Abstract
The development of a reliable procedure for the synthesis of the 16-glucoside and 16-sulfate of the resorcylic acid lactone (RAL) type compound zearalenone is presented. Different protective group strategies were considered and applied to enable the preparation of glucosides and sulfates that are difficult to access up to now. Acetyl and p-methoxybenzyl protection led to undesired results and were shown to be inappropriate. Finally, triisopropylsilyl-protected zearalenone was successfully used as intermediate for the first synthesis of the corresponding mycotoxin glucoside and sulfate that are highly valuable as reference materials for further studies in the emerging field of masked mycotoxins. Furthermore, high stability was observed for aryl sulfates prepared as tetrabutylammonium salts. Overall, these findings should be applicable for the synthesis of similar RAL type and natural product conjugates.

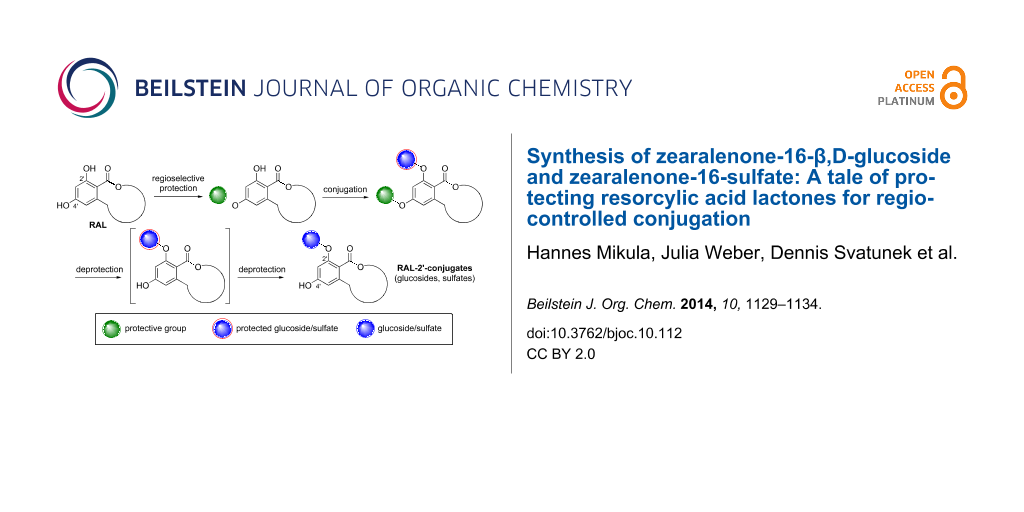
Graphical Abstract
Introduction
Resorcylic acid lactones (RALs, Figure 1), a compound class of benzannulated macrolides, are pharmacologically active secondary metabolites produced by a variety of different fungal species [1]. Zearalenone (ZEN, 1) is a well-known RAL type mycotoxin for which maximum tolerated levels in food and feed were enacted and recommended, respectively, in Europe [2,3]. ZEN is produced by several plant pathogenic Fusarium species, including Fusarium graminearum and Fusarium culmorum. These species, which are the most frequently occurring toxin-producing fungi of the northern temperate zone, are commonly found in cereals and crops throughout the world [4,5]. Significant levels of ZEN are prevalently found in grains such as maize, wheat, and rice [6]. It is known that ZEN can cause problems of the reproductive tract (e.g., impaired fertility) in animals [7]. Physiological studies revealed binding of ZEN to recombinant human estrogen receptors [8] and have furthermore shown a ZEN-induced stimulation of the growth of human breast cancer cells [9].
![[1860-5397-10-112-1]](/bjoc/content/figures/1860-5397-10-112-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of selected RAL type fungal secondary metabolites.
Figure 1: Structure of selected RAL type fungal secondary metabolites.
