Abstract
Developing highly active and durable visible-light-driven photocatalysts for the degradation of toxic pollutants is of vital significance. Herein, Ag2CO3 nanoparticles were in situ formed on Bi2MoO6 microflowers to produce Ag2CO3/Bi2MoO6 heterostructures via a facile procedure. The morphologies, phases, chemical compositions, and optical properties of Ag2CO3/Bi2MoO6 were examined by multiple characterization techniques. The Ag2CO3/Bi2MoO6 heterostructures exhibited substantially improved performance in the removal of industrial dyes (rhodamine B (RhB), methyl orange (MO), and methyl blue (MB)), and the antibiotic tetracycline hydrochloride (TC), compared with bare Bi2MoO6 and Ag2CO3 under visible-light irradiation. The enhancement of activity was attributed to the high charge-separation capacity, which results from the matched band alignment of the two components. The cycling experiments showed a good durability of Ag2CO3/Bi2MoO6. Holes were found to be the dominant active species accounting for the pollutant degradation. This compound is a promising candidate for wastewater treatment.
Introduction
Industrial pollutants, such as industrial dyes and antibiotics, in wastewaters pose a huge threat to the environment [1,2]. Thus, many methods for pollutant removal have been established. However, the conventional wastewater treatments are usually accompanied by high cost, low efficiency and other insufficiencies [2]. The decomposition and mineralization of pollutants under sunlight through photocatalysis has been demonstrated to be an effective and green technology for environmental remediation [3-6]. Crucial to photocatalysis is to obtain high-performance photocatalysts [7,8]. Obtaining excellent photocatalysts that can be excited by visible light (43% of the solar energy spectrum) is very important for practical applications [9-14].
Bi2MoO6 has been regarded as a promising visible-light-driven (VLD) photocatalyst because of its good activity, chemical stability and nontoxicity [15-17]. However, the low carrier-separation rate and narrow photo-response range of Bi2MoO6 substantially lower its photocatalytic performance [18,19]. To overcome this obstacle, various methods have been developed, including doping [20,21] and the construction of heterojunctions [22-33]. Particularly, the combination of Bi2MoO6 with other semiconductors to construct heterojunction photocatalysts leads to an enhanced activity of Bi2MoO6, which originates from the increased charge separation at the interface [22-30].
Recently, Ag-based compounds (e.g., Ag3PO4, Ag3VO4, Ag2CrO4, and Ag2CO3) [34-36] have emerged as good VLD photocatalysts for pollutant removal. Ag2CO3 exhibits a high visible-light photocatalytic activity [37]. However, it is unstable under illumination. Previous studies found that the good stability of Ag2CO3 could be achieved through the rational construction of heterojunctions, such as Ag2CO3/Bi2WO6 [37], Ag2O/Ag2CO3 [38], Ag2CO3/Bi2O2CO3 [39], Ag2CO3/C3N4 [40], Ag/Ag2CO3/BiVO4 [41] Ag2CO3/AgBr/ZnO [42], and Ag/Ag2CO3/Bi2MoO6 [32]. The band structure of Ag2CO3 matches well with that of Bi2MoO6 [32]. Moreover, morphology modulation is another significant way to enhance photocatalytic activity. Three-dimensional nanostructures endow materials with unique physicochemical properties, for instance, high specific surface area, good molecular diffusion/transport, and good recyclability and light harvesting ability. To the best of our knowledge, application of Ag2CO3 nanoparticles coupled with flower-like Bi2MoO6 for photocatalytic degradation of toxic pollutants remains unreported.
Herein, we synthesized flower-like Ag2CO3/Bi2MoO6 heterostructures, in which Ag2CO3 nanoparticles were evenly anchored on Bi2MoO6 microflowers to construct novel hierarchical heterojunction photocatalysts by via in situ precipitation. The photocatalytic properties of Ag2CO3/Bi2MoO6 was measured regarding the photocatalytic degradation of industrial dyes (rhodamine B (RhB), methyl orange (MO), and methyl blue (MB)), and the antibiotic tetracycline hydrochloride (TC) under visible light. The improved performance of the photocatalytic degradation was prominent, and the reasons were rationally analyzed. Also, the photocatalytic mechanism of pollutant degradation over Ag2CO3/Bi2MoO6 was discussed.
Results and Discussion
Characterization of catalysts
A series of flowerlike Ag2CO3/Bi2MoO6 heterostructures with different weight ratios (0.1/1, 0.2/1, 0.3/1, and 0.5/1) were constructed and labeled ACO/BMO-10, ACO/BMO-20, ACO/BMO-30, and ACO/BMO-50, respectively. The crystal structure of Bi2MoO6, Ag2CO3, and Ag2CO3/Bi2MoO6 heterostructures were determined by XRD technique (Figure 1). The diffraction peaks of Ag2CO3 and Bi2MoO6 were indexed as orthorhombic Bi2MoO6 (JCPDS 76-2388) and monoclinic Ag2CO3 (JCPDS 26-0399), respectively. The XRD pattern of these heterostructures show the characteristic peaks of both Ag2CO3 and Bi2MoO6, indicating the successful synthesis of Ag2CO3/Bi2MoO6 heterostructures. In another publication, Ag/Ag2CO3/Bi2MoO6 nanoplates, composed of three phases, have been described [32].
![[2190-4286-9-214-1]](/bjnano/content/figures/2190-4286-9-214-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: XRD patterns of Ag2CO3/Bi2MoO6 heterojunctions, bare Bi2MoO6 and Ag2CO3.
Figure 1: XRD patterns of Ag2CO3/Bi2MoO6 heterojunctions, bare Bi2MoO6 and Ag2CO3.
To visually study the microstructure and morphology of Ag2CO3/Bi2MoO6, SEM images of the as-prepared catalysts were taken. Bare Bi2MoO6 presents a hierarchical microsphere structure (diameter: 1.6–3.5 μm, Figure 2a,b). After Ag2CO3 was loaded onto Bi2MoO6, the resulting Ag2CO3/Bi2MoO6 retained the flower-like architecture. The representative ACO/BMO-30 displays the flower-like structure, the surface of which is decorated with Ag2CO3 nanoparticles (size: 10–50 nm, Figure 2c,d). In contrast, the previously reported Ag/Ag2CO3/Bi2MoO6 is composed of Bi2MoO6 nanoplates and Ag/Ag2CO3 nanorods/nanoparticles [32].
![[2190-4286-9-214-2]](/bjnano/content/figures/2190-4286-9-214-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: SEM images of (a, b) bare Bi2MoO6 and (c, d) ACO/BMO-30.
Figure 2: SEM images of (a, b) bare Bi2MoO6 and (c, d) ACO/BMO-30.
