Abstract
Although 2D layered nanomaterials have been intensively investigated towards their application in energy conversion and storage devices, their disadvantages have rarely been explored so far especially compared to their 3D counterparts. Herein, WO3·nH2O (n = 0, 1, 2), as the most common and important electrochemical and electrochromic active nanomaterial, is synthesized in 3D and 2D structures through a facile hydrothermal method, and the disadvantages of the corresponding 2D structures are examined. The weakness of 2D WO3·nH2O originates from its layered structure. X-ray diffraction and scanning electron microscopy analyses of as-grown WO3·nH2O samples suggest a structural transition from 2D to 3D upon temperature increase. 2D WO3·nH2O easily generates structural instabilities by 2D intercalation, resulting in a faster performance degradation, due to its weak interlayer van der Waals forces, even though it outranks the 3D network structure in terms of improved electronic properties. The structural transformation of 2D layered WO3·nH2O into 3D nanostructures is observed via ex situ Raman measurements under electrochemical cycling experiments. The proposed degradation mechanism is confirmed by the morphology changes. The work provides strong evidence for and in-depth understanding of the weakness of 2D layered nanomaterials and paves the way for further interlayer reinforcement, especially for 2D layered transition metal oxides.
Introduction
Within the less than 20 years since the successful exfoliation of atomically thin graphene, 2D layered nanomaterials have been contributing greatly to the advances of nanoscience and nanotechnology with their exotic properties and versatility of applications [1-10]. Among all 2D nanomaterials, 2D transition metal oxides (TMOs) are the group with the highest electrochemical activities for energy conversion and storage [11-15]. As the energy-related field is rapidly developing, the limits of 2D TMOs began to come clear and ways of chemical functionalization were developed to make them more suitable for practical applications [16-18]. However, comparably less attention has been paid to the comprehensive investigation of the disadvantage of 2D TMOs and their failure mechanism.
Tungsten trioxide (WO3) is one of the few TMOs with both excellent electrochemical and electrochromic properties [19-23]. It has a three-dimensional (3D) network lattice structure consisting of corner-sharing or edge-sharing WO6 octahedra [24-26]. Its phases (monoclinic, triclinic, orthorhombic and tetragonal) form trigonal, quadrangular, pentagonal, and hexagonal tunnels and cavities for 3D electrochemcial intercalation [27-29]. In contrast, its hydrates exhibit a 2D layered strcuture composed of WO5(OH2) single sheets in a corner-sharing arrangement with additional water molecules between layers, which is suitable for 2D intercalation chemistry [30-32].
Both WO3 and its hydrates have been fabricated via different methods and analyzed with regard to electrochemical, photocatalysis, sensing and electrochromic applications [24,33,34]. Oriented WO3·H2O sheets were hydrothermally grown in mixed acids at 80 °C for 17 h, followed by sintering at 500 °C in order to obtain crystalline WO3 for the photoelectrochemical water oxidation [35]. A 2D WO3 nanosheet sensor fabricated by high-temperature anodization of tungsten thin films displayed a maximum response of 80% for 1% of hydrogen gas at 250 °C [36]. 2D WO3·2H2O films developed by a facile dipping process exhibited a significantly improved response time as electrochromic electrodes compared to WO3 thin films [37]. The acidic precipitation reaction was also adopted to fabricate WO3·2H2O electrochemical energy storage electrodes with a higher rate capability than annealed WO3 [38]. The investigation of 2D sheets of WO3 and a rGO–WO3 composite prepared via a one-pot hydrothermal method suggested that the rGO–WO3 composite could be a promising material for photocatalytic and antibacterial applications [39]. Unfortunately, despite the great number of 2D WO3 compounds and their hydrates synthesized and utilized for energy conversion and storage applications, their weakness has not yet been investigated thoroughly.
In this study, WO3·nH2O (n = 0, 1, 2) was fabricated by a facile hydrothermal method for the first time at the different termperatures to investigate the disadvantages of 2D structures. The growth mechanism analyzed by X-ray diffraction (XRD) and scanning electron microscopy (SEM) suggested a 2D to 3D structural transition upon temperature increment, revealing that the weakness of layered 2D WO3·nH2O originates from weak interlayer van der Waals interactions. The faster performance degradation in electrochemical tests of 2D layered WO3·nH2O further indicated the structural instability of 2D nanostructures compared to 3D nanostructures. The structural transformation of 2D layered WO3·nH2O to 3D structures was observed via ex situ Raman measurements under electrochemical cycling experiments. The morphology change confirmed the degradation mechanism proposed in this work. Consequently, this work provides an in-depth understanding of the weakness of 2D layered nanomaterials and paves the way for the interlayer reinforcement of 2D TMOs.
Experimental
All nanostructured WO3 and their hydrates in this work were prepared on FTO/glass substrates through a facile hydrothermal reaction at different temperatures. All chemicals were purchased from chemical suppliers and were used without further purification.
Before the hydrothermal reaction, the seed solution was first spin-coated on the FTO/glass and annealed at 350 °C for 20 min. To prepare the seed solution, 0.824 g of sodium tungstate dihydrate (Na2WO4·2H2O) was initially dissolved into 10 mL deionized water under continuous stirring. After complete dissolution of the Na2WO4·2H2O powder, 0.416 mL of hydrochloric acid (HCl, 36–38 wt %) solution was added dropwise while stirring for 15 min at room temperature. Subsequently, 0.2241 g of oxalic acid (C2H2O4) was added to the solution and the solution was then diluted with deionized water to a total volume of 12.5 mL accompanied by another 15 min of stirring. The prepared seed solution was spin-coated onto FTO/glass at 3000 rpm for four times with each step consisting of 40 s spin-coating at room temperature followed by annealing at 350 °C for 20 min.
The as-prepared FTO/glasses were sealed in 25 mL Teflon-lined stainless autoclaves filled with 15 mL solution that was prepared by the same procedures as the seed solution mentioned above. However, its concentration was ten times lower than that of the the seed solution. Then the autoclaves were heated at temperatures of 80, 100, 120, 150 or 180 °C for 2 h. After heating, the autoclaves cooled down to the room temperature naturally. The FTO/glass substrates were taken out from autoclaves and dried at 60 °C for 1 h. Subsequently, the as-grown WO3 and its hydrates were measured and analyzed.
The crystal structures of the samples were identified with a high-resolution X-ray diffractometer (HR-XRD, SmartLab, Rigaku). The morphology of samples was investigated by field-emission scanning electron microscopy (FE-SEM, JSM-7100F, Jeol) together with energy-dispersive spectroscopy (EDS). Information about chemical composition and bonding was collected by X-ray photoelectron spectrometry (XPS, K-Alpha, Thermo Scientific) and Raman spectroscopy (EZRaman-N-785, TSI. Inc.), respectively.