Additionally, masked mycotoxins, especially altered derivatives formed through conjugation to sugar moieties or sulfate, emerge after metabolization by living plants. Due to changed chemical structures and properties compared to the parent mycotoxins, these conjugates can usually not be detected applying standard analytical techniques [10,11]. Responsible biochemical transformations are catalyzed usually by enzymes within detoxification processes [12]. Schneweis et al. reported the occurrence of ZEN-14-β,D-glucoside (5, Figure 2A) in wheat [13] and the first chemical synthesis of this compound applying phase transfer glycosylation has been reported by Grabley et al. [14]. ZEN-14-sulfate (6, Figure 2A) was first isolated from F. graminearum-inoculated rice [15] and both, the glucoside 5 and the sulfate 6, were identified as ZEN metabolites in Arabidopsis thaliana [16]. These conjugates are easily hydrolyzed back to the parental mycotoxin during digestion of contaminated grain, and should therefore be considered as masked mycotoxins [12]. Recently it has been shown that ZEN treated barley, wheat and Brachypodium distachyon cells produce both the ZEN-14 and the ZEN-16-glucoside, with up to 18-fold higher levels of ZEN-16-glucoside than ZEN-14-glucoside in barley roots [17]. We therefore intended to develop a synthetic method for regiocontrolled conjugation of ZEN. Basically, the RAL type moiety of ZEN contains two possible sites for glycosylation/sulfation, but due to the higher reactivity of the phenol group at position 14, reactions at this site are strongly favored compared to conjugation at C16–OH [18-20]. Although natural products containing a RAL type moiety conjugated at the phenol group in ortho position to the carboxyl group (2’-OH) were already detected and identified [21-25], to the best of our knowledge there are no reliable synthetic procedures and strategies towards this class of compounds described in the literature so far. The synthesis of the natural glucoside delphoside by Saeed was performed using methyl ether protection at O-6 of the isocoumarin core structure during glucosylation and rather harsh unfavorable demethylation with boron tribromide in the last step [26]. Without structure verification and characterization ZEN-16-β,D-glucoside (7, Figure 2B) was tentatively identified as a byproduct of the Königs–Knorr glucosylation of ZEN for preparation of ZEN-14-glucoside [27]. In the course of ongoing research in the emerging field of masked mycotoxins, we were able to prepare ZEN-16-β,D-glucoside (7) and ZEN-16-sulfate (8, Figure 2B) in reasonable amounts after the development of reliable procedures that should be generally applicable to resorcylic acid lactones. Additionally the first chemical synthesis of the ZEN-derivative 14-O-acetylzearalenone (14-AcZEN, produced by some Fusarium strains) [28] is reported.
![[1860-5397-10-112-2]](/bjoc/content/figures/1860-5397-10-112-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of zearalenone conjugates: (A) ZEN-14-β,D-glucoside (5) and ZEN-14-sulfate (6), (B) ZEN-16-β,D-glucoside (7) and ZEN-16-sulfate (8). Sulfates shown as sodium salts.
Figure 2: Structures of zearalenone conjugates: (A) ZEN-14-β,D-glucoside (5) and ZEN-14-sulfate (6), (B) ZEN-...
Results and Discussion
The general strategy for regiocontrolled conjugation at position 2’ of resorcylic acid esters and lactones is shown in Scheme 1. Regioselective protection of the more nucleophilic 4’-phenol and subsequent glucosylation or sulfation should lead to the desired products.
![[1860-5397-10-112-i1]](/bjoc/content/inline/1860-5397-10-112-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General strategy for the synthesis of RAL-2’-conjugation (Pg: protective group, pGlc: protected glucose moiety, pS: protected sulfate, Glc: β,D-glucoside).
Scheme 1: General strategy for the synthesis of RAL-2’-conjugation (Pg: protective group, pGlc: protected glu...