Further information about the structure of ACO/BMO-30 was collected from TEM images (Figure 3). The TEM images are in line with the SEM observations, i.e., ACO/BMO-30 exhibits a flower-like architecture loaded with Ag2CO3 nanoparticles (Figure 3a,b). The HRTEM displays two different lattice spacings of 0.32 and 0.43 nm, which match well with the (121) planes of orthorhombic Bi2MoO6 and the (110) planes of monoclinic Ag2CO3 (Figure 3c). Moreover, the energy-dispersive spectroscopy (EDS) pattern confirmed the existence of Ag, C, O, Bi, and Mo in ACO/BMO-30 (Figure 3d).
![[2190-4286-9-214-3]](/bjnano/content/figures/2190-4286-9-214-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a–c) TEM images of ACO/BMO-30; (d) EDS pattern of ACO/BMO-30.
Figure 3: (a–c) TEM images of ACO/BMO-30; (d) EDS pattern of ACO/BMO-30.
The optical absorption of Bi2MoO6, Ag2CO3 and Ag2CO3/Bi2MoO6 heterostructures were measured by using UV–vis diffuse reflection spectra (UV–vis DRS, Figure 4). The absorption edges of Ag2CO3 and Bi2MoO6 are around 570 nm and 470 nm, respectively, in line with already reported values [30,33,39]. Compared to pristine Bi2MoO6, the absorption of the Ag2CO3/Bi2MoO6 heterostructures was substantially improved owing through the introduction of Ag2CO3 nanoparticles. Ag/Ag2CO3/Bi2MoO6 [32], Ag2MoO4/Bi2MoO6 [22], and Ag2CO3/Bi2MoO6 heterostrutures are VLD photocatalysts.
![[2190-4286-9-214-4]](/bjnano/content/figures/2190-4286-9-214-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis diffuse reflection spectra of bare Bi2MoO6, Ag2CO3 and Ag2CO3/Bi2MoO6 heterostructures.
Figure 4: UV–vis diffuse reflection spectra of bare Bi2MoO6, Ag2CO3 and Ag2CO3/Bi2MoO6 heterostructures.
The band gap energy (Eg) can be estimated from the Tauc plot: (αhν) = A(hν − Eg)n/2. Where α, h, ν, and A are absorption coefficient, Planck’s constant, the frequency of light, and a constant, respectively. The value of n depends on the type of electronic transition and n is equal to 1 for Bi2MoO6 and Ag2CO3. The Tauc plots of Ag2CO3 and Bi2MoO6 converted from the UV–vis DRS measurements are shown in Figure S1 (Supporting Information File 1). The band gaps are determined to be 2.17 for Ag2CO3 and 2.66 eV for Bi2MoO6.
The band potentials of Ag2CO3 and Bi2MoO6 can be estimated by the empirical equations:
![[2190-4286-9-214-i1]](/bjnano/content/inline/2190-4286-9-214-i1.svg?max-width=590&scale=1.18182)
![[2190-4286-9-214-i2]](/bjnano/content/inline/2190-4286-9-214-i2.svg?max-width=590&scale=1.18182)
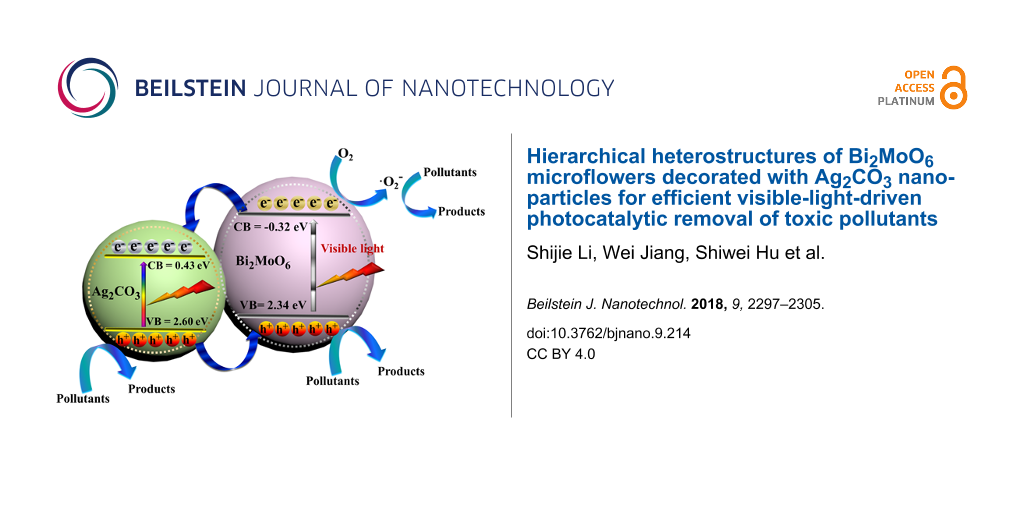
where the X value for Bi2MoO6 is 5.5 eV [43], and that for Ag2CO3 is 6.02 eV [44]. The value of E0 is ca. 4.5 eV. Hence, the values of EVB and ECB of Bi2MoO6 were calculated as −0.32 and 2.34 eV, and those of Ag2CO3 were calculated as 0.43 and 2.60 eV.
PL spectra were measured to analyze the electron–hole-separation efficiency [45-47]. Figure 5 shows the contrast between the PL spectra of bare Bi2MoO6 and of ACO/BMO-30. The emission peak of Bi2MoO6 is centered at ca. 465 nm under an excitation wavelength of 300 nm. Intriguingly, the PL emission intensity of Bi2MoO6 was reduced after the introduction of Ag2CO3. A similar phenomenon was also found in Ag/Ag2CO3/Bi2MoO6 [32] and Ag2MoO4/Bi2MoO6 [22]. This result shows that the charge separation efficiency is enhanced in ACO/BMO-30.
![[2190-4286-9-214-5]](/bjnano/content/figures/2190-4286-9-214-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: PL spectra of bare Bi2MoO6 and ACO/BMO-30.
Figure 5: PL spectra of bare Bi2MoO6 and ACO/BMO-30.
