Abstract
Using magnetron sputtering and heat treatment, Ag@TiO2 nanotubes are prepared. The effects of heat-treatment temperature and heating time on the evolution of Ag nanofilms on the surface of TiO2 nanotubes and microstructure of Ag nanofilms are investigated by X-ray diffraction, field emission scanning electron microscopy, and transmission electron microscopy. Ag atoms migrate mainly on the outmost surface of the TiO2 nanotubes, and fast diffusion of Ag atoms is observed. The diffusivity for the diffusion of Ag atoms on the outmost surface of the TiO2 nanotubes at 400 °C is 6.87 × 10−18 m2/s, which is three orders of magnitude larger than the diffusivities for the diffusion of Ag through amorphous TiO2 films. The activation energy for the diffusion of Ag atoms on the outmost surface of the TiO2 nanotubes in the temperature range of 300 to 500 °C is 157 kJ/mol, which is less than that for the lattice diffusion of Ag and larger than that for the grain boundary diffusion. The diffusion of Ag atoms leads to the formation of Ag nanocrystals on the outmost surface of TiO2 nanotubes. Probably there are hardly any Ag nanocrystals formed inside the TiO2 nanotubes through the migration of Ag.
Introduction
Titanium dioxide (TiO2) has gained great attention for various applications, such as energy storage [1-8] and photo-assisted reactions [9-14], due to its inherent semiconducting characteristics, chemical stability, high photo-conversion efficiency, non-toxicity and low cost [15,16]. TiO2 of low-dimensional structures, including nanotubes, nanoparticles, nanorods, mesoporous spheres, nanosheets, nanowires have been synthesized via anodic oxidation, template, and hydrothermal processing to increase the ratio of surface area to volume for the maximization of effective surface area [17]. Among the low-dimensional structures, TiO2 nanotubes (TNT) seem to be an ideal candidate for the applications in energy storage and photovoltaics.
The intrinsic poor electric conductivity and large bandgaps (approx. 3.4 eV for anatase TiO2 [18] and approx. 3.0 eV for rutile TiO2 [19,20]) have limited the applications of TiO2 of low-dimensional structures. To increase the electric performance of TiO2, TiO2-based materials have been developed by incorporating metal nanoparticles in TiO2 nanotube arrays, using electrochemical deposition [21], irradiation of microwave [22], reduction [23], and sol–gel process [24], which involve the use of aqueous solutions. In addition, the technique of magnetron sputtering has been used to deposit Ag nanostructures on the surface of TiO2 nanotube arrays. It is worth mentioning that Enachi et al. [25] heat-treated the TiO2 nanotube arrays after the deposition of Ag film of 50 nm on the top surface of TiO2 nanotube arrays and observed the formation of Ag nanodots on the top surface of the TiO2 nanotube arrays. However, they did not examine and discuss whether there exist Ag nanodots inside the TiO2 nanotubes or on the surface of the TiO2 nanotubes.
The microstructures of TiO2-based materials depend on the migration of atoms (dopants), which determine the functionality of TiO2-based materials. Although recent success in improving the photocatalytic activity and energy storage of Li ions in TiO2-based materials has been reported [26-29], the understanding of the migration of atoms (dopants) in TiO2 nanotube arrays, which determines the evolution of the microstructures of the TiO2-based materials, remains elusive. In this work, we report the discovery of the fast diffusion of silver through TiO2 nanotube arrays in forming Ag@TiO2 nanotube arrays. The Ag@TiO2 nanotube arrays are prepared by the heat treatment of TiO2 nanotube arrays coated with Ag nanofilms. The microstructures of the Ag@TiO2 nanotube arrays are characterized to determine the diffusivity and activation energy of the Ag diffusion on the surface of the TiO2 nanotubes.
Results
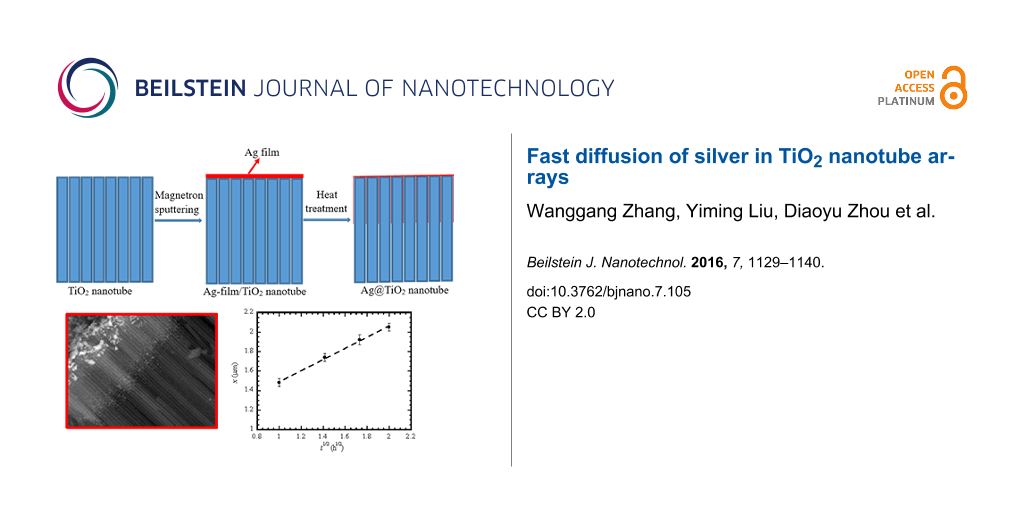
The preparation route of the Ag@TiO2 nanotubes is schematically illustrated in Figure 1. The pure TiO2 nanotubes are prepared by a simple two-step anodization process, and then a layer of Ag film is deposited on the top of the TiO2 nanotubes via sputtering magnetron. The heat treatment of the TiO2 nanotubes with Ag nanofilm leads to the formation of Ag@TiO2 nanotubes.
![[2190-4286-7-105-1]](/bjnano/content/figures/2190-4286-7-105-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic of the facile route for the preparation of Ag@TiO2 nanotubes.
Figure 1: Schematic of the facile route for the preparation of Ag@TiO2 nanotubes.
Figure 2 shows typical SEM images of the pure TiO2 nanotube arrays prepared by a two-step anodization process without heat treatment. It is evident that the prepared pure TiO2 nanotube arrays consist of regular tubes with a diameter of 75 ± 5 nm, a wall thickness of 7 ± 2 nm and an average length of about 6.5 μm. The outmost surface of the pure TiO2 nanotubes prepared by the two-step anodization process is relatively clean and smooth, as shown in Figure 2b. For comparison, typical SEM images of the pure TiO2 nanotube arrays prepared by a one-step anodization process are shown in Figure S1c in Supporting Information File 1. The outmost surface of the pure TiO2 nanotubes prepared by the two-step anodization process is much cleaner and smoother than those prepared by the one-step anodization process. No “bamboo-like” structures are present on the outmost surface of the pure TiO2 nanotubes. Only the pure TiO2 nanotubes prepared by the two-step anodization process were used in this work. The inserted image in Figure 2b shows the topology of the Ti surface after the nanotubes formed by the one-step anodization were ultrasonically removed. There are no TiO2 nanotubes observable, and well-ordered round imprints are observed on the surface of the Ti foil.