For the development of a reliable protective group strategy and subsequent reaction optimization, 2,4-dihydroxybenzoic acid isopropyl ester (9) [29] was used as a RAL mimic. For protection of the 4-OH group we first considered an acetyl group that could be regioselectively introduced by reaction of 9 with acetic anhydride and catalytic amounts of 4-(dimethylamino)pyridine (DMAP) to obtain the acetylated RAL mimic 10 (Scheme 2A). Different methods for glycosylation were investigated using acetyl-protected glucosyl donors since diastereoselective β-conjugation, which is needed for the preparation of glucosides formed during phase II metabolism, is commonly achieved applying the participation of acyl groups at O-2 of the glycosyl donor (anchimeric effect) [30]. Lewis acid-mediated glucosylation using the trichloroacetimidate donor 11 according to the procedure of Saeed [26] did not lead to the desired product, which can be explained by the weak nucleophilicity of the 2-OH group of the acceptor 10. This assumption was supported by detection of the glucosyl acetamide 12, which is known to be formed by rearrangement of 11 when activated in the presence of a weak acceptor (Scheme 2B) [31-34]. Königs–Knorr glucosylation, which in general is most frequently used for the glucosylation of phenols, using commercially available bromo sugar 13 activated by silver(I) salts or under phase transfer conditions led to complex product mixtures (Scheme 2C).
![[1860-5397-10-112-i2]](/bjoc/content/inline/1860-5397-10-112-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: (A) Regioselective acetylation of resorcylic acid ester 9. (B) Lewis acid mediated glucosylation; no conversion of 10, partially rearrangement of 11 to glucosyl acetamide 12. (C) Königs–Knorr glucosylation; Ag(I): Ag2CO3 or Ag2O, CH2Cl2 or MeCN; PTG: phase transfer glucosylation, NaOH, tetrabutylammonium bromide, pH 10–11, CHCl3/H2O.
Scheme 2: (A) Regioselective acetylation of resorcylic acid ester 9. (B) Lewis acid mediated glucosylation; n...
Nevertheless, the procedure for selective acetylation of resorcylic acid esters and lactones was applied for the first synthesis of 14-O-acetylzearalenone (14) (Scheme 3).
![[1860-5397-10-112-i3]](/bjoc/content/inline/1860-5397-10-112-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Regioselective acetylation of ZEN (1) affording 14-O-acetylzearalenone (14).
Scheme 3: Regioselective acetylation of ZEN (1) affording 14-O-acetylzearalenone (14).
To avoid undesired cleavage of the acetyl group during glucosylation of 10, p-methoxybenzyl (PMB) protection was applied instead, since the PMB group was considered to be inert to the reactions conditions of the Königs–Knorr procedure, thus forcing conjugation at position 2. Regioselective p-methoxybenzylation of 9 was achieved by reaction with PMB-Cl and Cs2CO3 in dry DMF after optimization in terms of base and solvent type (Scheme 4A). Königs–Knorr glucosylation of the PMB-protected mimic 15 afforded 16, which was deprotected using 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) for oxidative PMB cleavage and subsequent ester saponification to yield the desired glucoside 17 in an overall yield of 30% (Scheme 4B).
![[1860-5397-10-112-i4]](/bjoc/content/inline/1860-5397-10-112-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: (A) Regioselective p-methoxybenzylation of 9. (B) Synthesis of the ZEN-16-Glc mimic 17.
Scheme 4: (A) Regioselective p-methoxybenzylation of 9. (B) Synthesis of the ZEN-16-Glc mimic 17.
Applying this procedure to ZEN (1) afforded the glucosylated intermediate 19 after Königs–Knorr glucosylation of 14-PMB-ZEN (18), but subsequent deprotection using DDQ did not afford the desired product. Also an alternative procedure for oxidative PMB cleavage using cerium ammonium nitrate (CAN) did not lead to the formation of the deprotected compound 20 (Scheme 5). Beside unreacted 19, LC–MS/MS analysis showed the formation of a product with an m/z value 16 amu higher than calculated for 19 indicating oxidation of this intermediate but no deprotection.