Photocatalytic performance
The efficiency of Ag2CO3/Bi2MoO6 heterostructures in the photocatalytic degradation of industrial dyes (RhB, MO and MB), and the antibiotic TC of the under visible light was measured. Figure 6a displays the degradation of RhB as a function of the time. The RhB concentration remains unchanged in the absence of catalysts. In the presence of bare Bi2MoO6 and Ag2CO3 only 39.8% and 58.7% of RhB were degraded after 90 min of reaction time. The degradation of RhB was substantially enhanced when a combination of Bi2MoO6 and Ag2CO3 was used. For instance, the introduction of a low amount of Ag2CO3 (10 wt %) resulted in 74.9% degradation of RhB. Obviously, the RhB degradation performance is closely related to the loading amount of Ag2CO3. Among the heterostructures, ACO/BMO-30 achieves the best activity in the degradation of RhB, with 100% degradation efficiency after 60 min of reaction time. ACO/BMO-10, ACO/BMO-20 and ACO/BMO-50 showed degradation efficiencies of 60.2%, 69.6%, and 85.1%, respectively. Remarkably, a mixture of 23.1 wt % Ag2CO3 and 76.9 wt % Bi2MoO6 exhibited a much lower activity than ACO/BMO-30, verifying that the close contact between the components also has a significant influence on the photocatalytic performance of the heterostructures.
![[2190-4286-9-214-6]](/bjnano/content/figures/2190-4286-9-214-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (a) Photocatalytic degradation of RhB over different samples under visible light. (b) Rate constants of RhB degradation for different catalysts.
Figure 6: (a) Photocatalytic degradation of RhB over different samples under visible light. (b) Rate constant...
The pseudo-first-order kinetic plots and rate constants of RhB degradation for various catalysts are presented in Figure 6b. The degradation rate constant of ACO/BMO-30 is 0.0562 min−1, which 9.0-, 4.7- and 5.2-fold higher than that of bare Bi2MoO6 (0.0056 min−1), Ag2CO3 (0.0099 min−1), and the mixture (0.0091 min−1).
Figure 7 and Figure S2 (Supporting Information File 1) show the degradation of MO and TC as a function of the time. Again, ACO/BMO-30 displayed the highest activity, with degradation efficiencies of 94.9% for MO, 100% for MB, and 78.9% for TC. The photocatalytic activity in the degradation of TC of ACO/BMO-30 was further compared with that of Ag/Ag2CO3/Bi2MoO6 [32], and of Ag2MoO4/Bi2MoO6 [22]. As shown in Figure S3 (Supporting Information File 1), ACO/BMO-30 is much more active than Ag2MoO4/Bi2MoO6, but not as active as Ag/Ag2CO3/Bi2MoO6 due to the fact that Ag/Ag2CO3/Bi2MoO6 is a ternary composite.
![[2190-4286-9-214-7]](/bjnano/content/figures/2190-4286-9-214-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Photocatalytic degradation of (a) MO and (b) TC over different samples under visible light.
Figure 7: Photocatalytic degradation of (a) MO and (b) TC over different samples under visible light.
To examine the mineralization ability of ACO/BMO-30, the total organic carbon (TOC) was measured during the RhB degradation, and the result is presented in Figure S4 (Supporting Information File 1). Apparently, the TOC removal efficiency gradually goes up with the increase of reaction time and the final TOC removal efficiency is as high as 80.6% after 5 h of reaction. Hence, ACO/BMO-30 shows a decent mineralization ability.
To test the durability of ACO/BMO-30, repeated runs were carried out under unchanging conditions. As depicted in Figure 8a, after six consecutive runs, the RhB degradation efficiency is still about 92.2%. Additionally, no apparent change of the crystalline structure was found after the photocatalytic reactions, as shown in the XRD pattern (Figure 8b). However, the XPS pattern of the used ACO/BMO-30 suggests that some Ag+ is reduced to Ag(0) after the reaction (Figure S5, Supporting Information File 1). It has been recognized that the formation of Ag/Ag2CO3 shows a stable structure [32]. These results verify that ACO/BMO-30 is stable. It is rationally speculated that ACO/BMO-30 has great potential for the elimination of toxic pollutants.
![[2190-4286-9-214-8]](/bjnano/content/figures/2190-4286-9-214-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: (a) The cycling performance of ACO/BMO-30. (b) The XRD patterns of ACO/BMO-30 before and after five reaction cycles.
Figure 8: (a) The cycling performance of ACO/BMO-30. (b) The XRD patterns of ACO/BMO-30 before and after five...
Photocatalytic mechanism
In general, radicals and other active species (e.g., •O2−, •OH and h+) produced in the photocatalytic reaction system contribute to the decomposition of pollutant [48-52]. We performed radical-scavenger tests to determine the active species in the degradation of RhB. As displayed in Figure 9, the addition of isopropanol (IPA) resulted in a slight inhibition of the RhB degradation, signifying that •OH is not the major active species. The addition of p-benzoquinone (BQ) makes the RhB degradation efficiency decline from 100% to 67.3%, suggesting that •O2− plays a minor role. After the addition of ammonium oxalate (AO) the RhB degradation efficiency decreased from 100% to 19.3%, indicating that h+ plays a dominant role in the degradation of RhB over Ag2CO3/Bi2MoO6.
![[2190-4286-9-214-9]](/bjnano/content/figures/2190-4286-9-214-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Radical-scavenger tests over ACO/BMO-30.
Figure 9: Radical-scavenger tests over ACO/BMO-30.
Based on the above results, a possible schematic mechanism is presented in Figure 10. In the Ag2MoO4/Bi2MoO6 system [22], electron–hole pairs can only be generated by visible light in Bi2MoO6. In comparison, both Ag2CO3 and Bi2MoO6 are able to absorb visible light and, and charge carriers are photo-generated on the surface of Ag2CO3 and Bi2MoO6. The electrons in the CB of Bi2MoO6 can partially transfer into that of Ag2CO3. The electrons are then captured by the oxygen, producing •O2− radicals, which play a minor role in decomposing pollutants, as confirmed by the radical-scavenger test (Figure 9). At the same time, the holes in the VB of Ag2CO3 can partially flow into that of Bi2MoO6. The holes in both components mainly account for the pollutant degradation (Figure 9). Electron–hole recombination is severely inhibited, as verified by PL results (Figure 5) [53,54]. Although the VB potential of Ag2CO3 (2.6 eV) is more positive than that of H2O/•OH (2.38 eV/NHE at pH 7), the generated •OH radicals did not act as the major active species for pollutant degradation (Figure 9). In Ag/Ag2CO3/Bi2MoO6, Ag(0) can also be excited to generate electrons, reacting with O2 to form •O2− radicals due to the plasmon resonance (SPR) effect [32]. Based on above analysis, the rational design and construction of Bi2MoO6-based heterostructures is favorable for the separation of charges, leading to a superior activity in pollutant degradation.
![[2190-4286-9-214-10]](/bjnano/content/figures/2190-4286-9-214-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Proposed photocatalytic degradation mechanism of Ag2CO3/Bi2MoO6.
Figure 10: Proposed photocatalytic degradation mechanism of Ag2CO3/Bi2MoO6.