![[2190-4286-7-105-2]](/bjnano/content/figures/2190-4286-7-105-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: SEM images of the prepared pure TiO2 nanotubes by a two-step anodization process without heat treatment; (a) overview of the TiO2 nanotubes with an inserted image showing the average length of about 6.5 µm of the TiO2 nanotubes, (b) side view of the TiO2 nanotubes with an inserted image showing well-ordered round imprints on the Ti foil after removing the TiO2 nanotubes formed by a one-step anodization process, and (c) top view of the TiO2 nanotubes, and (d) bottom view of the TiO2 nanotubes.
Figure 2: SEM images of the prepared pure TiO2 nanotubes by a two-step anodization process without heat treat...
The SEM image of Figure 2d reveals the closed honeycomb-like structure of the bottom surface of the pure TiO2 nanotube arrays, similar to the structure prepared by the one-step anodization process (see Figure S1d in Supporting Information File 1). The prepared pure TiO2 nanotube arrays exhibit a highly ordered structure, and the TiO2 nanotubes are normal to the surface of the corresponding Ti foil.
Transmission electron microscopy (TEM) was used to analyze the microstructure of the prepared pure TiO2 nanotubes. Figure 3a shows the TEM images of the pure TiO2 nanotubes, which were heat-treated at 500 °C for 2 h. The wall thickness of the TiO2 nanotubes is relatively uniform along the length of the nanotubes. There is no significant change in the morphology of the pure TiO2 nanotubes after the heat treatment. The average inner diameter and wall thickness of the pure TiO2 nanotubes are ca. 75 and ca. 7 nm, respectively, in accord with the SEM observation.
![[2190-4286-7-105-3]](/bjnano/content/figures/2190-4286-7-105-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: TEM images of the pure TiO2 nanotubes heat-treated at 500 °C for 2 h; (a) TEM image of the pure TiO2 nanotubes, (b) SAED pattern showing the presence of TiO2 nanocrystals, and (c) HRTEM image of a pure TiO2 nanotube.
Figure 3: TEM images of the pure TiO2 nanotubes heat-treated at 500 °C for 2 h; (a) TEM image of the pure TiO2...
The selective area electron diffraction (SAED) pattern of the pure TiO2 nanotubes shown in Figure 3b depicts diffusion rings, suggesting the formation of TiO2 polycrystals after heat treatment. From the HRTEM (high-resolution TEM) analysis shown in Figure 3c, a width of 3.52 Å between neighboring lattice fringes is observed in agreement with the (101) lattice spacing of anatase TiO2 [30,31]. This result reveals the highly crystalline nature of the TiO2 nanotubes in accord with the SAED pattern. The heat treatment can lead to a transition of TiO2 nanotubes from the amorphous phase to a crystalline phase, which has been observed by Jarose et al. [32].
It must be emphasized that the heat treatment with a temperature less than and equal to 500 °C does not cause any morphological changes of the pure TiO2 nanotubes, as demonstrated in Figure 2 and Figure 3. The outmost surface of the pure TiO2 nanotubes remained relatively clean and smooth.
A Ag nanofilm of 230 ± 10 nm in thickness was deposited on top of the prepared pure TiO2 nanotube arrays. The pure TiO2 nanotube arrays with Ag nanofilm were heat-treated at three temperatures of 300, 400, and 500 °C in air for different periods of time. X-ray diffraction (XRD) was used to analyze the crystal structure of Ti and Ag in the heat-treated TiO2 nanotube arrays with Ag nanofilm. Figure 4 shows the XRD pattern of the TiO2 nanotube arrays with Ag nanofilm. No crystallinity of TiO2 is observable for the untreated TiO2 nanotube arrays with Ag nanofilm, and the coating of the Ag nanofilm does not cause any change of the microstructure of the TiO2 nanotube arrays. The XRD patterns of all the heat-treated TiO2 nanotube arrays with Ag nanofilm clearly display the peaks of TiO2, suggesting the transition of TiO2 from the amorphous phase to the crystalline phase in accord with the results observed from the TEM analysis. The diffraction peaks with the 2θ values of ca. 25.3°, ca. 37.8°, ca. 48.0°, ca. 54.0° and ca. 75.0° shown in Figure 4 correspond to the spacings of the (101), (004), (200), (105), and (215) crystal lattice planes of the anatase phase of TiO2 in accord with the values in the standard card (JCPDS NO.21-1272). No diffraction peaks for the rutile phase of TiO2 are detectable. The diffraction peaks with the 2θ values of ca. 38.1°, ca. 44.3°, ca. 64.4° and ca. 77.4° correspond to spacings of the (111), (200), (220) and (311) crystal lattice planes of cubic Ag with a lattice constant of 4.0861 Å (JCPDS NO.65-2871). The diffraction peaks with the 2θ values of ca. 40.2°, ca. 53.0°, ca. 63.0° and ca. 70.7° correspond to the spacings of the (101), (102), (110) and (103) crystal lattice planes of Ti substrate (JCPDS NO.65-6231).
![[2190-4286-7-105-4]](/bjnano/content/figures/2190-4286-7-105-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: XRD patterns of TiO2 nanotube arrays with Ag nanofilm heat treated at three different temperatures for different periods of time.
Figure 4: XRD patterns of TiO2 nanotube arrays with Ag nanofilm heat treated at three different temperatures ...
Figure 5 shows SEM images of the TiO2 nanotube arrays with Ag nanofilm after heat treatment at different temperatures for 2 h. It is evident that a Ag nanofilm of about 225 nm was deposited on the top of the TiO2 nanotube arrays. Without heat treatment, there is little Ag migrating through the TiO2 nanotubes, as shown in Figure 5a. The outmost surface of the TiO2 nanotubes remains relatively clean and smooth. The magnetron sputtering did not cause any significant migration/diffusion of Ag through the TiO2 nanotube arrays. The Ag nanofilm generally covered the top of the TiO2 nanotube arrays (see Figure S2a in Supporting Information File 1) and made the topology of the TiO2 nanotubes indistinguishable. With the heat treatment at 300 °C for 2 h, Ag migrated slightly into the TiO2 nanotube arrays, as shown in Figure 5b. There are Ag nanoparticles present on the outmost surface of the TiO2 nanotubes. The Ag nanofilm became irregular due to the migration of Ag through the TiO2 nanotube arrays and dewetting of the Ag nanofilm, and the topology of TiO2 nanotubes become visible (see Figure S2b in Supporting Information File 1). The increase of the temperature to 400 °C for the heat treatment caused a significant amount of Ag to migrate through the TiO2 nanotube arrays, as shown in Figure 5c, leading to the formation of Ag nanoparticles. There was a layer of Ag over the TiO2 nanotubes. Ag nanoparticles scattered around the propagation front, likely representing the nucleation and growth of Ag nanoparticles induced by the migration/diffusion of Ag during the heat treatment. For the segment of the TiO2 nanotubes not covered by the Ag layer and Ag nanoparticles, the outmost surface remained relatively clean and smooth. Figure S3 in Supporting Information File 1 shows the line scan of EDX of the cross section of the corresponding TiO2 nanotube arrays with Ag nanofilm, which supports the SEM image shown in Figure 5c. Further increase of the temperature to 500 °C for the heat treatment introduced an interesting phenomenon, as shown in Figure 5d. After heat treatment at 500 °C for 2 h, Ag migrated completely through the TiO2 nanotube arrays to the interface between the TiO2 nanotubes and the Ti foil and formed a “film” of Ag covering the outmost surface of the TiO2 nanotubes. The Ag “film” basically occupied the space between TiO2 nanotubes. Ag atoms migrated through the space between TiO2 nanotubes instead of migrating/diffusing through the inner surface of the TiO2 nanotubes. There are probably hardly any Ag nanoparticles formed inside the TiO2 nanotubes through the migration of Ag, where some Ag nanoparticles are present on the surface of the Ag film. As shown in Figure S2 in Supporting Information File 1, more TiO2 nanotubes become visible for the heat treatment at higher temperatures, which confirms the migration of Ag through the TiO2 nanotube arrays. The increase in the temperature for the heat treatment of the TiO2 nanotube arrays with Ag nanofilm enhances the migration of Ag atoms, suggesting that the migration of Ag atoms is a thermal activation process.