Electrochemical characterization of the samples was performed using an Autolab PGSTAT204 (Metrohm Autolab B.V.) with a three-electrode configuration in 1.0 M H2SO4 aqueous solution. The as-prepared sample, a Pt wire and a Ag/AgCl electrode acted as working, counter and reference electrode, respectively. Cyclic voltammetry (CV) was conducted in the potential range from −0.8 V to +0.8 V (vs Ag/AgCl).
Results and Discussion
The crystal structure of the as-prepared samples was initially investigated by XRD. As shown in the XRD patterns from the top to bottom of Figure 1a, the samples display different phase compositions depending on the synthesis temperature. The samples synthesized at 80 °C is composed of monoclinic WO3·2H2O (JCPDS No. 18-1420) [40] and orthorhombic WO3·H2O (JCPDS No. 43-0679) [41], which are both layered structures as illustrated in Figure 1b and Figure 1c, respectively. The structural difference between these two components is the number of interlayer water molecules. For the samples synthesized at 100 and 120 °C, only WO3·H2O (JCPDS no. 43-0679) was measured with two main peaks corresponding to (020) and (111) as some of the interlayer water disappeared [41]. However, the dominant facet changed from (020) to (111) as the synthesis temperature increased. Moreover, for the samples synthesized at 150 °C, the full width at half maximum (FWHM) of the (111) reflection of the main component WO3·H2O broadened, which indicates the instability of WO3·H2O at such a high temperature. A small hump on the left side of the (111) peak of WO3·H2O clearly shows the appearance of monoclinic WO3 (JCPDS no. 43-1035) [42,43], which also supports the dehydration tendency of WO3·H2O. When the samples were grown at 180 °C, only monoclinic WO3 (JCPDS no. 43-1035) with its dominant (002) facet existed in the sample. The crystal structure of monoclinic WO3 presented in Figure 1d displays 3D covalent bonding structures compared to the stacked 2D layers of WO3·2H2O and WO3·H2O with only weak interlayer van der Waals forces. It is also noteworthy that at temperatures lower than 80 °C, neither WO3·2H2O nor WO3·H2O could be synthesized using the hydrothermal process presented in this work. Both experimental results and the theoretical models mentioned above suggested that the layered WO3 hydrates are relatively unstable compared to WO3.
![[2190-4286-9-265-1]](/bjnano/content/figures/2190-4286-9-265-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) XRD patterns of the as-prepared samples at 80, 100, 120, 150 and 180 °C from top to bottom. Schematic illustration of the crystal structures: b) monoclinic WO3·2H2O, c) orthorhombic WO3·H2O and d) monoclinic WO3.
Figure 1: a) XRD patterns of the as-prepared samples at 80, 100, 120, 150 and 180 °C from top to bottom. Sche...
The following figures (Figures 2–4) show the morphology of the three typical samples synthesized at 80, 120 and 180 °C, examined by SEM. Figure 2a,b shows the relatively uniform growth of WO3 hydrates with flower-like balls on the upper layer and nanosheets beneath them as represented in Figure 2c. The nanosheets were ca. 1.3 μm square-shaped and almost vertically aligned on the substrate (Figure 2d,e). The image of a typical nanosheet shows the very low thickness of ca. 27 nm (Figure 2f). The upper layer with grouped flower-like balls is highlighted in Figure 2g,h. The images show that the flower-like balls have a diameter of ca. 2.5 μm and are composed of self-assembled square nanosheets with similar sizes as the nanosheets in the layer grown beneath. The magnified image in Figure 2i displays nanosheets with average thickness of ca. 37 nm in a flower-like ball crossing each other.
![[2190-4286-9-265-2]](/bjnano/content/figures/2190-4286-9-265-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: SEM images of the sample synthesized at 80 °C. a) Overview and b) magnified images of the WO3 hydrates grown on the substrate; c) image of a representative area; d) typical image of nanosheets and e) magnified view; f) image of a typical nanosheet; g) grouped flower-like balls; h) typical flower-like ball with i) magnified image.
Figure 2: SEM images of the sample synthesized at 80 °C. a) Overview and b) magnified images of the WO3 hydra...
Figure 3 shows SEM images of the sample synthesized at 120 °C. The as-grown pure WO3·H2O existed in form of square sheets and hexagonal plates as displayed in Figure 3a–c. The square sheets were measured to be ca. 6 μm long and ca. 1.5 μm thick, while the hexagonal plates were 0.5 μm thick with diagonal length of ca. 2.9 μm. Figure 3d,e shows the square sheets with opened layers, indicating the layered crystal structure of the as-grown material. The magnified image of the opened layers in Figure 3f clearly demonstrated the 2D layered nature of WO3·H2O. In addition to the square sheets, the hexagonal plates were stacked by nanoribbons in the direction parallel to the diagonals of the hexagons (Figure 3g). As shown in Figure 3g,h, the nanoribbons were wider (ca. 400 nm) at the center and narrower (ca. 100 nm) near the two ends, forming tips at their very ends (Figure 3i).
![[2190-4286-9-265-3]](/bjnano/content/figures/2190-4286-9-265-3.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: SEM images of the sample synthesized at 120 °C. a) Overview and b) magnified images of WO3·H2O grown on the substrate; c) image of a representative area; d) square sheets with opened layers; e) a typical square sheet with opened layers and f) its magnified view at the opened layers; g) a typical hexagonal plate; magnified views of h) the edge and i) the side face of a hexagonal plate.
Figure 3: SEM images of the sample synthesized at 120 °C. a) Overview and b) magnified images of WO3·H2O grow...
The SEM images of the sample synthesized at 180 °C are presented in Figure 4. The WO3 structures are assembled from square sheets with a few individual sheets beneath as indicated in Figure 4a and Figure 4b. The square sheets with a length of ca. 2.8 μm and a thickness of ca. 0.4 μm grew crossed with each other at all angles, forming a network structure (Figure 4c,d). Figure 4e clearly displays the crossed square nanosheets in the network structure. The individual square sheets were measured to be ca. 3.5 μm long and ca. 0.7 μm thick (Figure 4f,g). The EDS element mapping in Figure 4h,i demonstrates a homogeneous elemental distribution in the sheet and confirms the formation of WO3. The oxygen appearing outside the square comes from the substrate.