Conclusion
We synthesized a novel photocatalyst of Bi2MoO6 microflowers covered with Ag2CO3 nanoparticles by a facile procedure. The Ag2CO3/Bi2MoO6 heterostructures, especially ACO/BMO-30, showed a substantially enhanced photocatalytic performance in the removal of pollutants (RhB, MO, MB and TC) under visible light compared to bare Bi2MoO6 and Ag2CO3. Moreover, ACO/BMO-30 possesses good durability and stability. The enhanced photocatalytic performance is ascribed to the extended optical response and the matched band structure, reducing carrier recombination. This study offers a novel highly efficient VLD photocatalyst with promising applications in environmental remediation.
Experimental
Materials
Bi(NO3)3·5H2O, NaHCO3, NH3·H2O, Na2MoO4·2H2O, rhodamine B (RhB), methyl blue (MB), tetracycline hydrochloride (TC), methyl orange (MO), isopropanol (IPA), AgNO3, p-benzoquinone (BQ), and ammonium oxalate (AO) were obtained from Chemical Reagent factory (China). All the reagents were used directly without further treatment.
Synthesis
Flower-like Bi2MoO6 was synthesized using a solvothermal route. Typically, Na2MoO4·2H2O (1 mmol) was dissolved in a mixture of 40 mL of CH3CH2OH and 40 mL of ethylene glycol with the aid of ultrasonic treatment. Then Bi(NO3)3·5H2O (2 mmol) was also dissolved in the above solution in the same way. Subsequently, the solution was magnetically stirred for 1 h, and then loaded into a 100 mL Teflon container and reacted at 160 °C for 25 h. After the reaction system had cooled down to room temperature, the precipitants were washed thoroughly with deionized water and ethanol, dried, and calcined at 350 °C for 1 h.
Ag2CO3/Bi2MoO6 heterostructures were obtained by wet-chemical deposition. Typically, 0.6 g Bi2MoO6 was ultrasonically dispersed in 30 mL of deionized water. Then AgNO3 solution (0.1 M) was poured into the suspension and kept stirring for 1 h in the dark. Subsequently, NH3·H2O (0.05 M) was added into the above mixture. After that, NaHCO3 solution (0.1 M) was dropped into the above system slowly under magnetic stirring in the dark, followed by stirring for another 2 h. The collected solid was washed, and dried at 70 °C overnight. By varying the added amounts of AgNO3, NaHCO3, and NH3·H2O, the as-prepared Ag2CO3/Bi2MoO6 heterojunctions with various weight ratios (0. 1/1, 0.2/1, 0.3/1 and 0.5/1) were obtained and denoted as ACO/BMO-10, ACO/BMO-20, ACO/BMO-30, and ACO/BMO-50, respectively. For comparison, pure Ag2CO3 was prepared by the same method without the addition of Bi2MoO6. A mixture of Ag2CO3 and Bi2MoO6, symbolized as mixture, was prepared by simple physical mixing.
Characterization
The microstructure of the samples was observed with a Hitachi S-4800 scanning electron microscope (SEM) and a JEM-2010F transmission electron microscope (TEM). The corresponding chemical compositions were detected by energy-dispersive X-ray (EDX) spectroscopy equipped on the SEM. The crystal structure of catalysts was identified by X-ray diffractometry (XRD, Bruker D8 Advance) with a scanning range of 2θ from 20° to 80°. The UV–vis spectra were recorded using an UV-2600 UV–vis spectrophotometer (Shimadzu). The photoluminescence (PL) emission spectra of the samples were recorded on a Hitachi F-7000 spectrophotometer, employing an excitation wavelength of 300 nm.
Photocatalytic test
The photo-degradation of RhB, MO, TC and MB was carried out in a reactor containing 40 mg of sample, and 100 mL of RhB (10 mg·L−1), MO (10 mg·L−1), TC (20 mg·L−1) or MB (10 mg·L−1) under visible-light irradiation. A 300 W xenon lamp equipped with a UV-cutoff filter (λ > 400 nm) served as the light source. First, the suspension was stirred in the dark for half an hour. During reaction, 2 mL of solution was taken at certain intervals, and centrifuged to obtain the supernatant for UV–vis spectrophotometry measurements. Total organic carbon (TOC) tests were executed by the degradation of RhB (50 mg·L−1, 150 mL) solution with 300 mg of ACO/BMO-30 as the catalyst.
Supporting Information
| Supporting Information File 1: Additional experimental data. | ||
| Format: PDF | Size: 308.3 KB | Download |
Acknowledgements
This work has been financially supported by the National Natural Science Foundation of China (51708504, 31501573 and 51602049), the Public Projects of Zhejiang Province (2017C32079 and LGN18E080003), the Science and Technology project of Zhoushan (2017C41006, 2016C41012, 2015C21014, 2015C21013) and National Key Research and Development Program of China (2016YFC0400501/2016YFC0400509).