![[2190-4286-7-105-5]](/bjnano/content/figures/2190-4286-7-105-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: SEM images of the TiO2 nanotube arrays with Ag nanofilm after different heat treatment for 2 h; (a) without heat treatment, (b) heat treatment at 300 °C, (c) heat treatment at 400 °C, and (d) heat treatment at 500 °C.
Figure 5: SEM images of the TiO2 nanotube arrays with Ag nanofilm after different heat treatment for 2 h; (a)...
Figure 6 shows SEM images of the TiO2 nanotube arrays with Ag nanofilm, which were heat treated at 400 °C for 1, 2, 3 and 4 h. It is evident that the migration/diffusion length of Ag atoms into the TiO2 nanotube arrays increases with the increase of the heating time, as expected. Relatively uniform migration/diffusion lengths are observed for each individual heating time, even though there are some regions with larger migration/diffusion lengths. A significant amount of Ag nanoparticles near the propagation fronts was formed after all four periods of time.
![[2190-4286-7-105-6]](/bjnano/content/figures/2190-4286-7-105-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: SEM images of the heat-treated TiO2 nanotube arrays with Ag nanofilm at 400 °C for different periods of time: (a) 1 h, (b) 2 h, (c) 3 h, and (d) 4 h.
Figure 6: SEM images of the heat-treated TiO2 nanotube arrays with Ag nanofilm at 400 °C for different period...
TEM was used to analyze the microstructures of the heat-treated TiO2 nanotube arrays with Ag nanofilm. Figure 7 shows the TEM images of the TiO2 nanotube arrays with Ag nanofilm heat-treated at 500 °C for 2 h. The morphology of TiO2 nanotubes remained the same except the presence of a layer of Ag nanoparticles, as shown in Figure 7a, which is in accord with the images shown in Figure 5 and Figure 6. The migration/diffusion of Ag and heat treatment do not change the nano-tubular structure of TiO2. It is interesting to note that the Ag film, which covers the outmost surface of the TiO2 nanotubes, is not continuous and made of Ag nanoparticles of about 10 nm in size, as shown in the image embedded in Figure 7a.
![[2190-4286-7-105-7]](/bjnano/content/figures/2190-4286-7-105-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: TEM images of the TiO2 nanotube arrays with Ag nanofilm heat treated at 500 °C for 2 h; (a) TEM image of a TiO2 nanotube covered by a Ag nanofilm, which consists of numerous Ag nanoparticles, (b) SAED pattern showing the presence of Ag nanocrystals, and (c) HRTEM image of the Ag nanofilm showing characteristic lattice spacings of 3.52 Å for TiO2 nanotubes and 2.36 Å for Ag.
Figure 7: TEM images of the TiO2 nanotube arrays with Ag nanofilm heat treated at 500 °C for 2 h; (a) TEM ima...
The EDS result (see Figure S4 in Supporting Information File 1) reveals the presence of Ti, O, and Ag, confirming the existence of Ag around the TiO2 nanotubes. The SAED pattern of the Ag film on the outmost surface of TiO2 nanotubes, as shown in Figure 7b, displays diffuse rings, suggesting that Ag nanoparticles are present in the form of polycrystals. This result suggests that Ag was not oxidized during the heat treatment. The (111), (200), (220) and (311) crystal planes of cubic Ag are observed, in good accord with the XRD analysis. The HRTEM image shown in Figure 7c depicts the characteristic lattice fringe of 3.52 Å for TiO2 nanotubes and 2.36 Å for Ag, which correspond to the (101) plane of anatase TiO2 and {111} planes of face-centered cubic (fcc) structure of Ag [33], respectively. The Ag nanoparticles are Ag nanocrystals. There is no oxidation of Ag nanocrystals in contrast to the general statement that Ag nanoparticles are very reactive and are easily oxidized under the condition of UV irradiation, ambient atmosphere and room temperature [34]. Heat treatment at relatively high temperatures likely leads to the decomposition of silver oxide [34].
It is worth mentioning that the heat-treatment-induced migration/diffusion of Ag into the TiO2 nanotube arrays of anatase phase has also been observed. The SEM images in Figure S6 in Supporting Information File 1 for the TiO2 nanotube arrays of anatase phase with Ag film heat treated at 500 °C in air for 2 h demonstrates the presence of Ag nanoparticles on the outmost surface of the TiO2 nanotube arrays of anatase phase. Ag atoms can migrate into TiO2 nanotube arrays of either amorphous phase or anatase phase.
Discussion
As shown in Figure 5 and Figure 6, there are probably only very few Ag nanoparticles formed inside the TiO2 nanotubes through the migration of Ag. Such behavior reveals the curvature effect on the distribution or wetting of Ag on the surface of the TiO2 nanotubes. According to the Gibbs–Thomson relation [35,36], the equilibrium concentration of solute atoms on a curved surface is determined by the surface energy and the mean curvature. Thus, the equilibrium concentration of Ag atoms on the inner surface of a TiO2 nanotube is lower than that on the planar surface of TiO2 of the same structure, and on the outer surface it is higher than that on the planar surface. Ag atoms are preferred to migrate on the outmost surface of the TiO2 nanotubes and form a layer of Ag during the heat treatment, as observed in Figure 5 and Figure 6.