![[2190-4286-9-265-4]](/bjnano/content/figures/2190-4286-9-265-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: SEM images of the sample synthesized at 180 °C. a) Overview and b) magnified images of WO3 grown on the substrate; images of c) one part of the network structure and d) the magnified view; e) image of the crossed nanosheets; f) vertically standing and g) flat laying square sheets. EDS element mapping of the flat laying square sheet: h) W and i) O.
Figure 4: SEM images of the sample synthesized at 180 °C. a) Overview and b) magnified images of WO3 grown on...
Three typical sample structures acquired with SEM are presented in Figure 5a. It clearly shows that the feature structure size of the nanostructures synthesized at 80 °C are much smaller compared to similar feature structures obtained in the other two samples, especially in terms of the longitudinal size. Besides the lower synthesis temperature, this could be mainly due to the interlayer water molecules in WO3·2H2O, which caused the slow growth of the samples. The structural features of the WO3 sample synthesized at 180 °C are also smaller than those of the WO3·H2O sample synthesized at 120 °C. Although a higher synthesis temperature produces more energy for the sample growth, the layered WO3·H2O still grew faster than the 3D WO3, as the formation of van der Waals interactions consumes less energy than the formation of covalent bonds. From both the structural and energy consumption point of view, WO3·2H2O and WO3·H2O are kinetically and thermodynamically less stable than WO3.
![[2190-4286-9-265-5]](/bjnano/content/figures/2190-4286-9-265-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: a) The structural feature sizes of three typical samples synthesized at 80, 120 and 180 °C, respectively. High-resolution XPS core-level b) W4f and c) O1s spectra of the three as-synthesized samples.
Figure 5: a) The structural feature sizes of three typical samples synthesized at 80, 120 and 180 °C, respect...
Figure 5b,c depicts the high-resolution XPS core-level W4f and O1s spectra, respectively. The W 4f orbitals in Figure 5b are almost identical in the three samples and can be resolved into W 4f5/2 and W 4f7/2. The two main peaks correspond to the W4f7/2 and W4f5/2 of the tungsten atoms in a +6 oxidation state [44,45]. The two peaks from the two samples synthesized with lower temperatures are both located at 35.9 eV and 38.0 eV, which is ca. 0.2 eV lower than the peak locations (ca. 36.1 eV and ca. 38.2 eV) in the WO3 sample synthesized at 180 °C. The very small peaks at ca. 34.6 eV in all three samples originate from W5+ ions in the lattice, which reveals the formation of a few oxygen vacancies [29,46]. The O 1s spectra for all samples in Figure 5c depict two peaks with one main peak from the lattice oxygen, OL, and another from the oxygen in water molecules, OH2O [47,48]. With increasing temperature, the lattice oxygen in the three as-synthesized samples shifts to lower binding energies from 530.8 eV over 530.6 eV to 520.3 eV. The area of the OH2O peaks indicates the structural difference between layered WO3·2H2O, layered WO3·H2O and 3D WO3. The decreasing area of OH2O peaks with higher synthesis temperature demonstrated the increasing stability of the samples as the interlayer water and coordinated water molecules disappeared successively with only a few unavoidable surface-absorbed water molecules left [49,50]. The above XPS results confirm the SEM analysis.
To get further inside of the electrochemical performance degradation of the three samples, CV tests were carried out at a scan rate of 50 mV·s−1 within the potential range from −0.8 V to +0.8 V (vs Ag/AgCl). The electrochemical energy conversion and storage of WO3·nH2O in H2SO4 electrolyte are based on the intercalation of protons and injection of electrons as described in the following equation [38]:
![[Graphic 1]](/bjnano/content/inline/2190-4286-9-265-i1.svg?max-width=637&scale=1.18182)
The transition of W between the valence states of W6+ and W5+ is the basis of both electrochemical energy storage and electrochromic behavior. As shown in Figure 6a, in all cases the cathodic current rises when the potential was scanned to negative values because of the intercalation of protons into the samples. Figure 6c–e illustrates the intercalation of ions into WO3·nH2O. The reduction of W6+ to W5+ in the process also resulted in coloration. In the reverse process, the rise of anodic current indicates the deintercalation of protons and oxidation of tungsten ions with concomitant bleaching. The video presented in Supporting Information File 1 shows the coloration/bleaching processes of the samples during cycling. With a growing number of cycles, the samples exhibited a strong performance degradation as displayed in Figure 6b. The integrated areas of the CV curves in Figure 6b suggested much larger drops of the specific capacitance of the WO3 hydrates compared to those of WO3. As shown in Figure 6c–e, the intercalated ions in WO3 hydrates are surrounded by weak hydrogen bonds, coordination bonds, and van der Waals forces, while the ions in WO3 are surrounded by a strong 3D covalent bond network. The intercalation/deintercalation process in WO3 may cause less distortions and destructions of the structure than in WO3 hydrates, leading to a better electrochemical stability.
![[2190-4286-9-265-6]](/bjnano/content/figures/2190-4286-9-265-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: CV curves of the three samples at a) the 1st cycle and b) the 500th cycle; schematic illustration of ion intercalation into c) WO3·2H2O, d) WO3·H2O, and e) WO3.
Figure 6: CV curves of the three samples at a) the 1st cycle and b) the 500th cycle; schematic illustration o...
The SEM images of the samples taken after 500 cycles of CV support the performance degradation. Specifically, the nanostructured flower-like balls and the nanosheets in the samples synthesized at 80 °C seem to be glued together in Figure 7a, forming a thin film-like structure as indicated in the inset of Figure 7a, which could be a result of strong reaction between the interlayer water molecules and the electrolyte. For the WO3·H2O sample synthesized at 120 °C (Figure 7b), the sheets were found to swell heavily due to the 2D intercalation/deintercalation processes in the CV cycles, turning almost twice as thick as their original thickness. Under the strong effect of 2D intercalation/deintercalation, the hexagonal plates were transformed into very thin nanosheets, which were composed of nanoribbons (Inset of Figure 7b). In contrast to the two samples mentioned above, the WO3 sample synthesized at 180 °C still kept some of the features from the original sample, as depicted in Figure 7c. Although the original network was broken, a considerable number of the sheet components in the samples remained almost unchanged in their original shape (Inset of Figure 7c).
![[2190-4286-9-265-7]](/bjnano/content/figures/2190-4286-9-265-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: SEM images of the three typical samples synthesized at a) 80 °C, b) 120 °C and c) 180 °C after 500 CV cycles.
Figure 7: SEM images of the three typical samples synthesized at a) 80 °C, b) 120 °C and c) 180 °C after 500 ...