References
-
Kumar, S.; Kumar, A.; Bahuguna, A.; Sharma, V.; Krishnan, V. Beilstein J. Nanotechnol. 2017, 8, 1571–1600. doi:10.3762/bjnano.8.159
Return to citation in text: [1] -
Hong, Y.; Li, C.; Zhang, G.; Meng, Y.; Yin, B.; Zhao, Y.; Shi, W. Chem. Eng. J. 2016, 299, 74–84. doi:10.1016/j.cej.2016.04.092
Return to citation in text: [1] [2] -
Wang, F.; Li, Q.; Xu, D. Adv. Energy Mater. 2017, 7, 1700529. doi:10.1002/aenm.201700529
Return to citation in text: [1] -
Wang, W.; Li, G.; Xia, D.; An, T.; Zhao, H.; Wong, P. K. Environ. Sci.: Nano 2017, 4, 782–799. doi:10.1039/C7EN00063D
Return to citation in text: [1] -
Bora, L. V.; Mewada, R. K. Renewable Sustainable Energy Rev. 2017, 76, 1391–1421. doi:10.1016/j.rser.2017.01.130
Return to citation in text: [1] -
Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, Y.; Zhou, Y.; Mo, L.; Liu, J. Beilstein J. Nanotechnol. 2018, 9, 1308–1316. doi:10.3762/bjnano.9.123
Return to citation in text: [1] -
Adhikari, S. P.; Hood, Z. D.; Wang, H.; Peng, R.; Krall, A.; Li, H.; Chen, V. W.; More, K. L.; Wu, Z.; Geyer, S.; Lachgar, A. Appl. Catal., B 2017, 217, 448–458. doi:10.1016/j.apcatb.2017.05.092
Return to citation in text: [1] -
Adhikari, S. P.; Hood, Z. D.; More, K. L.; Chen, V. W.; Lachgar, A. ChemSusChem 2016, 9, 1869–1879. doi:10.1002/cssc.201600424
Return to citation in text: [1] -
Ong, W.-J.; Tan, L.-L.; Ng, Y. H.; Yong, S.-T.; Chai, S.-P. Chem. Rev. 2016, 116, 7159–7329. doi:10.1021/acs.chemrev.6b00075
Return to citation in text: [1] -
Moniz, S. J. A.; Shevlin, S. A.; Martin, D. J.; Guo, Z.-X.; Tang, J. Energy Environ. Sci. 2015, 8, 731–759. doi:10.1039/C4EE03271C
Return to citation in text: [1] -
Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, J.; Wang, Z. J. Colloid Interface Sci. 2017, 501, 156–163. doi:10.1016/j.jcis.2017.04.057
Return to citation in text: [1] -
Chen, C.; Ma, W.; Zhao, J. Chem. Soc. Rev. 2010, 39, 4206–4219. doi:10.1039/b921692h
Return to citation in text: [1] -
Li, S.; Hu, S.; Xu, K.; Jiang, W.; Liu, Y.; Leng, Z.; Liu, J. J. Colloid Interface Sci. 2017, 504, 561–569. doi:10.1016/j.jcis.2017.06.018
Return to citation in text: [1] -
Zhang, G.; Lan, Z.-A.; Wang, X. Angew. Chem., Int. Ed. 2016, 55, 15712–15727. doi:10.1002/anie.201607375
Return to citation in text: [1] -
Wang, Q.; Sun, K.; Lu, Q.; Wei, M.; Yao, L.; Guo, E. Dyes Pigm. 2018, 155, 194–201. doi:10.1016/j.dyepig.2018.03.048
Return to citation in text: [1] -
Dai, W.; Yu, J.; Xu, H.; Hu, X.; Luo, X.; Yang, L.; Tu, X. CrystEngComm 2016, 18, 3472–3480. doi:10.1039/c6ce00248j
Return to citation in text: [1] -
Long, J.; Wang, S.; Chang, H.; Zhao, B.; Liu, B.; Zhou, Y.; Wei, W.; Wang, X.; Huang, L.; Huang, W. Small 2014, 10, 2791–2795. doi:10.1002/smll.201302950
Return to citation in text: [1] -
Guo, C.; Xu, J.; Wang, S.; Li, L.; Zhang, Y.; Li, X. CrystEngComm 2012, 14, 3602–3608. doi:10.1039/c2ce06757a
Return to citation in text: [1] -
Ma, Y.; Jia, Y.; Wang, L.; Yang, M.; Bi, Y.; Qi, Y. J. Power Sources 2016, 331, 481–486. doi:10.1016/j.jpowsour.2016.09.084
Return to citation in text: [1] -
Yu, C.; Wu, Z.; Liu, R.; Dionysiou, D. D.; Yang, K.; Wang, C.; Liu, H. Appl. Catal., B 2017, 209, 1–11. doi:10.1016/j.apcatb.2017.02.057
Return to citation in text: [1] -
Dutta, D. P.; Ballal, A.; Chopade, S.; Kumar, A. J. Photochem. Photobiol., A 2017, 346, 105–112. doi:10.1016/j.jphotochem.2017.05.044
Return to citation in text: [1] -
Zhang, J. L.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2017, 71, 156–164. doi:10.1016/j.jtice.2016.11.030
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Meng, Q.; Zhou, Y.; Chen, G.; Hu, Y.; Lv, C.; Qiang, L.; Xing, W. Chem. Eng. J. 2018, 334, 334–343. doi:10.1016/j.cej.2017.07.134
Return to citation in text: [1] [2] -
Sun, Y.; Wu, J.; Ma, T.; Wang, P.; Cui, C.; Ma, D. Appl. Surf. Sci. 2017, 403, 141–150. doi:10.1016/j.apsusc.2017.01.130
Return to citation in text: [1] [2] -
Ma, D.; Wu, J.; Gao, M.; Xin, Y.; Sun, Y.; Ma, T. Chem. Eng. J. 2017, 313, 1567–1576. doi:10.1016/j.cej.2016.11.036
Return to citation in text: [1] [2] -
Liang, J.; Liu, F.; Deng, J.; Li, M.; Tong, M. Water Res. 2017, 123, 632–641. doi:10.1016/j.watres.2017.06.060
Return to citation in text: [1] [2] -
Zhang, M.; Shao, C.; Mu, J.; Zhang, Z.; Guo, Z.; Zhang, P.; Liu, Y. CrystEngComm 2012, 14, 605–612. doi:10.1039/C1CE05974B
Return to citation in text: [1] [2] -
Li, S.; Shen, X.; Liu, J.; Zhang, L. Environ. Sci.: Nano 2017, 4, 1155–1167. doi:10.1039/C6EN00706F
Return to citation in text: [1] [2] -
Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Zhou, Y.; Liu, Y.; Mo, L. J. Colloid Interface Sci. 2018, 521, 42–49. doi:10.1016/j.jcis.2018.03.033
Return to citation in text: [1] [2] -