According to the theory of thermodynamics [37], the melting point of a material is a function of the size of the material. The size dependence of the melting point of a material in spherical shape can be expressed as [38]
![[2190-4286-7-105-i1]](/bjnano/content/inline/2190-4286-7-105-i1.svg?max-width=590&scale=1.18182)
with
![[2190-4286-7-105-i2]](/bjnano/content/inline/2190-4286-7-105-i2.svg?max-width=590&scale=1.18182)
Here, Tm(r) is the melting temperature of the material in a spherical shape of radius r, Tm(∞) is the melting temperature of the material in bulk shape, R is the gas constant, Svib(∞) is the vibrational melting entropy of the material, h is the atomic radius of the material, and d = 0 for nanoparticles. As shown in Figure S7 of Supporting Information File 1, the radius of Ag nanoparticles is in the range from 2.6 ± 0.2 to 5 ± 2 nm. From Equation 1 and Equation 2, one can note that the smaller the size of nanoparticle, the lower is the melting point. Using the parameters of h = 0.2898 nm [39], Tm(∞) = 1234 K [40], Svib(∞) = 7.98 J mol−1·K−1 [38] in Equation 1, one obtains a melting temperature of 857.5 K (584.5 °C) for the smallest size of Ag crystalline nanoparticles (2.4 nm), which is much higher than 300–500 °C used in the heat treatment. The formation of Ag nanocrystals on the outmost surface of the TiO2 nanotubes involves the nucleation and growth processes controlled by the migration/diffusion of Ag atoms.
Generally, the migration of Ag through the TiO2 nanotube arrays involves diffusion, which is controlled by the gradient of concentration. With the deposition of a Ag film on the top of highly ordered TiO2 nanotube arrays, the diffusion of Ag on the outmost surface of the TiO2 nanotubes can be approximated as a one-dimensional problem. The equation describing the diffusion of Ag along the outmost surface of the TiO2 nanotubes is
![[2190-4286-7-105-i3]](/bjnano/content/inline/2190-4286-7-105-i3.svg?max-width=590&scale=1.18182)
Here, c is an average concentration of Ag over the cross-sectional area confined by three adjacent TiO2 nanotubes that are in contact with each other. The condition of zero flux in the direction normal to the outmost surface was used to derive Equation 3. The concentration at the top of the TiO2 nanotubes can be approximated as constant, i.e.,
![[2190-4286-7-105-i4]](/bjnano/content/inline/2190-4286-7-105-i4.svg?max-width=590&scale=1.18182)
Here, c is the concentration of Ag atoms, c0 is the concentration of Ag atoms at the top of the TiO2 nanotubes, t is the time, D is the diffusivity of Ag atoms, and x is the migration distance of Ag on the outmost surface of the TiO2 nanotubes. The initial condition is
![[2190-4286-7-105-i5]](/bjnano/content/inline/2190-4286-7-105-i5.svg?max-width=590&scale=1.18182)
The solution of Equations 3–5 can be found as
![[2190-4286-7-105-i6]](/bjnano/content/inline/2190-4286-7-105-i6.svg?max-width=590&scale=1.18182)
For c(x, t)/c0 = 0.01, one obtains
![[2190-4286-7-105-i7]](/bjnano/content/inline/2190-4286-7-105-i7.svg?max-width=590&scale=1.18182)
which shows that the migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes is proportional to the square root of time, as expected for random diffusion. Using the SEM images shown in Figure 6, the temporal variation of the migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes for the heat treatment at 400 °C is depicted in Figure 8. It is evident that there is a linear relation between the square root of the migration time and the migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes, which is in accord with Equation 7. Using the linear regression to fit the experimental data in Figure 8, one obtains the diffusivity of 6.87 × 10−18 m2/s.
![[2190-4286-7-105-8]](/bjnano/content/figures/2190-4286-7-105-8.png?scale=1.76&max-width=1024&background=FFFFFF)
Figure 8: Migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes as a function of the time during the heat treatment at 400 °C.
Figure 8: Migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes as a function of the ti...
It is known that the temperature dependence of diffusivity follows an Arrhenius relation as D = D0·exp(−Q/RT) with D0 being a pre-exponential constant, Q is the activation energy for the rate process, R is the gas constant, and T is the absolute temperature. Using the Arrhenius relation, the temperature dependence of the migration distance of Ag on the outmost surface of the TiO2 nanotubes during the heat treatment can be expressed as
![[2190-4286-7-105-i8]](/bjnano/content/inline/2190-4286-7-105-i8.svg?max-width=590&scale=1.18182)
As shown in Figure 5d, Ag migrated completely through the TiO2 nanotube arrays to the interface between the TiO2 nanotubes and the Ti foil during the heat treatment at 500 °C for 2 h. One can approximate the migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes as about 6.5 μm of the length of the TiO2 nanotubes for the heat treatment at 500 °C for 2 h. Figure 9 shows the temperature dependence of the migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes for a heat treatment of 2 h. Using Equation 8 to fit the experimental data shown in Figure 9, one obtains the activation energy of 157 kJ/mol for the migration/diffusion of Ag atoms on the outmost surface of the TiO2 nanotubes in the temperature range of 300 to 500 °C.
![[2190-4286-7-105-9]](/bjnano/content/figures/2190-4286-7-105-9.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 9: Temperature dependence of the migration distance of Ag atoms on the outmost surface of the TiO2 nanotubes for a heat treatment of 2 h.
Figure 9: Temperature dependence of the migration distance of Ag atoms on the outmost surface of the TiO2 nan...
There are studies on the diffusion of Ag, including self-diffusion, grain boundary diffusion, and diffusion of Ag in amorphous TiO2 films. Table 1 summarises the data available in literature for the diffusivity and activation energy of the diffusion of Ag. For comparison, the results obtained in this work are also included in Table 1. It is evident that the diffusivity of Ag on the outmost surface of the TiO2 nanotubes is three orders of magnitude larger than the results reported by Kulczyk-Malecka et al. [41] for the sandwich structure of TiO2/Ag/TiO2 coatings, and the activation energy is also larger than that given by Kulczyk-Malecka et al. [41]. Such differences reveal the effect of the microstructure on the diffusion behavior of Ag. The TiO2 in TiO2/Ag/TiO2 coatings was present in the form of thin film and amorphous phase. Also, the time of the heat treatment for the measurement of the diffusivity was 5 min [41], which is much less than the time used in this work. It is interesting to note that the diffusivity obtained in this work is compatible with the diffusivity for the grain boundary diffusion of Ag with the activation energy being less than that for the lattice diffusion of Ag and larger than that for the grain boundary diffusion. Such a result suggests that both the grain boundary diffusion of Ag and the interface diffusion on the surface of the TiO2 nanotubes play important roles in controlling the migration/diffusion of Ag into the TiO2 nanotube arrays.
