doi:10.1021/ja2025454 |
| 20. | Dempsey, J. L.; Brunschwig, B. S.; Winkler, J. R.; Gray, H. B. Acc. Chem. Res. 2009, 42, 1995–2004. doi:10.1021/ar900253e |
| 23. | Li, Z.-J.; Li, X.-B.; Wang, J.-J.; Yu, S.; Li, C.-B.; Tung, C.-H.; Wu, L.-Z. Energy Environ. Sci. 2013, 6, 465–469. doi:10.1039/C2EE23898E |
| 34. | Xie, G.; Zhang, K.; Guo, B.; Liu, Q.; Fang, L.; Gong, J. R. Adv. Mater. 2013, 25, 3820–3839. doi:10.1002/adma.201301207 |
| 66. | Bai, S.; Shen, X.; Zhu, G.; Li, M.; Xi, H.; Chen, K. ACS Appl. Mater. Interfaces 2012, 4, 2378–2386. doi:10.1021/am300310d |
| 21. | Huang, J.; Mulfort, K. L.; Du, P.; Chen, L. X. J. Am. Chem. Soc. 2012, 134, 16472–16475. doi:10.1021/ja3062584 |
| 22. | Cao, S.-W.; Liu, X.-F.; Yuan, Y.-P.; Zhang, Z.-Y.; Fang, J.; Loo, S. C. J.; Barber, J.; Sum, T. C.; Xue, C. Phys. Chem. Chem. Phys. 2013, 15, 18363–18366. doi:10.1039/c3cp53350f |
| 35. | Zhang, X.-Y.; Li, H.-P.; Cui, X.-L.; Lin, Y. J. Mater. Chem. 2010, 20, 2801–2806. doi:10.1039/b917240h |
| 36. | Fan, W.; Lai, Q.; Zhang, Q.; Wang, Y. J. Phys. Chem. C 2011, 115, 10694–10701. doi:10.1021/jp2008804 |
| 37. | Pei, F.; Liu, Y.; Xu, S.; Lü, J.; Wang, C.; Cao, S. Int. J. Hydrogen Energy 2013, 38, 2670–2677. doi:10.1016/j.ijhydene.2012.12.045 |
| 38. | Jiang, B.; Tian, C.; Pan, Q.; Jiang, Z.; Wang, J.-Q.; Yan, W.; Fu, H. J. Phys. Chem. C 2011, 115, 23718–23725. doi:10.1021/jp207624x |
| 67. | Sun, Y.; Bigi, J. P.; Piro, N. A.; Tang, M. L.; Long, J. R.; Chang, C. J. J. Am. Chem. Soc. 2011, 133, 9212–9215. doi:10.1021/ja202743r |
| 20. | Dempsey, J. L.; Brunschwig, B. S.; Winkler, J. R.; Gray, H. B. Acc. Chem. Res. 2009, 42, 1995–2004. doi:10.1021/ar900253e |
| 51. | Zhang, H.-H.; Feng, K.; Chen, B.; Meng, Q.-Y.; Li, Z.-J.; Tung, C.-H.; Wu, L.-Z. Catal. Sci. Technol. 2013, 3, 1815–1821. doi:10.1039/c3cy00098b |
| 18. | Bachmann, C.; Guttentag, M.; Spingler, B.; Alberto, R. Inorg. Chem. 2013, 52, 6055–6061. doi:10.1021/ic4004017 |
| 19. | Guttentag, M.; Rodenberg, A.; Bachmann, C.; Senn, A.; Hamm, P.; Alberto, R. Dalton Trans. 2013, 42, 334–337. doi:10.1039/c2dt31699d |
| 24. | Chen, D.; Zhang, H.; Liu, Y.; Li, J. Energy Environ. Sci. 2013, 6, 1362–1387. doi:10.1039/c3ee23586f |
| 25. | Li, D.; Kaner, R. B. Science 2008, 320, 1170–1171. doi:10.1126/science.1158180 |
| 26. | Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Science 2004, 306, 666–669. doi:10.1126/science.1102896 |
| 27. | Loh, K. P.; Bao, Q.; Ang, P. K.; Yang, J. J. Mater. Chem. 2010, 20, 2277–2289. doi:10.1039/b920539j |
| 28. | Bolotin, K. I.; Sikes, K. J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H. L. Solid State Commun. 2008, 146, 351–355. doi:10.1016/j.ssc.2008.02.024 |