Li, S.; Hu, S.; Zhang, J.; Jiang, W.; Liu, J. J. Colloid Interface Sci. 2017, 497, 93–101. doi:10.1016/j.jcis.2017.02.069
Return to citation in text: [1] [2] [3] -
Li, S.; Hu, S.; Jiang, W.; Zhou, Y.; Liu, J.; Wang, Z. J. Colloid Interface Sci. 2018, 530, 171–178. doi:10.1016/j.jcis.2018.06.084
Return to citation in text: [1] -
Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Zhang, J.; Liu, H.; Ma, Z. J. Mol. Catal. A: Chem. 2016, 424, 37–44. doi:10.1016/j.molcata.2016.08.009
Return to citation in text: [1] [2] -
Jiao, Z.; Zhang, Y.; Yu, H.; Lu, G.; Ye, J.; Bi, Y. Chem. Commun. 2013, 49, 636–638. doi:10.1039/C2CC37324F
Return to citation in text: [1] -
Xu, D.; Cheng, B.; Zhang, J.; Wang, W.; Yu, J.; Ho, W. J. Mater. Chem. A 2015, 3, 20153–20166. doi:10.1039/C5TA05248C
Return to citation in text: [1] -
Yan, M.; Wu, Y.; Yan, Y.; Yan, X.; Zhu, F.; Hua, Y.; Shi, W. ACS Sustainable Chem. Eng. 2016, 4, 757–766. doi:10.1021/acssuschemeng.5b00690
Return to citation in text: [1] -
Bao, J.; Guo, S.; Gao, J.; Hu, T.; Yang, L.; Liu, C.; Peng, J.; Jiang, C. RSC Adv. 2015, 5, 97195–97204. doi:10.1039/C5RA18938A
Return to citation in text: [1] [2] -
Zhao, X.; Su, Y.; Qi, X.; Han, X. ACS Sustainable Chem. Eng. 2017, 5, 6148–6158. doi:10.1021/acssuschemeng.7b01040
Return to citation in text: [1] -
Li, T.; Hu, X.; Liu, C.; Tang, C.; Wang, X.; Luo, S. J. Mol. Catal. A: Chem. 2016, 425, 124–135. doi:10.1016/j.molcata.2016.10.001
Return to citation in text: [1] [2] -
Yu, C.; Wei, L.; Zhou, W.; Dionysiou, D. D.; Zhu, L.; Shu, Q.; Liu, H. Chemosphere 2016, 157, 250–261. doi:10.1016/j.chemosphere.2016.05.021
Return to citation in text: [1] -
Liu, Y.; Kong, J.; Yuan, J.; Zhao, W.; Zhu, X.; Sun, C.; Xie, J. Chem. Eng. J. 2018, 331, 242–254. doi:10.1016/j.cej.2017.08.114
Return to citation in text: [1] -
Pirhashemi, M.; Habibi-Yangjeh, A. J. Colloid Interface Sci. 2016, 474, 103–113. doi:10.1016/j.jcis.2016.04.022
Return to citation in text: [1] -
Li, H.; Liu, J.; Hou, W.; Du, N.; Zhang, R.; Tao, X. Appl. Catal., B 2014, 160–161, 89–97. doi:10.1016/j.apcatb.2014.05.019
Return to citation in text: [1] -
Yuan, X.; Jiang, L.; Chen, X.; Leng, L.; Wang, H.; Wu, Z.; Xiong, T.; Liang, J.; Zeng, G. Environ. Sci.: Nano 2017, 4, 2175–2185. doi:10.1039/C7EN00713B
Return to citation in text: [1] -
Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, J.; Wang, Z. Mol. Catal. 2017, 435, 135–143. doi:10.1016/j.mcat.2017.03.027
Return to citation in text: [1] -
Liu, Y.; Yang, Z.-H.; Song, P.-P.; Xu, R.; Wang, H. Appl. Surf. Sci. 2018, 430, 561–570. doi:10.1016/j.apsusc.2017.06.231
Return to citation in text: [1] -
Liu, J.; Li, Y.; Ke, J.; Wang, S.; Wang, L.; Xiao, H. Appl. Catal., B 2018, 224, 705–714. doi:10.1016/j.apcatb.2017.11.028
Return to citation in text: [1] -
Li, S.; Hu, S.; Xu, K.; Jiang, W.; Liu, J.; Wang, Z. Nanomaterials 2017, 7, 22–34. doi:10.3390/nano7010022
Return to citation in text: [1] -
Feizpoor, S.; Habibi-Yangjeh, A. J. Colloid Interface Sci. 2018, 524, 325–336. doi:10.1016/j.jcis.2018.03.069
Return to citation in text: [1] -
Li, S.; Zhang, J.; Hu, S.; Xu, K.; Jiang, W.; Liu, J. J. Alloys Compd. 2017, 695, 1137–1144. doi:10.1016/j.jallcom.2016.10.240
Return to citation in text: [1] -
Adhikari, S. P.; Dean, H.; Hood, Z. D.; Peng, R.; More, K. L.; Ivanov, I.; Wu, Z.; Lachgar, A. RSC Adv. 2015, 5, 91094–91102. doi:10.1039/C5RA13579F
Return to citation in text: [1] -
Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, Y.; Zhou, Y.; Mo, L.; Liu, J. Front. Chem. (Lausanne, Switz.) 2018, 6, No. 255. doi:10.3389/fchem.2018.00255
Return to citation in text: [1] -
Ye, R.; Zhao, J.; Wickemeyer, B. B.; Toste, F. D.; Somorjai, G. A. Nat. Catal. 2018, 1, 318–325. doi:10.1038/s41929-018-0052-2
Return to citation in text: [1] -
Zhang, P.; Wang, T.; Chang, X.; Gong, J. Acc. Chem. Res. 2016, 49, 911–921. doi:10.1021/acs.accounts.6b00036
Return to citation in text: [1]
| 45. | Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, J.; Wang, Z. Mol. Catal. 2017, 435, 135–143. doi:10.1016/j.mcat.2017.03.027 |
| 46. | Liu, Y.; Yang, Z.-H.; Song, P.-P.; Xu, R.; Wang, H. Appl. Surf. Sci. 2018, 430, 561–570. doi:10.1016/j.apsusc.2017.06.231 |
| 47. | Liu, J.; Li, Y.; Ke, J.; Wang, S.; Wang, L.; Xiao, H. Appl. Catal., B 2018, 224, 705–714. doi:10.1016/j.apcatb.2017.11.028 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 22. | Zhang, J. L.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2017, 71, 156–164. doi:10.1016/j.jtice.2016.11.030 |
| 1. | Kumar, S.; Kumar, A.; Bahuguna, A.; Sharma, V.; Krishnan, V. Beilstein J. Nanotechnol. 2017, 8, 1571–1600. doi:10.3762/bjnano.8.159 |