Table 1: Diffusivity and activation energy for the diffusion of Ag.
| temperature (°C) | diffusivity (m2/s) | activation energy (kJ/mol) | |
|---|---|---|---|
| diffusion of Ag in TiO2/Ag/TiO2 coatings [41] | 100–400 | (3.5–5.8) × 10−21 | 127.5 |
| diffusion of Ag in TiO2/Ag/TiO2 coatings [41] | 600 | 5.93 × 10−20 | 3.5 |
| lattice diffusion of Ag [42] | 400 | 1.1 × 10−19 | 192.0 |
| grain boundary diffusion of Ag [42] | 400 | 6.0 × 10−18 | 111 |
| Ag diffusion into Au core [43] | 27 | (7.5–21) × 10−25 | |
| Ag on surface of TiO2 nanotubes (this work) | 400 | 6.87 × 10−18 | 157 |
Conclusion
In summary, an approach has been developed to coat the outmost surface of TiO2 nanotubes in nanotube arrays with Ag via the deposition of Ag films on top of the TiO2 nanotube arrays by magnetron sputtering and subsequent heat treatment at a temperature between 300 and 500 °C. A two-step anodization process was used to prepare the TiO2 nanotube arrays. The migration of Ag atoms through the TiO2 nanotube arrays was investigated ex situ by using SEM and TEM. In contrast to the Ag@TiO2 nanotubes prepared by the methods involving aqueous solutions, Ag nanocrystals were formed on the outmost surface of TiO2 nanotubes. There probably were hardly any Ag nanocrystals formed inside the TiO2 nanotubes through the migration of Ag. Such a result reveals that Ag atoms prefer to migrate/diffuse on the convex surface of the TiO2 nanotubes. The average migration/diffusion length of Ag atoms on the outmost surface of the TiO2 nanotubes along the longitudinal direction of the TiO2 nanotubes increases with the increase of the heat treatment temperature and the heating time. During the heat treatment at 500 °C for 2 h, Ag atoms migrated completely through the TiO2 nanotube arrays and formed an Ag nanofilm, which consists of numerous Ag nanocrystals, covering the outmost surface of the TiO2 nanotubes.
Using the theory of diffusion, the migration/diffusion of Ag atoms on the outmost surface of the TiO2 nanotubes was analyzed. The diffusivity for the diffusion of Ag atoms on the outmost surface of the TiO2 nanotubes at 400 °C is 6.87 × 10−18 m2/s, which is three orders of magnitude larger than the results reported by Kulczyk-Malecka et al. [41] for the sandwich structure of TiO2/Ag/TiO2 coatings. Such a result demonstrates the fast diffusion of Ag atoms on the surface of the TiO2 nanotubes. The activation energy for the migration/diffusion of Ag on the outmost surface of the TiO2 nanotubes in the temperature range of 300 to 500 °C is 157 kJ/mol, which is less than that for the lattice diffusion of Ag and larger than that for the grain boundary diffusion. Both the grain boundary diffusion of Ag and the interface diffusion on the outmost surface of the TiO2 nanotubes play important roles in the migration/diffusion of Ag into the TiO2 nanotube arrays.
Experimental
Preparation of TiO2 nanotubes
TiO2 nanotubes on Ti foils (99.9 atom %) were fabricated via a two-step anodization process [44]. Briefly, commercially available pure titanium foils (30 × 10 × 0.1 mm3, China Research Institute of Nonferrous Metals, China) were ultrasonically cleaned with acetone, ethanol, and deionized water for 15 min each. Anodization of the Ti foils was performed in a mixture of water, ethylene glycol and 0.13 M NH4F, using a conventional two-electrode cell system. The volume ratio of water to ethylene glycol was 2:100. The cleaned Ti foil was used as the working electrode, and a platinum foil was used as the counter electrode. The anodization voltage was 50 V, and the anodization time was 1 h. TiO2 nanotubes with “bamboo-like” structure at the outer layer were obtained (Figure S1c in Supporting Information File 1). The TiO2 nanotubes with “bamboo-like” structure were completely removed ultrasonically from the as-anodized Ti foils, resulting in well-ordered round imprints on the surface of the Ti foils (see the inserted image in Figure 2b). After being cleaned ultrasonically, the Ti foils were anodized for another 30 min under the same condition to obtain the two-step TiO2 nanotube arrays with relatively clean outmost surface (Figure 2b). The prepared TiO2 nanotube arrays were rinsed with water and dried in air.
Deposition of Ag nanofilms on the top of TiO2 nanotube arrays
Ag nanofilm of 230 ± 10 nm in thickness was deposited on the top of the TiO2 nanotube arrays at room temperature (25 °C) via magnetron sputtering in a multifunctional magnetron sputtering instrument (JGP560B), using an Ag target. Argon gas was flowed into the sputtering chamber after the pressure of the sputtering chamber reached 8 × 10−4 Pa. The surface residuals were removed via pre-sputtering. During the sputtering, argon gas flowed through the chamber at a rate of 30 sccm and a chamber pressure of 6 Pa. The separation between the top of the TiO2 nanotubes and the target was about 30 mm.
The TiO2 nanotube arrays with Ag nanofilms were heat treated at temperatures of 300, 400 and 500 °C, respectively, for different holding times (1, 2 and 3 h) at a ramping rate of 2 °C·min−1 in a tube furnace in air. The heat treatment allowed Ag to migrate through the TiO2 nanotubes. Figure 1 shows schematically the facile route for the preparation of Ag@TiO2 nanotubes.
Materials characterization
The morphology and selected area electron diffraction (SAED) patterns of the prepared structures were examined using field-emission scanning electron microscopy (FESEM) (Tescan MIRA3 LMH) at 10 kV and transmission electron microscopy (TEM) (JEOL 2100-F) at 200 kV. The composition of the prepared structures was determined using energy dispersive spectroscopy (EDS) (OXFORD X-Max 80), and the crystal structure of the prepared structures was analyzed with Cu Kα radiation on a Rigaku D/max 2500 X-ray diffractometer with patterns recorded in a range of 20–80°.
To prepare the TEM specimens, the as-prepared Ag@TiO2 nanotubes, which were scratched from the Ag@TiO2 nanotube arrays using a pair of sharp tweezers, were treated ultrasonically in ethanol for 5–8 min to form a suspension. A drop of the suspension was then drippled on a carbon film supported by a copper grid and dried in air before the TEM analysis.
Supporting Information
| Supporting Information File 1: Additional experimental results. | ||
| Format: PDF | Size: 1.2 MB | Download |
Acknowledgements
LW is grateful for the financial support from the National Natural Science Foundation of China (51301118, 51274149) and the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi province (2013108). WGZ is grateful for the support from China Scholarship Council and the Fundamental Research Funds for the Central Universities. FY is grateful for the support from the “Hundred-People-Plan” Program of Shanxi (2014). LYM is grateful for the support from Natural Science Foundation of Shanxi province (2015021066).