![[1860-5397-10-112-i5]](/bjoc/content/inline/1860-5397-10-112-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Regioselective protection of ZEN (1) and failed PMB cleavage of the glucosylated intermediate 19.
Scheme 5: Regioselective protection of ZEN (1) and failed PMB cleavage of the glucosylated intermediate 19.
Since the DDQ-promoted cleavage of phenolic PMB ethers can be complicated by overoxidation, especially with electron-rich phenolic compounds [35], we assume a significant effect of the conjugated olefinic double bond at C-6’ of the resorcylic acid moiety being responsible for the observed different behavior of ZEN (1) and the ZEN mimic 9.
After this second setback the protective group strategy was changed again within a third approach. Considering steric hindrance and methods for selective deprotection under relatively mild conditions led us to the use of triisopropylsilyl (TIPS) protection for regiocontrolled glucosylation of resorcylic acid esters and lactones. Regioselective silylation of 9 and ZEN (1) was readily achieved affording compounds 21 and 22 (14-TIPS-ZEN), respectively, in nearly quantitative yields by reaction with TIPS-Cl and imidazole in dry CH2Cl2. Applying this strategy we were finally able to accomplish the synthesis of the ZEN mimic glucoside 17 (Scheme 6A) as well as of the target compound ZEN-16-β,D-glucoside (7) as shown in Scheme 6B. Reasonable yields of 41% (17) and 34% (7), respectively, were obtained applying an optimized purification protocol.
![[1860-5397-10-112-i6]](/bjoc/content/inline/1860-5397-10-112-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of (A) ZEN mimic glucoside 17 and (B) ZEN-16-β,D-glucoside (7); a: TIPS-Cl, imidazole, 16 h, rt, >99% for 21 and 22; b: 13, Ag2O, MeCN, 96 h, rt; c: TBAF, AcOH, THF, 24 h; d: KOH, THF/H2O, 4 h, rt, 41% for 17, 34% for 7 (3 steps).
Scheme 6: Synthesis of (A) ZEN mimic glucoside 17 and (B) ZEN-16-β,D-glucoside (7); a: TIPS-Cl, imidazole, 16...
Additionally, TIPS protection was applied for the synthesis of ZEN-16-sulfate (8) using a procedure that was successfully applied for the synthesis of ZEN-14-sulfate (6) as described recently [20]. Reaction of 22 with the 2,2,2-trichloroethyl (TCE) protected sulfuryl imidazolium salt 23 [36,37] gave the protected sulfate 24. TIPS cleavage and subsequent TCE deprotection using Zn/ammonium formate (HCOONH4) yielded the desired product. Interestingly, when using the crude intermediate after TIPS deprotection without purification directly in the second step, we obtained the tetrabutylammonium salt of ZEN-16-sulfate (NBu4-8) in good yield (65% over 2 steps) as shown in Scheme 7. The stability of this compound was significantly increased compared to the corresponding sodium salt, which is of great importance in terms of preparation of reference material and long term stability of appropriate standard solutions for further investigations.
![[1860-5397-10-112-i7]](/bjoc/content/inline/1860-5397-10-112-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Chemical sulfation using the 2,2,2-trichloroethyl (TCE)-protected sulfuryl imidazolium salt 23 yielding ZEN-16-sulfate (8) as tetrabutylammonium salt; a: 23, 1,2-dimethylimidazole, CH2Cl2, 12 h, rt, 91%, b: TBAF, AcOH, THF, 3 h, −10 °C, c: Zn, HCOONH4, MeOH, 16 h, rt, 65% (2 steps).
Scheme 7: Chemical sulfation using the 2,2,2-trichloroethyl (TCE)-protected sulfuryl imidazolium salt 23 yiel...