| 2. | Hong, Y.; Li, C.; Zhang, G.; Meng, Y.; Yin, B.; Zhao, Y.; Shi, W. Chem. Eng. J. 2016, 299, 74–84. doi:10.1016/j.cej.2016.04.092 |
| 9. | Ong, W.-J.; Tan, L.-L.; Ng, Y. H.; Yong, S.-T.; Chai, S.-P. Chem. Rev. 2016, 116, 7159–7329. doi:10.1021/acs.chemrev.6b00075 |
| 10. | Moniz, S. J. A.; Shevlin, S. A.; Martin, D. J.; Guo, Z.-X.; Tang, J. Energy Environ. Sci. 2015, 8, 731–759. doi:10.1039/C4EE03271C |
| 11. | Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, J.; Wang, Z. J. Colloid Interface Sci. 2017, 501, 156–163. doi:10.1016/j.jcis.2017.04.057 |
| 12. | Chen, C.; Ma, W.; Zhao, J. Chem. Soc. Rev. 2010, 39, 4206–4219. doi:10.1039/b921692h |
| 13. | Li, S.; Hu, S.; Xu, K.; Jiang, W.; Liu, Y.; Leng, Z.; Liu, J. J. Colloid Interface Sci. 2017, 504, 561–569. doi:10.1016/j.jcis.2017.06.018 |
| 14. | Zhang, G.; Lan, Z.-A.; Wang, X. Angew. Chem., Int. Ed. 2016, 55, 15712–15727. doi:10.1002/anie.201607375 |
| 39. | Li, T.; Hu, X.; Liu, C.; Tang, C.; Wang, X.; Luo, S. J. Mol. Catal. A: Chem. 2016, 425, 124–135. doi:10.1016/j.molcata.2016.10.001 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 7. | Adhikari, S. P.; Hood, Z. D.; Wang, H.; Peng, R.; Krall, A.; Li, H.; Chen, V. W.; More, K. L.; Wu, Z.; Geyer, S.; Lachgar, A. Appl. Catal., B 2017, 217, 448–458. doi:10.1016/j.apcatb.2017.05.092 |
| 8. | Adhikari, S. P.; Hood, Z. D.; More, K. L.; Chen, V. W.; Lachgar, A. ChemSusChem 2016, 9, 1869–1879. doi:10.1002/cssc.201600424 |
| 40. | Yu, C.; Wei, L.; Zhou, W.; Dionysiou, D. D.; Zhu, L.; Shu, Q.; Liu, H. Chemosphere 2016, 157, 250–261. doi:10.1016/j.chemosphere.2016.05.021 |
| 3. | Wang, F.; Li, Q.; Xu, D. Adv. Energy Mater. 2017, 7, 1700529. doi:10.1002/aenm.201700529 |
| 4. | Wang, W.; Li, G.; Xia, D.; An, T.; Zhao, H.; Wong, P. K. Environ. Sci.: Nano 2017, 4, 782–799. doi:10.1039/C7EN00063D |
| 5. | Bora, L. V.; Mewada, R. K. Renewable Sustainable Energy Rev. 2017, 76, 1391–1421. doi:10.1016/j.rser.2017.01.130 |
| 6. | Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, Y.; Zhou, Y.; Mo, L.; Liu, J. Beilstein J. Nanotechnol. 2018, 9, 1308–1316. doi:10.3762/bjnano.9.123 |
| 37. | Bao, J.; Guo, S.; Gao, J.; Hu, T.; Yang, L.; Liu, C.; Peng, J.; Jiang, C. RSC Adv. 2015, 5, 97195–97204. doi:10.1039/C5RA18938A |
| 22. | Zhang, J. L.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2017, 71, 156–164. doi:10.1016/j.jtice.2016.11.030 |
| 2. | Hong, Y.; Li, C.; Zhang, G.; Meng, Y.; Yin, B.; Zhao, Y.; Shi, W. Chem. Eng. J. 2016, 299, 74–84. doi:10.1016/j.cej.2016.04.092 |
| 38. | Zhao, X.; Su, Y.; Qi, X.; Han, X. ACS Sustainable Chem. Eng. 2017, 5, 6148–6158. doi:10.1021/acssuschemeng.7b01040 |
| 53. | Ye, R.; Zhao, J.; Wickemeyer, B. B.; Toste, F. D.; Somorjai, G. A. Nat. Catal. 2018, 1, 318–325. doi:10.1038/s41929-018-0052-2 |
| 54. | Zhang, P.; Wang, T.; Chang, X.; Gong, J. Acc. Chem. Res. 2016, 49, 911–921. doi:10.1021/acs.accounts.6b00036 |
| 22. | Zhang, J. L.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2017, 71, 156–164. doi:10.1016/j.jtice.2016.11.030 |
| 23. | Meng, Q.; Zhou, Y.; Chen, G.; Hu, Y.; Lv, C.; Qiang, L.; Xing, W. Chem. Eng. J. 2018, 334, 334–343. doi:10.1016/j.cej.2017.07.134 |
| 24. | Sun, Y.; Wu, J.; Ma, T.; Wang, P.; Cui, C.; Ma, D. Appl. Surf. Sci. 2017, 403, 141–150. doi:10.1016/j.apsusc.2017.01.130 |
| 25. | Ma, D.; Wu, J.; Gao, M.; Xin, Y.; Sun, Y.; Ma, T. Chem. Eng. J. 2017, 313, 1567–1576. doi:10.1016/j.cej.2016.11.036 |
| 26. | Liang, J.; Liu, F.; Deng, J.; Li, M.; Tong, M. Water Res. 2017, 123, 632–641. doi:10.1016/j.watres.2017.06.060 |
| 27. | Zhang, M.; Shao, C.; Mu, J.; Zhang, Z.; Guo, Z.; Zhang, P.; Liu, Y. CrystEngComm 2012, 14, 605–612. doi:10.1039/C1CE05974B |
| 28. | Li, S.; Shen, X.; Liu, J.; Zhang, L. Environ. Sci.: Nano 2017, 4, 1155–1167. doi:10.1039/C6EN00706F |
| 29. | Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Zhou, Y.; Liu, Y.; Mo, L. J. Colloid Interface Sci. 2018, 521, 42–49. doi:10.1016/j.jcis.2018.03.033 |
| 30. | Li, S.; Hu, S.; Zhang, J.; Jiang, W.; Liu, J. J. Colloid Interface Sci. 2017, 497, 93–101. doi:10.1016/j.jcis.2017.02.069 |
| 31. | Li, S.; Hu, S.; Jiang, W.; Zhou, Y.; Liu, J.; Wang, Z. J. Colloid Interface Sci. 2018, 530, 171–178. doi:10.1016/j.jcis.2018.06.084 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 33. | Zhang, J.; Liu, H.; Ma, Z. J. Mol. Catal. A: Chem. 2016, 424, 37–44. doi:10.1016/j.molcata.2016.08.009 |
| 34. | Jiao, Z.; Zhang, Y.; Yu, H.; Lu, G.; Ye, J.; Bi, Y. Chem. Commun. 2013, 49, 636–638. doi:10.1039/C2CC37324F |
| 35. | Xu, D.; Cheng, B.; Zhang, J.; Wang, W.; Yu, J.; Ho, W. J. Mater. Chem. A 2015, 3, 20153–20166. doi:10.1039/C5TA05248C |