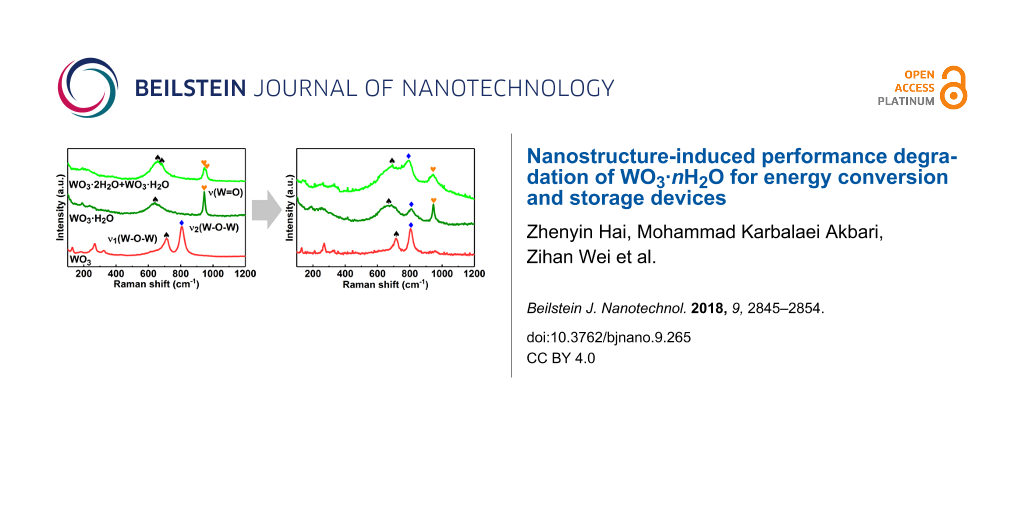
Figure 8 compares the Raman spectra of the three samples before and after CV tests. As displayed in Figure 8a, the initial WO3 sample is characterized by two main peaks at ca. 713 and ca. 807 cm−1, which are associated with two types of W–O–W stretching vibration modes [51,52]. The ν1(W–O–W) mode also appeared in the other two initial samples. The Raman spectra of the initial WO3·H2O and WO3·2H2O samples were characterized by the stretching vibration mode of their terminal W=O bonds [53,54]. In spite of their high similarity, the peaks of WO3·2H2O are shifted slightly to higher wavenumbers compared to those of WO3·H2O as indicated in the sample containing both WO3·2H2O and WO3·H2O. The Raman spectra presented in Figure 8b reflect the structural transformation of the samples after CV tests. The Raman spectra of both the samples synthesized at 80 and 120 °C showed a new peak from the W–O–W stretching vibration mode (ν2(W–O–W)), while the other peaks were broadened. The peaks of the ν1(W–O–W) modes in these two samples shifted to higher wavenumbers and became relatively stronger in comparison to the W=O peaks. These results clearly suggest the break of W=O bonds and the formation of W–O–W bonds, which leads to dehydration of WO3 hydrates. In contrast, the Raman spectra of the WO3 samples synthesized at 180 °C remains almost unaffected after CV tests. The investigation of Raman spectra further supports the relative electrochemical instability of WO3·2H2O and WO3·H2O compared to WO3.
![[2190-4286-9-265-8]](/bjnano/content/figures/2190-4286-9-265-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Raman spectra of the three samples a) before and b) after the CV tests.
Figure 8: Raman spectra of the three samples a) before and b) after the CV tests.
Conclusion
In summary, WO3·nH2O (n = 0, 1, 2) synthesized in 2D and 3D nanostructures by a facile hydrothermal method, and the disadvantages of the 2D structures were thoroughly examined. The weakness of 2D WO3·nH2O originates from the layered structure. XRD and SEM characterizations of the as-grown WO3·nH2O samples suggested a structural transition from 2D to 3D upon temperature increase. The independent electrochemical tests of three typical samples confirmed a faster performance degradation in the 2D nanostructures compared to 3D nanostructures, supported by the SEM investigation and further explained by subsequent ex situ Raman measurements. Although 2D layered WO3·nH2O nanostructures outranks the 3D network counterparts in terms of the improved electronic properties, they can easily generate the structural instability by 2D intercalation owing to its weak interlayer van der Waals interactions. Their morphology change confirms the degradation mechanism proposed in this work. Consequently, this work provides in-depth understanding the weakness of 2D layered nanomaterials and paves the way for further interlayer reinforcement of 2D TMOs.
Supporting Information
The video shows the typical coloration/bleaching process of the samples during electrochemical cycling.
| Supporting Information File 1: Coloration/bleaching during electrochemical cycling. | ||
| Format: MP4 | Size: 8.6 MB | Download |
Acknowledgements
The work was supported by the Research and Development Program of the Ghent University Global Campus, South Korea. This work was supported in part by the Melbourne Centre for Nanofabrication (MCN) in the Victoria Node of the Australian National Fabrication Facility (ANFF). F.V. acknowledges the support from the Tomsk Polytechnic University Competitiveness Enhancement Program grant (VIU-316/2017). S.Z. acknowledges the support from the “100 Talents Program” of Shanxi Province, P.R. China.