Conclusion
In summary, different methods for the regioselective protection of resorcylic acid esters and lactones were investigated for subsequent regiocontrolled glucosylation and sulfation. Whereas acetyl and p-methoxybenzyl protection led to undesired products, TIPS-protected RALs were successfully used as intermediates for the preparation of corresponding glucosides and sulfates applying the Königs–Knorr glucosylation and chemical sulfation using TCE-protected sulfuryl imidazolium salt 23, respectively. These methods were used for the first chemical synthesis of the ZEN-16-conjugates 7 and 8 in reasonable amounts for ongoing research and further investigations in the field of conjugated/masked mycotoxins.
Supporting Information
| Supporting Information File 1: Experimental details (including remarks and general procedures), characterization data, copies of NMR spectra of new compounds, 2D NMR spectra of glucoside 7. | ||
| Format: PDF | Size: 1.5 MB | Download |
Acknowledgements
The Theodor Körner fund (Vienna, Austria), the Austrian Federal Ministry of Economy, Family and Youth as well as the National Foundation for Research, Technology and Development are gratefully acknowledged for financial support. GA and RK were supported by the Austrian Science Fund (FWF) special research project SFB Fusarium (F3706 and F3708).
References
-
Winssinger, N.; Barluenga, S. Chem. Commun. 2007, 22–36. doi:10.1039/b610344h
Return to citation in text: [1] -
European legislation (EC 1881/2006). Commission Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuff – consolidated version 03.12.2012. European Community (EC): Brussels, Belgium. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:2006R1881:20121203:EN:PDF (accessed April 2, 2014).
Return to citation in text: [1] -
European commission recommendation (2006/576/EC) on certain mycotoxins in animal feed – 17.08.2006.. European Community (EC): Brussels (Belgium). http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:32006H0576 (accessed April 2, 2014).
Return to citation in text: [1] -
Creppy, E. E. Toxicol. Lett. 2002, 127, 19–28. doi:10.1016/S0378-4274(01)00479-9
Return to citation in text: [1] -
Sudakin, D. L. Toxicol. Lett. 2003, 143, 97–107. doi:10.1016/S0378-4274(03)00116-4
Return to citation in text: [1] -
Krska, R.; Josephs, R. Fresenius' J. Anal. Chem. 2001, 369, 469–476. doi:10.1007/s002160100715
Return to citation in text: [1] -
Tiemann, U.; Tomek, W.; Schneider, F.; Vanselow, J. Reprod. Toxicol. 2003, 17, 673–681. doi:10.1016/j.reprotox.2003.07.001
Return to citation in text: [1] -
Miksicek, R. J. J. Steroid Biochem. Mol. Biol. 1994, 49, 153–160. doi:10.1016/0960-0760(94)90005-1
Return to citation in text: [1] -
Shier, W. T.; Shier, A. C.; Xie, W.; Mirocha, C. J. Toxicon 2001, 39, 1435–1438. doi:10.1016/S0041-0101(00)00259-2
Return to citation in text: [1] -
Berthiller, F.; Schuhmacher, R.; Adam, G.; Krska, R. Anal. Bioanal. Chem. 2009, 395, 1243–1252. doi:10.1007/s00216-009-2874-x
Return to citation in text: [1] -
Tran, S. T.; Smith, T. K. Anim. Feed Sci. Technol. 2011, 163, 84–92. doi:10.1016/j.anifeedsci.2010.10.008
Return to citation in text: [1] -
Berthiller, F.; Crews, C.; Dall'Asta, C.; De Saeger, S.; Haesaert, G.; Karlovsky, P.; Oswald, I. P.; Seefelder, W.; Speijers, G.; Stroka, J. Mol. Nutr. Food Res. 2013, 57, 165–186. doi:10.1002/mnfr.201100764
Return to citation in text: [1] [2] -
Schneweis, I.; Meyer, K.; Engelhardt, G.; Bauer, J. J. Agric. Food Chem. 2002, 50, 1736–1738. doi:10.1021/jf010802t
Return to citation in text: [1] -
Grabley, S.; Gareis, M.; Böckers, W.; Thiem, J. Synthesis 1992, 1078–1080. doi:10.1055/s-1992-26306
Return to citation in text: [1] -
Plasencia, J.; Mirocha, C. J. Appl. Environ. Microbiol. 1991, 57, 146–150.