References
-
Kim, M. S.; Lee, T.-W.; Parka, J. H. J. Electrochem. Soc. 2009, 156, A584–A588. doi:10.1149/1.3129682
Return to citation in text: [1] -
Deng, D.; Kim, M. G.; Lee, J. Y.; Cho, J. Energy Environ. Sci. 2009, 2, 818–837. doi:10.1039/b823474d
Return to citation in text: [1] -
Zeng, P.; Liu, Z.; Hu, Z.; Zhai, J.; Jiang, L. RSC Adv. 2013, 3, 22853–22856. doi:10.1039/c3ra43906b
Return to citation in text: [1] -
Zhao, B. T.; Jiang, S. M.; Su, C.; Cai, R.; Ran, R.; Tadé, M. O.; Shao, Z. J. Mater. Chem. A 2013, 1, 12310–12320. doi:10.1039/c3ta12770b
Return to citation in text: [1] -
Weng, Z.; Guo, H.; Liu, X.; Wu, S.; Yeung, K. W. K.; Chu, P. K. RSC Adv. 2013, 3, 24758–24775. doi:10.1039/c3ra44031a
Return to citation in text: [1] -
Hemalatha, K.; Prakash, A. S.; Guruprakash, K.; Jayakumar, M. J. Mater. Chem. A 2014, 2, 1757–1766. doi:10.1039/C3TA13352D
Return to citation in text: [1] -
Lee, E. J.; Nam, I.; Yi, J.; Bang, J. H. J. Mater. Chem. A 2015, 3, 3500–3510. doi:10.1039/C4TA05988C
Return to citation in text: [1] -
Wu, Q. L.; Li, J.; Deshpande, R. D.; Subramanian, N.; Rankin, S. E.; Yang, F.; Cheng, Y.-T. J. Phys. Chem. C 2012, 116, 18669–18677. doi:10.1021/jp3072266
Return to citation in text: [1] -
Zhang, W.; Liu, Y.; Zhou, D.; Wen, J.; Liang, W.; Yang, F. RSC Adv. 2015, 5, 57155–57163. doi:10.1039/C5RA08802J
Return to citation in text: [1] -
Kraeutler, B.; Bard, A. J. J. Am. Chem. Soc. 1977, 99, 7729–7731. doi:10.1021/ja00465a065
Return to citation in text: [1] -
Nakato, Y.; Tsumura, A.; Tsubomura, H. J. Phys. Chem. 1983, 87, 2402–2405. doi:10.1021/j100236a032
Return to citation in text: [1] -
Nakato, Y.; Akanuma, H.; Shimizu, J.-i.; Magari, Y. J. Electroanal. Chem. 1995, 396, 35–39. doi:10.1016/0022-0728(95)04007-B
Return to citation in text: [1] -
Bauer, R.; Waldner, G.; Fallmann, H.; Hager, S.; Klare, M.; Krutzler, T.; Malato, S.; Maletzky, P. Catal. Today 1999, 53, 131–144. doi:10.1016/S0920-5861(99)00108-X
Return to citation in text: [1] -
Ma, D.; Yan, Y.; Ji, H.; Chen, C.; Zhao, J. Chem. Commun. 2015, 51, 17451–17454. doi:10.1039/C5CC07123B
Return to citation in text: [1] -
Linsebigler, A. L.; Lu, G.; Yates, J. T., Jr. Chem. Rev. 1995, 95, 735–758. doi:10.1021/cr00035a013
Return to citation in text: [1] -
Shankar, K.; Basham, J. I.; Allam, N. K.; Varghese, O. K.; Mor, G. K.; Feng, X. J.; Paulose, M.; Seabold, J. A.; Choi, K.-S.; Grimes, C. A. J. Phys. Chem. C 2009, 113, 6327–6359. doi:10.1021/jp809385x
Return to citation in text: [1] -
Rolison, D. R. Science 2003, 299, 1698–1701. doi:10.1126/science.1082332
Return to citation in text: [1] -
Tang, H.; Lévy, F.; Berger, H.; Schmid, P. E. Phys. Rev. B 1995, 52, 7771–7774. doi:10.1103/PhysRevB.52.7771
Return to citation in text: [1] -
Pascual, J.; Camassel, J.; Mathieu, H. Phys. Rev. B 1978, 18, 5606–5614. doi:10.1103/PhysRevB.18.5606
Return to citation in text: [1] -
Amtout, A.; Leonelli, R. Phys. Rev. B 1995, 51, 6842–6851. doi:10.1103/PhysRevB.51.6842
Return to citation in text: [1] -
Huang, L.; Peng, F.; Wang, H.; Yu, H.; Geng, W.; Yang, J.; Zhang, S.; Zhao, H. Mater. Chem. Phys. 2011, 130, 316–322. doi:10.1016/j.matchemphys.2011.06.052
Return to citation in text: [1] -
Guo, G.; Yu, B.; Yu, P.; Chen, X. Talanta 2009, 79, 570–575. doi:10.1016/j.talanta.2009.04.036
Return to citation in text: [1] -
Yan, J.; Song, H.; Yang, S.; Yan, J.; Chen, X. Electrochim. Acta 2008, 53, 6351–6355. doi:10.1016/j.electacta.2008.04.048
Return to citation in text: [1] -
Amin, S. A.; Pazouki, M.; Hosseinnia, A. Powder Technol. 2009, 196, 241–245. doi:10.1016/j.powtec.2009.07.021
Return to citation in text: [1] -
Enachi, M.; Guix, M.; Braniste, T.; Postolache, V.; Ciobanu, V.; Ursaki, V.; Schmidt, O. G.; Tiginyanu, I. Surf. Eng. Appl. Electrochem. 2015, 51, 3–8. doi:10.3103/S1068375515010044
Return to citation in text: [1] -
He, B.-L.; Dong, B.; Li, H.-L. Electrochem. Commun. 2007, 9, 425–430. doi:10.1016/j.elecom.2006.10.008
Return to citation in text: [1] -
Nam, S. H.; Shim, H.-S.; Kim, Y.-S.; Dar, M. A.; Kim, J. G.; Kim, W. B. ACS Appl. Mater. Interfaces 2010, 2, 2046–2052. doi:10.1021/am100319u
Return to citation in text: [1] -
Wei, W.; Oltean, G.; Tai, C.-W.; Edström, K.; Björefors, F.; Nyholm, L. J. Mater. Chem. A 2013, 1, 8160–8169. doi:10.1039/c3ta11273j
Return to citation in text: [1] -
Zhu, K.; Wang, Q.; Kim, J.-H.; Pesaran, A. A.; Frank, A. J. J. Phys. Chem. C 2012, 116, 11895–11899. doi:10.1021/jp301884x
Return to citation in text: [1] -
Liu, E.; Kang, L.; Wu, F.; Sun, T.; Hu, X.; Yang, Y.; Liu, H.; Fan, J. Plasmonics 2014, 9, 61–70. doi:10.1007/s11468-013-9598-7
Return to citation in text: [1] -
Xiao, F. J. Phys. Chem. C 2012, 116, 16487–16498. doi:10.1021/jp3034984
Return to citation in text: [1] -
Jarosz, M.; Syrek, K.; Kapusta-Kołodziej, J.; Mech, J.; Małek, K.; Hnida, K.; Łojewski, T.; Jaskuła, M.; Sulka, G. D. J. Phys. Chem. C 2015, 119, 24182–24191. doi:10.1021/acs.jpcc.5b08403
Return to citation in text: [1] -
Yang, D.; Sun, Y.; Tong, Z.; Tian, Y.; Li, Y.; Jiang, Z. J. Phys. Chem. C 2015, 119, 5827–5835. doi:10.1021/jp511948p
Return to citation in text: [1] -
Shpak, A. P.; Gorbik, P. P., Eds. Nanomaterials and Supramolecular Structures – Physics, Chemistry, and Applications; Springer: Dordrecht, Netherlands, 2010. doi:10.1007/978-90-481-2309-4
Return to citation in text: [1] [2] -
Yang, F. Thin Solid Films 2004, 446, 313–317. doi:10.1016/j.tsf.2003.10.021
Return to citation in text: [1] -
Gusak, A. M.; Lyashenko, Y. A.; Kornienko, S. V.; Pasichnyy, M. O.; Shirinyan, A. S.; Zaporozhets, T. V., Eds. Diffusion-controlled solid state reactions: in alloys, thin-films, and nanosystems; John Wiley & Sons: New York, NY, U.S.A., 2010. doi:10.1002/9783527631025
Return to citation in text: [1] -
Ragone, D. V. Thermodynamics of materials; Wiley: Hoboken, NJ, U.S.A., 1995.