References
-
Xia, F.; Wang, H.; Xiao, D.; Dubey, M.; Ramasubramaniam, A. Nat. Photonics 2014, 8, 899–907. doi:10.1038/nphoton.2014.271
Return to citation in text: [1] -
Niu, T.; Li, A. Prog. Surf. Sci. 2015, 90, 21–45. doi:10.1016/j.progsurf.2014.11.001
Return to citation in text: [1] -
Li, H.; Shi, Y.; Chiu, M.-H.; Li, L.-J. Nano Energy 2015, 18, 293–305. doi:10.1016/j.nanoen.2015.10.023
Return to citation in text: [1] -
Di, J.; Xia, J.; Li, H.; Liu, Z. Nano Energy 2017, 35, 79–91. doi:10.1016/j.nanoen.2017.03.030
Return to citation in text: [1] -
Chakravarty, D.; Late, D. J. Eur. J. Inorg. Chem. 2015, 1973–1980. doi:10.1002/ejic.201500039
Return to citation in text: [1] -
Li, Z.-F.; Liu, Q.; Liu, Y.; Yang, F.; Xin, L.; Zhou, Y.; Zhang, H.; Stanciu, L.; Xie, J. ACS Appl. Mater. Interfaces 2015, 7, 27087–27095. doi:10.1021/acsami.5b05819
Return to citation in text: [1] -
Khandare, L.; Late, D. J. Appl. Surf. Sci. 2017, 418, 2–8. doi:10.1016/j.apsusc.2016.11.199
Return to citation in text: [1] -
Suryawanshi, S. R.; Kaware, V.; Chakravarty, D.; Walke, P. S.; More, M. A.; Joshi, K.; Rout, C. S.; Late, D. J. RSC Adv. 2015, 5, 80990–80997. doi:10.1039/C5RA12364J
Return to citation in text: [1] -
Sahoo, S.; Mondal, R.; Late, D. J.; Rout, C. S. Microporous Mesoporous Mater. 2017, 244, 101–108. doi:10.1016/j.micromeso.2017.02.043
Return to citation in text: [1] -
Sahoo, S.; Naik, K. K.; Late, D. J.; Rout, C. S. J. Alloys Compd. 2017, 695, 154–161. doi:10.1016/j.jallcom.2016.10.163
Return to citation in text: [1] -
Kalantar-zadeh, K.; Ou, J. Z.; Daeneke, T.; Mitchell, A.; Sasaki, T.; Fuhrer, M. S. Appl. Mater. Today 2016, 5, 73–89. doi:10.1016/j.apmt.2016.09.012
Return to citation in text: [1] -
Salanne, M.; Rotenberg, B.; Naoi, K.; Kaneko, K.; Taberna, P.-L.; Grey, C. P.; Dunn, B.; Simon, P. Nat. Energy 2016, 1, No. 16070. doi:10.1038/nenergy.2016.70
Return to citation in text: [1] -
Haque, F.; Daeneke, T.; Kalantar-zadeh, K.; Ou, J. Z. Nano-Micro Lett. 2018, 10, No. 23. doi:10.1007/s40820-017-0176-y
Return to citation in text: [1] -
Osada, M.; Sasaki, T. J. Mater. Chem. 2009, 19, 2503–2511. doi:10.1039/b820160a
Return to citation in text: [1] -
Kisu, K.; Iijima, M.; Iwama, E.; Saito, M.; Orikasa, Y.; Naoi, W.; Naoi, K. J. Mater. Chem. A 2014, 2, 13058–13068. doi:10.1039/C4TA01994F
Return to citation in text: [1] -
Peng, L.; Zhu, Y.; Chen, D.; Ruoff, R. S.; Yu, G. Adv. Energy Mater. 2016, 6, 1600025. doi:10.1002/aenm.201600025
Return to citation in text: [1] -
Wang, D.; Li, X.-B.; Han, D.; Tian, W. Q.; Sun, H.-B. Nano Today 2017, 16, 30–45. doi:10.1016/j.nantod.2017.07.001
Return to citation in text: [1] -
Hai, Z.; Zhuiykov, S. Adv. Mater. Interfaces 2018, 5, 1701385. doi:10.1002/admi.201701385
Return to citation in text: [1] -
Zheng, H.; Ou, J. Z.; Strano, M. S.; Kaner, R. B.; Mitchell, A.; Kalantar-zadeh, K. Adv. Funct. Mater. 2011, 21, 2175–2196. doi:10.1002/adfm.201002477
Return to citation in text: [1] -
Yan, C.; Kang, W.; Wang, J.; Cui, M.; Wang, X.; Foo, C. Y.; Chee, K. J.; Lee, P. S. ACS Nano 2014, 8, 316–322. doi:10.1021/nn404061g
Return to citation in text: [1] -
Yang, P.; Sun, P.; Chai, Z.; Huang, L.; Cai, X.; Tan, S.; Song, J.; Mai, W. Angew. Chem., Int. Ed. 2014, 53, 11935–11939. doi:10.1002/anie.201407365
Return to citation in text: [1] -
Yuan, S.-J.; Li, W.-W.; Cheng, Y.-Y.; He, H.; Chen, J.-J.; Tong, Z.-H.; Lin, Z.-Q.; Zhang, F.; Sheng, G.-P.; Yu, H.-Q. Nat. Protoc. 2014, 9, 112–119. doi:10.1038/nprot.2013.173
Return to citation in text: [1] -
Baeck, S.-H.; Choi, K.-S.; Jaramillo, T. F.; Stucky, G. D.; McFarland, E. W. Adv. Mater. 2003, 15, 1269–1273. doi:10.1002/adma.200304669
Return to citation in text: [1] -
Wang, J.; Liu, C.-j. ChemBioEng Rev. 2015, 2, 335–350. doi:10.1002/cben.201500014
Return to citation in text: [1] [2] -
Zhang, J.; Zhang, P.; Wang, T.; Gong, J. Nano Energy 2015, 11, 189–195. doi:10.1016/j.nanoen.2014.10.021
Return to citation in text: [1] -
Sasidharan, M.; Gunawardhana, N.; Yoshio, M.; Nakashima, K. Nano Energy 2012, 1, 503–508. doi:10.1016/j.nanoen.2012.03.003
Return to citation in text: [1] -
Cong, S.; Geng, F.; Zhao, Z. Adv. Mater. 2016, 28, 10518–10528. doi:10.1002/adma.201601109
Return to citation in text: [1] -