| 36. | Yan, M.; Wu, Y.; Yan, Y.; Yan, X.; Zhu, F.; Hua, Y.; Shi, W. ACS Sustainable Chem. Eng. 2016, 4, 757–766. doi:10.1021/acssuschemeng.5b00690 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 20. | Yu, C.; Wu, Z.; Liu, R.; Dionysiou, D. D.; Yang, K.; Wang, C.; Liu, H. Appl. Catal., B 2017, 209, 1–11. doi:10.1016/j.apcatb.2017.02.057 |
| 21. | Dutta, D. P.; Ballal, A.; Chopade, S.; Kumar, A. J. Photochem. Photobiol., A 2017, 346, 105–112. doi:10.1016/j.jphotochem.2017.05.044 |
| 37. | Bao, J.; Guo, S.; Gao, J.; Hu, T.; Yang, L.; Liu, C.; Peng, J.; Jiang, C. RSC Adv. 2015, 5, 97195–97204. doi:10.1039/C5RA18938A |
| 48. | Li, S.; Hu, S.; Xu, K.; Jiang, W.; Liu, J.; Wang, Z. Nanomaterials 2017, 7, 22–34. doi:10.3390/nano7010022 |
| 49. | Feizpoor, S.; Habibi-Yangjeh, A. J. Colloid Interface Sci. 2018, 524, 325–336. doi:10.1016/j.jcis.2018.03.069 |
| 50. | Li, S.; Zhang, J.; Hu, S.; Xu, K.; Jiang, W.; Liu, J. J. Alloys Compd. 2017, 695, 1137–1144. doi:10.1016/j.jallcom.2016.10.240 |
| 51. | Adhikari, S. P.; Dean, H.; Hood, Z. D.; Peng, R.; More, K. L.; Ivanov, I.; Wu, Z.; Lachgar, A. RSC Adv. 2015, 5, 91094–91102. doi:10.1039/C5RA13579F |
| 52. | Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Liu, Y.; Zhou, Y.; Mo, L.; Liu, J. Front. Chem. (Lausanne, Switz.) 2018, 6, No. 255. doi:10.3389/fchem.2018.00255 |
| 18. | Guo, C.; Xu, J.; Wang, S.; Li, L.; Zhang, Y.; Li, X. CrystEngComm 2012, 14, 3602–3608. doi:10.1039/c2ce06757a |
| 19. | Ma, Y.; Jia, Y.; Wang, L.; Yang, M.; Bi, Y.; Qi, Y. J. Power Sources 2016, 331, 481–486. doi:10.1016/j.jpowsour.2016.09.084 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 15. | Wang, Q.; Sun, K.; Lu, Q.; Wei, M.; Yao, L.; Guo, E. Dyes Pigm. 2018, 155, 194–201. doi:10.1016/j.dyepig.2018.03.048 |
| 16. | Dai, W.; Yu, J.; Xu, H.; Hu, X.; Luo, X.; Yang, L.; Tu, X. CrystEngComm 2016, 18, 3472–3480. doi:10.1039/c6ce00248j |
| 17. | Long, J.; Wang, S.; Chang, H.; Zhao, B.; Liu, B.; Zhou, Y.; Wei, W.; Wang, X.; Huang, L.; Huang, W. Small 2014, 10, 2791–2795. doi:10.1002/smll.201302950 |
| 22. | Zhang, J. L.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2017, 71, 156–164. doi:10.1016/j.jtice.2016.11.030 |
| 23. | Meng, Q.; Zhou, Y.; Chen, G.; Hu, Y.; Lv, C.; Qiang, L.; Xing, W. Chem. Eng. J. 2018, 334, 334–343. doi:10.1016/j.cej.2017.07.134 |
| 24. | Sun, Y.; Wu, J.; Ma, T.; Wang, P.; Cui, C.; Ma, D. Appl. Surf. Sci. 2017, 403, 141–150. doi:10.1016/j.apsusc.2017.01.130 |
| 25. | Ma, D.; Wu, J.; Gao, M.; Xin, Y.; Sun, Y.; Ma, T. Chem. Eng. J. 2017, 313, 1567–1576. doi:10.1016/j.cej.2016.11.036 |
| 26. | Liang, J.; Liu, F.; Deng, J.; Li, M.; Tong, M. Water Res. 2017, 123, 632–641. doi:10.1016/j.watres.2017.06.060 |
| 27. | Zhang, M.; Shao, C.; Mu, J.; Zhang, Z.; Guo, Z.; Zhang, P.; Liu, Y. CrystEngComm 2012, 14, 605–612. doi:10.1039/C1CE05974B |
| 28. | Li, S.; Shen, X.; Liu, J.; Zhang, L. Environ. Sci.: Nano 2017, 4, 1155–1167. doi:10.1039/C6EN00706F |
| 29. | Li, S.; Hu, S.; Jiang, W.; Liu, Y.; Zhou, Y.; Liu, Y.; Mo, L. J. Colloid Interface Sci. 2018, 521, 42–49. doi:10.1016/j.jcis.2018.03.033 |
| 30. | Li, S.; Hu, S.; Zhang, J.; Jiang, W.; Liu, J. J. Colloid Interface Sci. 2017, 497, 93–101. doi:10.1016/j.jcis.2017.02.069 |
| 22. | Zhang, J. L.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2017, 71, 156–164. doi:10.1016/j.jtice.2016.11.030 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 41. | Liu, Y.; Kong, J.; Yuan, J.; Zhao, W.; Zhu, X.; Sun, C.; Xie, J. Chem. Eng. J. 2018, 331, 242–254. doi:10.1016/j.cej.2017.08.114 |
| 42. | Pirhashemi, M.; Habibi-Yangjeh, A. J. Colloid Interface Sci. 2016, 474, 103–113. doi:10.1016/j.jcis.2016.04.022 |
| 43. | Li, H.; Liu, J.; Hou, W.; Du, N.; Zhang, R.; Tao, X. Appl. Catal., B 2014, 160–161, 89–97. doi:10.1016/j.apcatb.2014.05.019 |
| 44. | Yuan, X.; Jiang, L.; Chen, X.; Leng, L.; Wang, H.; Wu, Z.; Xiong, T.; Liang, J.; Zeng, G. Environ. Sci.: Nano 2017, 4, 2175–2185. doi:10.1039/C7EN00713B |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 22. | Zhang, J. L.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2017, 71, 156–164. doi:10.1016/j.jtice.2016.11.030 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 30. | Li, S.; Hu, S.; Zhang, J.; Jiang, W.; Liu, J. J. Colloid Interface Sci. 2017, 497, 93–101. doi:10.1016/j.jcis.2017.02.069 |
| 33. | Zhang, J.; Liu, H.; Ma, Z. J. Mol. Catal. A: Chem. 2016, 424, 37–44. doi:10.1016/j.molcata.2016.08.009 |
| 39. | Li, T.; Hu, X.; Liu, C.; Tang, C.; Wang, X.; Luo, S. J. Mol. Catal. A: Chem. 2016, 425, 124–135. doi:10.1016/j.molcata.2016.10.001 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
| 32. | Zhang, J.; Ma, Z. J. Taiwan Inst. Chem. Eng. 2018, 88, 121–129. doi:10.1016/j.jtice.2018.03.043 |
© 2018 Li et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (https://www.beilstein-journals.org/bjnano)