Return to citation in text: [1] -
Jiang, Q.; Zhang, S.; Zhao, M. Mater. Chem. Phys. 2003, 82, 225–227. doi:10.1016/S0254-0584(03)00201-3
Return to citation in text: [1] [2] -
King, H. W. In Physical Metallurgy, 3rd ed.; Cahn, R. W.; Haasen, P., Eds.; North Holland: Amsterdam, Netherlands, 1983; p 63.
Return to citation in text: [1] -
Sargent, W. Table of periodic properties of the elements; Sargent-Welch Scientific: Skokie, IL, U.S.A., 1980.
Return to citation in text: [1] -
Kulczyk-Malecka, J.; Kelly, P. J.; West, G.; Clarke, G. C. B.; Ridealgh, J. A.; Almtoft, K. P.; Greer, A. L.; Barber, Z. H. Acta Mater. 2014, 66, 396–404. doi:10.1016/j.actamat.2013.11.030
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Mitchell, B. S. An introduction to materials engineering and science for chemical and materials engineers; John Wiley & Sons: New York, NY, U.S.A., 2004.
Return to citation in text: [1] [2] -
Jiang, Q.; Zhang, S. H.; Li, J. C. Solid State Commun. 2004, 130, 581–584. doi:10.1016/j.ssc.2004.03.033
Return to citation in text: [1] -
Yang, Y.; Wang, X.; Li, L. Mater. Sci. Eng., B 2008, 149, 58–62. doi:10.1016/j.mseb.2007.12.006
Return to citation in text: [1]
| 41. | Kulczyk-Malecka, J.; Kelly, P. J.; West, G.; Clarke, G. C. B.; Ridealgh, J. A.; Almtoft, K. P.; Greer, A. L.; Barber, Z. H. Acta Mater. 2014, 66, 396–404. doi:10.1016/j.actamat.2013.11.030 |
| 42. | Mitchell, B. S. An introduction to materials engineering and science for chemical and materials engineers; John Wiley & Sons: New York, NY, U.S.A., 2004. |
| 42. | Mitchell, B. S. An introduction to materials engineering and science for chemical and materials engineers; John Wiley & Sons: New York, NY, U.S.A., 2004. |
| 1. | Kim, M. S.; Lee, T.-W.; Parka, J. H. J. Electrochem. Soc. 2009, 156, A584–A588. doi:10.1149/1.3129682 |
| 2. | Deng, D.; Kim, M. G.; Lee, J. Y.; Cho, J. Energy Environ. Sci. 2009, 2, 818–837. doi:10.1039/b823474d |
| 3. | Zeng, P.; Liu, Z.; Hu, Z.; Zhai, J.; Jiang, L. RSC Adv. 2013, 3, 22853–22856. doi:10.1039/c3ra43906b |
| 4. | Zhao, B. T.; Jiang, S. M.; Su, C.; Cai, R.; Ran, R.; Tadé, M. O.; Shao, Z. J. Mater. Chem. A 2013, 1, 12310–12320. doi:10.1039/c3ta12770b |
| 5. | Weng, Z.; Guo, H.; Liu, X.; Wu, S.; Yeung, K. W. K.; Chu, P. K. RSC Adv. 2013, 3, 24758–24775. doi:10.1039/c3ra44031a |
| 6. | Hemalatha, K.; Prakash, A. S.; Guruprakash, K.; Jayakumar, M. J. Mater. Chem. A 2014, 2, 1757–1766. doi:10.1039/C3TA13352D |
| 7. | Lee, E. J.; Nam, I.; Yi, J.; Bang, J. H. J. Mater. Chem. A 2015, 3, 3500–3510. doi:10.1039/C4TA05988C |
| 8. | Wu, Q. L.; Li, J.; Deshpande, R. D.; Subramanian, N.; Rankin, S. E.; Yang, F.; Cheng, Y.-T. J. Phys. Chem. C 2012, 116, 18669–18677. doi:10.1021/jp3072266 |
| 18. | Tang, H.; Lévy, F.; Berger, H.; Schmid, P. E. Phys. Rev. B 1995, 52, 7771–7774. doi:10.1103/PhysRevB.52.7771 |
| 33. | Yang, D.; Sun, Y.; Tong, Z.; Tian, Y.; Li, Y.; Jiang, Z. J. Phys. Chem. C 2015, 119, 5827–5835. doi:10.1021/jp511948p |
| 34. | Shpak, A. P.; Gorbik, P. P., Eds. Nanomaterials and Supramolecular Structures – Physics, Chemistry, and Applications; Springer: Dordrecht, Netherlands, 2010. doi:10.1007/978-90-481-2309-4 |
| 15. | Linsebigler, A. L.; Lu, G.; Yates, J. T., Jr. Chem. Rev. 1995, 95, 735–758. doi:10.1021/cr00035a013 |
| 16. | Shankar, K.; Basham, J. I.; Allam, N. K.; Varghese, O. K.; Mor, G. K.; Feng, X. J.; Paulose, M.; Seabold, J. A.; Choi, K.-S.; Grimes, C. A. J. Phys. Chem. C 2009, 113, 6327–6359. doi:10.1021/jp809385x |
| 30. | Liu, E.; Kang, L.; Wu, F.; Sun, T.; Hu, X.; Yang, Y.; Liu, H.; Fan, J. Plasmonics 2014, 9, 61–70. doi:10.1007/s11468-013-9598-7 |
| 31. | Xiao, F. J. Phys. Chem. C 2012, 116, 16487–16498. doi:10.1021/jp3034984 |
| 9. | Zhang, W.; Liu, Y.; Zhou, D.; Wen, J.; Liang, W.; Yang, F. RSC Adv. 2015, 5, 57155–57163. doi:10.1039/C5RA08802J |
| 10. | Kraeutler, B.; Bard, A. J. J. Am. Chem. Soc. 1977, 99, 7729–7731. doi:10.1021/ja00465a065 |
| 11. | Nakato, Y.; Tsumura, A.; Tsubomura, H. J. Phys. Chem. 1983, 87, 2402–2405. doi:10.1021/j100236a032 |
| 12. | Nakato, Y.; Akanuma, H.; Shimizu, J.-i.; Magari, Y. J. Electroanal. Chem. 1995, 396, 35–39. doi:10.1016/0022-0728(95)04007-B |