Sun, W.; Yeung, M. T.; Lech, A. T.; Lin, C.-W.; Lee, C.; Li, T.; Duan, X.; Zhou, J.; Kaner, R. B. Nano Lett. 2015, 15, 4834–4838. doi:10.1021/acs.nanolett.5b02013
Return to citation in text: [1] -
Wu, X.; Yao, S. Nano Energy 2017, 42, 143–150. doi:10.1016/j.nanoen.2017.10.058
Return to citation in text: [1] [2] -
Kuti, L. M.; Bhella, S. S.; Thangadurai, V. Inorg. Chem. 2009, 48, 6804–6811. doi:10.1021/ic900738m
Return to citation in text: [1] -
Amano, F.; Tian, M.; Wu, G.; Ohtani, B.; Chen, A. ACS Appl. Mater. Interfaces 2011, 3, 4047–4052. doi:10.1021/am200897n
Return to citation in text: [1] -
Chen, D.; Gao, L.; Yasumori, A.; Kuroda, K.; Sugahara, Y. Small 2008, 4, 1813–1822. doi:10.1002/smll.200800205
Return to citation in text: [1] -
Jadkar, V.; Pawbake, A.; Waykar, R.; Jadhavar, A.; Date, A.; Late, D.; Pathan, H.; Gosavi, S.; Jadkar, S. Phys. Status Solidi A 2017, 214, 1600717. doi:10.1002/pssa.201600717
Return to citation in text: [1] -
Late, D. J.; Kashid, R. V.; Rout, C. S.; More, M. A.; Joag, D. S. Appl. Phys. A: Mater. Sci. Process. 2010, 98, 751–756. doi:10.1007/s00339-009-5536-0
Return to citation in text: [1] -
Qin, D.-D.; Tao, C.-L.; Friesen, S. A.; Wang, T.-H.; Varghese, O. K.; Bao, N.-Z.; Yang, Z.-Y.; Mallouk, T. E.; Grimes, C. A. Chem. Commun. 2012, 48, 729–731. doi:10.1039/C1CC15691H
Return to citation in text: [1] -
Rahmani, M. B.; Yaacob, M. H.; Sabri, Y. M. Sens. Actuators, B 2017, 251, 57–64. doi:10.1016/j.snb.2017.05.029
Return to citation in text: [1] -
Xie, Z.; Gao, L.; Liang, B.; Wang, X.; Chen, G.; Liu, Z.; Chao, J.; Chen, D.; Shen, G. J. Mater. Chem. 2012, 22, 19904–19910. doi:10.1039/c2jm33622g
Return to citation in text: [1] -
Mitchell, J. B.; Lo, W. C.; Genc, A.; Lebeau, J.; Augustyn, V. Chem. Mater. 2017, 29, 3928–3937. doi:10.1021/acs.chemmater.6b05485
Return to citation in text: [1] [2] -
Ahmed, B.; Ojha, A. K.; Singh, A.; Hirsch, F.; Fischer, I.; Patrice, D.; Materny, A. J. Hazard. Mater. 2018, 347, 266–278. doi:10.1016/j.jhazmat.2017.12.069
Return to citation in text: [1] -
Liang, L.; Zhang, J.; Zhou, Y.; Xie, J.; Zhang, X.; Guan, M.; Pan, B.; Xie, Y. Sci. Rep. 2013, 3, No. 1936. doi:10.1038/srep01936
Return to citation in text: [1] -
Zheng, J. Y.; Haider, Z.; Van, T. K.; Pawar, A. U.; Kang, M. J.; Kim, C. W.; Kang, Y. S. CrystEngComm 2015, 17, 6070–6093. doi:10.1039/C5CE00900F
Return to citation in text: [1] [2] -
Zheng, J. Y.; Song, G.; Kim, C. W.; Kang, Y. S. Nanoscale 2013, 5, 5279–5282. doi:10.1039/c3nr00964e
Return to citation in text: [1] -
Hai, Z.; Karbalaei Akbari, M.; Xue, C.; Xu, H.; Solano, E.; Detavernier, C.; Hu, J.; Zhuiykov, S. Compos. Commun. 2017, 5, 31–35. doi:10.1016/j.coco.2017.06.001
Return to citation in text: [1] -
Bai, S.; Zhang, K.; Wang, L.; Sun, J.; Luo, R.; Li, D.; Chen, A. J. Mater. Chem. A 2014, 2, 7927–7934. doi:10.1039/C4TA00053F
Return to citation in text: [1] -
Hai, Z.; Akbari, M. K.; Xue, C.; Xu, H.; Hyde, L.; Zhuiykov, S. Appl. Surf. Sci. 2017, 405, 169–177. doi:10.1016/j.apsusc.2017.02.031
Return to citation in text: [1] -
Qiu, M.; Zhu, D.; Bao, X.; Wang, J.; Wang, X.; Yang, R. J. Mater. Chem. A 2016, 4, 894–900. doi:10.1039/C5TA08898D
Return to citation in text: [1] -
Khan, H.; Zavabeti, A.; Wang, Y.; Harrison, C. J.; Carey, B. J.; Mohiuddin, M.; Chrimes, A. F.; Alves De Castro, I.; Zhang, B. Y.; Sabri, Y. M.; Bhargava, S. K.; Ou, J. Z.; Daeneke, T.; Russo, S. P.; Li, Y.; Kalantar-zadeh, K. Nanoscale 2017, 9, 19161–19175. doi:10.1039/C7NR05403C
Return to citation in text: [1] -
Hai, Z.; Karbalaei Akbari, M.; Wei, Z.; Xue, C.; Xu, H.; Hu, J.; Hyde, L.; Zhuiykov, S. Mater. Today Commun. 2017, 12, 55–62. doi:10.1016/j.mtcomm.2017.06.006
Return to citation in text: [1] -
Salmaoui, S.; Sediri, F.; Gharbi, N.; Perruchot, C.; Jouini, M. Electrochim. Acta 2013, 108, 634–643. doi:10.1016/j.electacta.2013.07.086
Return to citation in text: [1] -
Hai, Z.; Karbalaei Akbari, M.; Wei, Z.; Xue, C.; Xu, H.; Hu, J.; Zhuiykov, S. Electrochim. Acta 2017, 246, 625–633. doi:10.1016/j.electacta.2017.06.095
Return to citation in text: [1] -
Wang, R.; Chung, C. C.; Liu, Y.; Jones, J. L.; Augustyn, V. Langmuir 2017, 33, 9314–9323. doi:10.1021/acs.langmuir.7b00705
Return to citation in text: [1] -
Zhuiykov, S.; Hyde, L.; Hai, Z.; Karbalaei Akbari, M.; Kats, E.; Detavernier, C.; Xue, C.; Xu, H. Appl. Mater. Today 2017, 6, 44–53. doi:10.1016/j.apmt.2016.12.004