| 29. | Stoller, M. D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R. S. Nano Lett. 2008, 8, 3498–3502. doi:10.1021/nl802558y |
| 30. | Park, S.; Ruoff, R. S. Nat. Nanotechnol. 2009, 4, 217–224. doi:10.1038/nnano.2009.58 |
| 31. | Huang, X.; Qi, X.; Boey, F.; Zhang, H. Chem. Soc. Rev. 2012, 41, 666–686. doi:10.1039/c1cs15078b |
| 32. | Liu, Y.; Dong, X.; Chen, P. Chem. Soc. Rev. 2012, 41, 2283–2307. doi:10.1039/c1cs15270j |
| 33. | Xu, C.; Xu, B.; Gu, Y.; Xiong, Z.; Sun, J.; Zhao, X. Energy Environ. Sci. 2013, 6, 1388–1414. doi:10.1039/c3ee23870a |
| 65. | Biesinger, M. C.; Payne, B. P.; Grosvenor, A. P.; Lau, L. W. M.; Gerson, A. R.; Smart, R. S. C. Appl. Surf. Sci. 2011, 257, 2717–2730. doi:10.1016/j.apsusc.2010.10.051 |
| 51. | Zhang, H.-H.; Feng, K.; Chen, B.; Meng, Q.-Y.; Li, Z.-J.; Tung, C.-H.; Wu, L.-Z. Catal. Sci. Technol. 2013, 3, 1815–1821. doi:10.1039/c3cy00098b |
| 47. | Iwase, A.; Ng, Y. H.; Ishiguro, Y.; Kudo, A.; Amal, R. J. Am. Chem. Soc. 2011, 133, 11054–11057. doi:10.1021/ja203296z |
| 48. | Kaniyankandy, S.; Rawalekar, S.; Ghosh, H. N. J. Phys. Chem. C 2012, 116, 16271–16275. doi:10.1021/jp303712y |
| 49. | Lightcap, I. V.; Kamat, P. V. J. Am. Chem. Soc. 2012, 134, 7109–7116. doi:10.1021/ja3012929 |
| 50. | Cao, A.; Liu, Z.; Chu, S.; Wu, M.; Ye, Z.; Cai, Z.; Chang, Y.; Wang, S.; Gong, Q.; Liu, Y. Adv. Mater. 2010, 22, 103–106. doi:10.1002/adma.200901920 |
| 57. | Dong, J. F.; Wang, M.; Li, X. Q.; Chen, L.; He, Y.; Sun, L. C. ChemSusChem 2012, 5, 2133–2138. doi:10.1002/cssc.201200490 |
| 51. | Zhang, H.-H.; Feng, K.; Chen, B.; Meng, Q.-Y.; Li, Z.-J.; Tung, C.-H.; Wu, L.-Z. Catal. Sci. Technol. 2013, 3, 1815–1821. doi:10.1039/c3cy00098b |
| 55. | Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. J. Phys. Chem. C 2011, 115, 17009–17019. doi:10.1021/jp203741y |
| 56. | Hughes, M. N.; Rutt, K. J. J. Chem. Soc. A 1968, 0, 2788–2790. doi:10.1039/j19680002788 |
| 54. | Stankovich, S.; Dikin, D. A.; Piner, R. D.; Kohlhaas, K. A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S. T.; Ruoff, R. S. Carbon 2007, 45, 1558–1565. doi:10.1016/j.carbon.2007.02.034 |
| 52. | Huang, W.; Ouyang, X.; Lee, L. J. ACS Nano 2012, 6, 10178–10185. doi:10.1021/nn303917p |
| 53. | Si, Y.; Samulski, E. T. Nano Lett. 2008, 8, 1679–1682. doi:10.1021/nl080604h |
| 41. | Lv, X. J.; Fu, W. F.; Chang, H. X.; Zhang, H.; Cheng, J. S.; Zhang, G. J.; Song, Y.; Hu, C. Y.; Li, J. H. J. Mater. Chem. 2012, 22, 1539–1546. doi:10.1039/c1jm14502a |
| 51. | Zhang, H.-H.; Feng, K.; Chen, B.; Meng, Q.-Y.; Li, Z.-J.; Tung, C.-H.; Wu, L.-Z. Catal. Sci. Technol. 2013, 3, 1815–1821. doi:10.1039/c3cy00098b |
© 2014 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)