| 13. | Bauer, R.; Waldner, G.; Fallmann, H.; Hager, S.; Klare, M.; Krutzler, T.; Malato, S.; Maletzky, P. Catal. Today 1999, 53, 131–144. doi:10.1016/S0920-5861(99)00108-X |
| 14. | Ma, D.; Yan, Y.; Ji, H.; Chen, C.; Zhao, J. Chem. Commun. 2015, 51, 17451–17454. doi:10.1039/C5CC07123B |
| 32. | Jarosz, M.; Syrek, K.; Kapusta-Kołodziej, J.; Mech, J.; Małek, K.; Hnida, K.; Łojewski, T.; Jaskuła, M.; Sulka, G. D. J. Phys. Chem. C 2015, 119, 24182–24191. doi:10.1021/acs.jpcc.5b08403 |
| 23. | Yan, J.; Song, H.; Yang, S.; Yan, J.; Chen, X. Electrochim. Acta 2008, 53, 6351–6355. doi:10.1016/j.electacta.2008.04.048 |
| 25. | Enachi, M.; Guix, M.; Braniste, T.; Postolache, V.; Ciobanu, V.; Ursaki, V.; Schmidt, O. G.; Tiginyanu, I. Surf. Eng. Appl. Electrochem. 2015, 51, 3–8. doi:10.3103/S1068375515010044 |
| 44. | Yang, Y.; Wang, X.; Li, L. Mater. Sci. Eng., B 2008, 149, 58–62. doi:10.1016/j.mseb.2007.12.006 |
| 22. | Guo, G.; Yu, B.; Yu, P.; Chen, X. Talanta 2009, 79, 570–575. doi:10.1016/j.talanta.2009.04.036 |
| 26. | He, B.-L.; Dong, B.; Li, H.-L. Electrochem. Commun. 2007, 9, 425–430. doi:10.1016/j.elecom.2006.10.008 |
| 27. | Nam, S. H.; Shim, H.-S.; Kim, Y.-S.; Dar, M. A.; Kim, J. G.; Kim, W. B. ACS Appl. Mater. Interfaces 2010, 2, 2046–2052. doi:10.1021/am100319u |
| 28. | Wei, W.; Oltean, G.; Tai, C.-W.; Edström, K.; Björefors, F.; Nyholm, L. J. Mater. Chem. A 2013, 1, 8160–8169. doi:10.1039/c3ta11273j |
| 29. | Zhu, K.; Wang, Q.; Kim, J.-H.; Pesaran, A. A.; Frank, A. J. J. Phys. Chem. C 2012, 116, 11895–11899. doi:10.1021/jp301884x |
| 21. | Huang, L.; Peng, F.; Wang, H.; Yu, H.; Geng, W.; Yang, J.; Zhang, S.; Zhao, H. Mater. Chem. Phys. 2011, 130, 316–322. doi:10.1016/j.matchemphys.2011.06.052 |
| 43. | Jiang, Q.; Zhang, S. H.; Li, J. C. Solid State Commun. 2004, 130, 581–584. doi:10.1016/j.ssc.2004.03.033 |
| 19. | Pascual, J.; Camassel, J.; Mathieu, H. Phys. Rev. B 1978, 18, 5606–5614. doi:10.1103/PhysRevB.18.5606 |
| 20. | Amtout, A.; Leonelli, R. Phys. Rev. B 1995, 51, 6842–6851. doi:10.1103/PhysRevB.51.6842 |
| 24. | Amin, S. A.; Pazouki, M.; Hosseinnia, A. Powder Technol. 2009, 196, 241–245. doi:10.1016/j.powtec.2009.07.021 |
| 41. | Kulczyk-Malecka, J.; Kelly, P. J.; West, G.; Clarke, G. C. B.; Ridealgh, J. A.; Almtoft, K. P.; Greer, A. L.; Barber, Z. H. Acta Mater. 2014, 66, 396–404. doi:10.1016/j.actamat.2013.11.030 |
| 34. | Shpak, A. P.; Gorbik, P. P., Eds. Nanomaterials and Supramolecular Structures – Physics, Chemistry, and Applications; Springer: Dordrecht, Netherlands, 2010. doi:10.1007/978-90-481-2309-4 |
| 35. | Yang, F. Thin Solid Films 2004, 446, 313–317. doi:10.1016/j.tsf.2003.10.021 |
| 36. | Gusak, A. M.; Lyashenko, Y. A.; Kornienko, S. V.; Pasichnyy, M. O.; Shirinyan, A. S.; Zaporozhets, T. V., Eds. Diffusion-controlled solid state reactions: in alloys, thin-films, and nanosystems; John Wiley & Sons: New York, NY, U.S.A., 2010. doi:10.1002/9783527631025 |
| 41. | Kulczyk-Malecka, J.; Kelly, P. J.; West, G.; Clarke, G. C. B.; Ridealgh, J. A.; Almtoft, K. P.; Greer, A. L.; Barber, Z. H. Acta Mater. 2014, 66, 396–404. doi:10.1016/j.actamat.2013.11.030 |
| 41. | Kulczyk-Malecka, J.; Kelly, P. J.; West, G.; Clarke, G. C. B.; Ridealgh, J. A.; Almtoft, K. P.; Greer, A. L.; Barber, Z. H. Acta Mater. 2014, 66, 396–404. doi:10.1016/j.actamat.2013.11.030 |
| 41. | Kulczyk-Malecka, J.; Kelly, P. J.; West, G.; Clarke, G. C. B.; Ridealgh, J. A.; Almtoft, K. P.; Greer, A. L.; Barber, Z. H. Acta Mater. 2014, 66, 396–404. doi:10.1016/j.actamat.2013.11.030 |
| 41. | Kulczyk-Malecka, J.; Kelly, P. J.; West, G.; Clarke, G. C. B.; Ridealgh, J. A.; Almtoft, K. P.; Greer, A. L.; Barber, Z. H. Acta Mater. 2014, 66, 396–404. doi:10.1016/j.actamat.2013.11.030 |
| 40. | Sargent, W. Table of periodic properties of the elements; Sargent-Welch Scientific: Skokie, IL, U.S.A., 1980. |
| 38. | Jiang, Q.; Zhang, S.; Zhao, M. Mater. Chem. Phys. 2003, 82, 225–227. doi:10.1016/S0254-0584(03)00201-3 |
| 38. | Jiang, Q.; Zhang, S.; Zhao, M. Mater. Chem. Phys. 2003, 82, 225–227. doi:10.1016/S0254-0584(03)00201-3 |
| 39. | King, H. W. In Physical Metallurgy, 3rd ed.; Cahn, R. W.; Haasen, P., Eds.; North Holland: Amsterdam, Netherlands, 1983; p 63. |
© 2016 Zhang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)