Return to citation in text: [1] -
He, Y.; Zhao, Y. J. Phys. Chem. C 2008, 112, 61–68. doi:10.1021/jp076898x
Return to citation in text: [1] -
Nayak, A. K.; Lee, S.; Choi, Y. I.; Yoon, H. J.; Sohn, Y.; Pradhan, D. ACS Sustainable Chem. Eng. 2017, 5, 2741–2750. doi:10.1021/acssuschemeng.6b03084
Return to citation in text: [1]
| 29. | Wu, X.; Yao, S. Nano Energy 2017, 42, 143–150. doi:10.1016/j.nanoen.2017.10.058 |
| 46. | Qiu, M.; Zhu, D.; Bao, X.; Wang, J.; Wang, X.; Yang, R. J. Mater. Chem. A 2016, 4, 894–900. doi:10.1039/C5TA08898D |
| 42. | Zheng, J. Y.; Song, G.; Kim, C. W.; Kang, Y. S. Nanoscale 2013, 5, 5279–5282. doi:10.1039/c3nr00964e |
| 43. | Hai, Z.; Karbalaei Akbari, M.; Xue, C.; Xu, H.; Solano, E.; Detavernier, C.; Hu, J.; Zhuiykov, S. Compos. Commun. 2017, 5, 31–35. doi:10.1016/j.coco.2017.06.001 |
| 44. | Bai, S.; Zhang, K.; Wang, L.; Sun, J.; Luo, R.; Li, D.; Chen, A. J. Mater. Chem. A 2014, 2, 7927–7934. doi:10.1039/C4TA00053F |
| 45. | Hai, Z.; Akbari, M. K.; Xue, C.; Xu, H.; Hyde, L.; Zhuiykov, S. Appl. Surf. Sci. 2017, 405, 169–177. doi:10.1016/j.apsusc.2017.02.031 |
| 1. | Xia, F.; Wang, H.; Xiao, D.; Dubey, M.; Ramasubramaniam, A. Nat. Photonics 2014, 8, 899–907. doi:10.1038/nphoton.2014.271 |
| 2. | Niu, T.; Li, A. Prog. Surf. Sci. 2015, 90, 21–45. doi:10.1016/j.progsurf.2014.11.001 |
| 3. | Li, H.; Shi, Y.; Chiu, M.-H.; Li, L.-J. Nano Energy 2015, 18, 293–305. doi:10.1016/j.nanoen.2015.10.023 |
| 4. | Di, J.; Xia, J.; Li, H.; Liu, Z. Nano Energy 2017, 35, 79–91. doi:10.1016/j.nanoen.2017.03.030 |
| 5. | Chakravarty, D.; Late, D. J. Eur. J. Inorg. Chem. 2015, 1973–1980. doi:10.1002/ejic.201500039 |
| 6. | Li, Z.-F.; Liu, Q.; Liu, Y.; Yang, F.; Xin, L.; Zhou, Y.; Zhang, H.; Stanciu, L.; Xie, J. ACS Appl. Mater. Interfaces 2015, 7, 27087–27095. doi:10.1021/acsami.5b05819 |
| 7. | Khandare, L.; Late, D. J. Appl. Surf. Sci. 2017, 418, 2–8. doi:10.1016/j.apsusc.2016.11.199 |
| 8. | Suryawanshi, S. R.; Kaware, V.; Chakravarty, D.; Walke, P. S.; More, M. A.; Joshi, K.; Rout, C. S.; Late, D. J. RSC Adv. 2015, 5, 80990–80997. doi:10.1039/C5RA12364J |
| 9. | Sahoo, S.; Mondal, R.; Late, D. J.; Rout, C. S. Microporous Mesoporous Mater. 2017, 244, 101–108. doi:10.1016/j.micromeso.2017.02.043 |
| 10. | Sahoo, S.; Naik, K. K.; Late, D. J.; Rout, C. S. J. Alloys Compd. 2017, 695, 154–161. doi:10.1016/j.jallcom.2016.10.163 |
| 24. | Wang, J.; Liu, C.-j. ChemBioEng Rev. 2015, 2, 335–350. doi:10.1002/cben.201500014 |
| 25. | Zhang, J.; Zhang, P.; Wang, T.; Gong, J. Nano Energy 2015, 11, 189–195. doi:10.1016/j.nanoen.2014.10.021 |
| 26. | Sasidharan, M.; Gunawardhana, N.; Yoshio, M.; Nakashima, K. Nano Energy 2012, 1, 503–508. doi:10.1016/j.nanoen.2012.03.003 |
| 41. | Zheng, J. Y.; Haider, Z.; Van, T. K.; Pawar, A. U.; Kang, M. J.; Kim, C. W.; Kang, Y. S. CrystEngComm 2015, 17, 6070–6093. doi:10.1039/C5CE00900F |
| 19. | Zheng, H.; Ou, J. Z.; Strano, M. S.; Kaner, R. B.; Mitchell, A.; Kalantar-zadeh, K. Adv. Funct. Mater. 2011, 21, 2175–2196. doi:10.1002/adfm.201002477 |
| 20. | Yan, C.; Kang, W.; Wang, J.; Cui, M.; Wang, X.; Foo, C. Y.; Chee, K. J.; Lee, P. S. ACS Nano 2014, 8, 316–322. doi:10.1021/nn404061g |
| 21. | Yang, P.; Sun, P.; Chai, Z.; Huang, L.; Cai, X.; Tan, S.; Song, J.; Mai, W. Angew. Chem., Int. Ed. 2014, 53, 11935–11939. doi:10.1002/anie.201407365 |
| 22. | Yuan, S.-J.; Li, W.-W.; Cheng, Y.-Y.; He, H.; Chen, J.-J.; Tong, Z.-H.; Lin, Z.-Q.; Zhang, F.; Sheng, G.-P.; Yu, H.-Q. Nat. Protoc. 2014, 9, 112–119. doi:10.1038/nprot.2013.173 |
| 23. | Baeck, S.-H.; Choi, K.-S.; Jaramillo, T. F.; Stucky, G. D.; McFarland, E. W. Adv. Mater. 2003, 15, 1269–1273. doi:10.1002/adma.200304669 |
| 41. | Zheng, J. Y.; Haider, Z.; Van, T. K.; Pawar, A. U.; Kang, M. J.; Kim, C. W.; Kang, Y. S. CrystEngComm 2015, 17, 6070–6093. doi:10.1039/C5CE00900F |
| 16. | Peng, L.; Zhu, Y.; Chen, D.; Ruoff, R. S.; Yu, G. Adv. Energy Mater. 2016, 6, 1600025. doi:10.1002/aenm.201600025 |
| 17. | Wang, D.; Li, X.-B.; Han, D.; Tian, W. Q.; Sun, H.-B. Nano Today 2017, 16, 30–45. doi:10.1016/j.nantod.2017.07.001 |
| 18. | Hai, Z.; Zhuiykov, S. Adv. Mater. Interfaces 2018, 5, 1701385. doi:10.1002/admi.201701385 |