Return to citation in text: [1] -
Berthiller, F.; Werner, U.; Sulyok, M.; Krska, R.; Hauser, M.-T.; Schuhmacher, R. Food Addit. Contam. 2006, 23, 1194–1200. doi:10.1080/02652030600778728
Return to citation in text: [1] -
Kovalsky Paris, M. P.; Schweiger, W.; Hametner, C.; Stückler, R.; Muehlbauer, G. J.; Varga, E.; Krska, R.; Berthiller, F.; Adam, G. J. Agric. Food Chem. 2014, 62, 1181–1189. doi:10.1021/jf405627d
Return to citation in text: [1] -
Mikula, H.; Hametner, C.; Berthiller, E.; Warth, B.; Krska, R.; Adam, G.; Fröhlich, J. World Mycotoxin J. 2012, 5, 289–296. doi:10.3920/WMJ2012.1404
Return to citation in text: [1] -
Mikula, H.; Weber, J.; Lexmüller, S.; Bichl, G.; Schwartz, H.; Varga, E.; Berthiller, F.; Hametner, C.; Krska, R.; Fröhlich, J. Carbohydr. Res. 2013, 373, 59–63. doi:10.1016/j.carres.2013.03.002
Return to citation in text: [1] -
Mikula, H.; Sohr, B.; Skrinjar, P.; Weber, J.; Hametner, C.; Berthiller, F.; Krska, R.; Adam, G.; Fröhlich, J. Tetrahedron Lett. 2013, 54, 3290–3293. doi:10.1016/j.tetlet.2013.04.059
Return to citation in text: [1] [2] -
Bunyapaiboonsri, T.; Yoiprommarat, S.; Khonsanit, A.; Komwijit, S. J. Nat. Prod. 2008, 71, 891–894. doi:10.1021/np070689m
Return to citation in text: [1] -
Paraschos, S.; Magiatis, P.; Kalpoutzakis, E.; Harvala, C.; Skaltsounis, A.-L. J. Nat. Prod. 2001, 64, 1585–1587. doi:10.1021/np0103665
Return to citation in text: [1] -
Zidorn, C.; Lohwasser, U.; Pschorr, S.; Salvenmoser, D.; Ongania, K.-H.; Ellmerer, E. P.; Börner, A.; Stuppner, H. Phytochemistry 2005, 66, 1691–1697. doi:10.1016/j.phytochem.2005.05.004
Return to citation in text: [1] -
Xu, M.; Zhang, M.; Wang, D.; Yang, C.-R.; Zhang, Y.-J. Chem. Biodiversity 2011, 8, 1891–1900. doi:10.1002/cbdv.201000220
Return to citation in text: [1] -
Morikawa, T.; Wang, L.-B.; Ninomiya, K.; Nakamura, S.; Matsuda, H.; Muraoka, O.; Wu, L.-J.; Yoshikawa, M. Chem. Pharm. Bull. 2009, 57, 853–859. doi:10.1248/cpb.57.853
Return to citation in text: [1] -
Aamer, S. Chin. J. Chem. 2005, 23, 762–766. doi:10.1002/cjoc.200590762
Return to citation in text: [1] [2] -
Versilovskis, A.; Huybrecht, B.; Tangni, E. K.; Pussemier, L.; De Saeger, S.; Callebaut, A. Food Addit. Contam. 2011, 28, 1687–1693.