| 39. | Ahmed, B.; Ojha, A. K.; Singh, A.; Hirsch, F.; Fischer, I.; Patrice, D.; Materny, A. J. Hazard. Mater. 2018, 347, 266–278. doi:10.1016/j.jhazmat.2017.12.069 |
| 53. | He, Y.; Zhao, Y. J. Phys. Chem. C 2008, 112, 61–68. doi:10.1021/jp076898x |
| 54. | Nayak, A. K.; Lee, S.; Choi, Y. I.; Yoon, H. J.; Sohn, Y.; Pradhan, D. ACS Sustainable Chem. Eng. 2017, 5, 2741–2750. doi:10.1021/acssuschemeng.6b03084 |
| 11. | Kalantar-zadeh, K.; Ou, J. Z.; Daeneke, T.; Mitchell, A.; Sasaki, T.; Fuhrer, M. S. Appl. Mater. Today 2016, 5, 73–89. doi:10.1016/j.apmt.2016.09.012 |
| 12. | Salanne, M.; Rotenberg, B.; Naoi, K.; Kaneko, K.; Taberna, P.-L.; Grey, C. P.; Dunn, B.; Simon, P. Nat. Energy 2016, 1, No. 16070. doi:10.1038/nenergy.2016.70 |
| 13. | Haque, F.; Daeneke, T.; Kalantar-zadeh, K.; Ou, J. Z. Nano-Micro Lett. 2018, 10, No. 23. doi:10.1007/s40820-017-0176-y |
| 14. | Osada, M.; Sasaki, T. J. Mater. Chem. 2009, 19, 2503–2511. doi:10.1039/b820160a |
| 15. | Kisu, K.; Iijima, M.; Iwama, E.; Saito, M.; Orikasa, Y.; Naoi, W.; Naoi, K. J. Mater. Chem. A 2014, 2, 13058–13068. doi:10.1039/C4TA01994F |
| 40. | Liang, L.; Zhang, J.; Zhou, Y.; Xie, J.; Zhang, X.; Guan, M.; Pan, B.; Xie, Y. Sci. Rep. 2013, 3, No. 1936. doi:10.1038/srep01936 |
| 35. | Qin, D.-D.; Tao, C.-L.; Friesen, S. A.; Wang, T.-H.; Varghese, O. K.; Bao, N.-Z.; Yang, Z.-Y.; Mallouk, T. E.; Grimes, C. A. Chem. Commun. 2012, 48, 729–731. doi:10.1039/C1CC15691H |
| 37. | Xie, Z.; Gao, L.; Liang, B.; Wang, X.; Chen, G.; Liu, Z.; Chao, J.; Chen, D.; Shen, G. J. Mater. Chem. 2012, 22, 19904–19910. doi:10.1039/c2jm33622g |
| 38. | Mitchell, J. B.; Lo, W. C.; Genc, A.; Lebeau, J.; Augustyn, V. Chem. Mater. 2017, 29, 3928–3937. doi:10.1021/acs.chemmater.6b05485 |
| 24. | Wang, J.; Liu, C.-j. ChemBioEng Rev. 2015, 2, 335–350. doi:10.1002/cben.201500014 |
| 33. | Jadkar, V.; Pawbake, A.; Waykar, R.; Jadhavar, A.; Date, A.; Late, D.; Pathan, H.; Gosavi, S.; Jadkar, S. Phys. Status Solidi A 2017, 214, 1600717. doi:10.1002/pssa.201600717 |
| 34. | Late, D. J.; Kashid, R. V.; Rout, C. S.; More, M. A.; Joag, D. S. Appl. Phys. A: Mater. Sci. Process. 2010, 98, 751–756. doi:10.1007/s00339-009-5536-0 |
| 38. | Mitchell, J. B.; Lo, W. C.; Genc, A.; Lebeau, J.; Augustyn, V. Chem. Mater. 2017, 29, 3928–3937. doi:10.1021/acs.chemmater.6b05485 |
| 51. | Wang, R.; Chung, C. C.; Liu, Y.; Jones, J. L.; Augustyn, V. Langmuir 2017, 33, 9314–9323. doi:10.1021/acs.langmuir.7b00705 |
| 52. | Zhuiykov, S.; Hyde, L.; Hai, Z.; Karbalaei Akbari, M.; Kats, E.; Detavernier, C.; Xue, C.; Xu, H. Appl. Mater. Today 2017, 6, 44–53. doi:10.1016/j.apmt.2016.12.004 |
| 30. | Kuti, L. M.; Bhella, S. S.; Thangadurai, V. Inorg. Chem. 2009, 48, 6804–6811. doi:10.1021/ic900738m |
| 31. | Amano, F.; Tian, M.; Wu, G.; Ohtani, B.; Chen, A. ACS Appl. Mater. Interfaces 2011, 3, 4047–4052. doi:10.1021/am200897n |
| 32. | Chen, D.; Gao, L.; Yasumori, A.; Kuroda, K.; Sugahara, Y. Small 2008, 4, 1813–1822. doi:10.1002/smll.200800205 |
| 47. | Khan, H.; Zavabeti, A.; Wang, Y.; Harrison, C. J.; Carey, B. J.; Mohiuddin, M.; Chrimes, A. F.; Alves De Castro, I.; Zhang, B. Y.; Sabri, Y. M.; Bhargava, S. K.; Ou, J. Z.; Daeneke, T.; Russo, S. P.; Li, Y.; Kalantar-zadeh, K. Nanoscale 2017, 9, 19161–19175. doi:10.1039/C7NR05403C |
| 48. | Hai, Z.; Karbalaei Akbari, M.; Wei, Z.; Xue, C.; Xu, H.; Hu, J.; Hyde, L.; Zhuiykov, S. Mater. Today Commun. 2017, 12, 55–62. doi:10.1016/j.mtcomm.2017.06.006 |
| 27. | Cong, S.; Geng, F.; Zhao, Z. Adv. Mater. 2016, 28, 10518–10528. doi:10.1002/adma.201601109 |
| 28. | Sun, W.; Yeung, M. T.; Lech, A. T.; Lin, C.-W.; Lee, C.; Li, T.; Duan, X.; Zhou, J.; Kaner, R. B. Nano Lett. 2015, 15, 4834–4838. doi:10.1021/acs.nanolett.5b02013 |
| 29. | Wu, X.; Yao, S. Nano Energy 2017, 42, 143–150. doi:10.1016/j.nanoen.2017.10.058 |
| 36. | Rahmani, M. B.; Yaacob, M. H.; Sabri, Y. M. Sens. Actuators, B 2017, 251, 57–64. doi:10.1016/j.snb.2017.05.029 |
| 49. | Salmaoui, S.; Sediri, F.; Gharbi, N.; Perruchot, C.; Jouini, M. Electrochim. Acta 2013, 108, 634–643. doi:10.1016/j.electacta.2013.07.086 |
| 50. | Hai, Z.; Karbalaei Akbari, M.; Wei, Z.; Xue, C.; Xu, H.; Hu, J.; Zhuiykov, S. Electrochim. Acta 2017, 246, 625–633. doi:10.1016/j.electacta.2017.06.095 |
© 2018 Hai et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (https://www.beilstein-journals.org/bjnano)