Return to citation in text: [1] -
Muñoz, L.; Castro, J. L.; Cardelle, M.; Castedo, L.; Riguera, R. Phytochemistry 1989, 28, 83–85. doi:10.1016/0031-9422(89)85014-9
Return to citation in text: [1] -
Mikula, H.; Hametner, C.; Fröhlich, J. Synth. Commun. 2013, 43, 1939–1946. doi:10.1080/00397911.2012.681827
Return to citation in text: [1] -
Mydock, L. K.; Demchenko, A. V. Org. Biomol. Chem. 2010, 8, 497–510. doi:10.1039/b916088d
Return to citation in text: [1] -
Wegmann, B.; Schmidt, R. R. J. Carbohydr. Chem. 1987, 6, 357–375. doi:10.1080/07328308708057926
Return to citation in text: [1] -
Damager, I.; Olsen, C. E.; Møller, B. L.; Motawia, M. S. Carbohydr. Res. 1999, 320, 19–30. doi:10.1016/S0008-6215(99)00131-7
Return to citation in text: [1] -
Motawia, M. S.; Olsen, C. E.; Enevoldsen, K.; Marcussen, J.; Møller, B. L. Carbohydr. Res. 1995, 277, 109–123. doi:10.1016/0008-6215(95)00203-6
Return to citation in text: [1] -
Motawia, M. S.; Olsen, C. E.; Møller, B. L.; Marcussen, J. Carbohydr. Res. 1994, 252, 69–84. doi:10.1016/0008-6215(94)90006-X
Return to citation in text: [1] -
Green, T. W.; Wuts, P. G. W. Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: New York, 1999; pp 269–270. doi:10.1002/0471220574
Return to citation in text: [1] -
Ingram, L. J.; Desoky, A.; Ali, A. M.; Taylor, S. D. J. Org. Chem. 2009, 74, 6479–6485. doi:10.1021/jo9014112
Return to citation in text: [1] -
Liu, Y.; Lien, I.-F. F.; Ruttgaizer, S.; Dove, P.; Taylor, S. D. Org. Lett. 2004, 6, 209–212. doi:10.1021/ol036157o
Return to citation in text: [1]
| 7. | Tiemann, U.; Tomek, W.; Schneider, F.; Vanselow, J. Reprod. Toxicol. 2003, 17, 673–681. doi:10.1016/j.reprotox.2003.07.001 |
| 17. | Kovalsky Paris, M. P.; Schweiger, W.; Hametner, C.; Stückler, R.; Muehlbauer, G. J.; Varga, E.; Krska, R.; Berthiller, F.; Adam, G. J. Agric. Food Chem. 2014, 62, 1181–1189. doi:10.1021/jf405627d |
| 6. | Krska, R.; Josephs, R. Fresenius' J. Anal. Chem. 2001, 369, 469–476. doi:10.1007/s002160100715 |
| 18. | Mikula, H.; Hametner, C.; Berthiller, E.; Warth, B.; Krska, R.; Adam, G.; Fröhlich, J. World Mycotoxin J. 2012, 5, 289–296. doi:10.3920/WMJ2012.1404 |
| 19. | Mikula, H.; Weber, J.; Lexmüller, S.; Bichl, G.; Schwartz, H.; Varga, E.; Berthiller, F.; Hametner, C.; Krska, R.; Fröhlich, J. Carbohydr. Res. 2013, 373, 59–63. doi:10.1016/j.carres.2013.03.002 |
| 20. | Mikula, H.; Sohr, B.; Skrinjar, P.; Weber, J.; Hametner, C.; Berthiller, F.; Krska, R.; Adam, G.; Fröhlich, J. Tetrahedron Lett. 2013, 54, 3290–3293. doi:10.1016/j.tetlet.2013.04.059 |
| 4. | Creppy, E. E. Toxicol. Lett. 2002, 127, 19–28. doi:10.1016/S0378-4274(01)00479-9 |
| 5. | Sudakin, D. L. Toxicol. Lett. 2003, 143, 97–107. doi:10.1016/S0378-4274(03)00116-4 |
| 16. | Berthiller, F.; Werner, U.; Sulyok, M.; Krska, R.; Hauser, M.-T.; Schuhmacher, R. Food Addit. Contam. 2006, 23, 1194–1200. doi:10.1080/02652030600778728 |
| 2. | European legislation (EC 1881/2006). Commission Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuff – consolidated version 03.12.2012. European Community (EC): Brussels, Belgium. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:2006R1881:20121203:EN:PDF (accessed April 2, 2014). |
| 3. | European commission recommendation (2006/576/EC) on certain mycotoxins in animal feed – 17.08.2006.. European Community (EC): Brussels (Belgium). http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:32006H0576 (accessed April 2, 2014). |
| 12. | Berthiller, F.; Crews, C.; Dall'Asta, C.; De Saeger, S.; Haesaert, G.; Karlovsky, P.; Oswald, I. P.; Seefelder, W.; Speijers, G.; Stroka, J. Mol. Nutr. Food Res. 2013, 57, 165–186. doi:10.1002/mnfr.201100764 |
| 12. | Berthiller, F.; Crews, C.; Dall'Asta, C.; De Saeger, S.; Haesaert, G.; Karlovsky, P.; Oswald, I. P.; Seefelder, W.; Speijers, G.; Stroka, J. Mol. Nutr. Food Res. 2013, 57, 165–186. doi:10.1002/mnfr.201100764 |
| 14. | Grabley, S.; Gareis, M.; Böckers, W.; Thiem, J. Synthesis 1992, 1078–1080. doi:10.1055/s-1992-26306 |
| 10. | Berthiller, F.; Schuhmacher, R.; Adam, G.; Krska, R. Anal. Bioanal. Chem. 2009, 395, 1243–1252. doi:10.1007/s00216-009-2874-x |
| 11. | Tran, S. T.; Smith, T. K. Anim. Feed Sci. Technol. 2011, 163, 84–92. doi:10.1016/j.anifeedsci.2010.10.008 |
| 9. | Shier, W. T.; Shier, A. C.; Xie, W.; Mirocha, C. J. Toxicon 2001, 39, 1435–1438. doi:10.1016/S0041-0101(00)00259-2 |
| 8. | Miksicek, R. J. J. Steroid Biochem. Mol. Biol. 1994, 49, 153–160. doi:10.1016/0960-0760(94)90005-1 |
| 13. | Schneweis, I.; Meyer, K.; Engelhardt, G.; Bauer, J. J. Agric. Food Chem. 2002, 50, 1736–1738. doi:10.1021/jf010802t |
| 27. | Versilovskis, A.; Huybrecht, B.; Tangni, E. K.; Pussemier, L.; De Saeger, S.; Callebaut, A. Food Addit. Contam. 2011, 28, 1687–1693. |
| 21. | Bunyapaiboonsri, T.; Yoiprommarat, S.; Khonsanit, A.; Komwijit, S. J. Nat. Prod. 2008, 71, 891–894. doi:10.1021/np070689m |
| 22. | Paraschos, S.; Magiatis, P.; Kalpoutzakis, E.; Harvala, C.; Skaltsounis, A.-L. J. Nat. Prod. 2001, 64, 1585–1587. doi:10.1021/np0103665 |
| 23. | Zidorn, C.; Lohwasser, U.; Pschorr, S.; Salvenmoser, D.; Ongania, K.-H.; Ellmerer, E. P.; Börner, A.; Stuppner, H. Phytochemistry 2005, 66, 1691–1697. doi:10.1016/j.phytochem.2005.05.004 |
| 24. | Xu, M.; Zhang, M.; Wang, D.; Yang, C.-R.; Zhang, Y.-J. Chem. Biodiversity 2011, 8, 1891–1900. doi:10.1002/cbdv.201000220 |
| 25. | Morikawa, T.; Wang, L.-B.; Ninomiya, K.; Nakamura, S.; Matsuda, H.; Muraoka, O.; Wu, L.-J.; Yoshikawa, M. Chem. Pharm. Bull. 2009, 57, 853–859. doi:10.1248/cpb.57.853 |
| 20. | Mikula, H.; Sohr, B.; Skrinjar, P.; Weber, J.; Hametner, C.; Berthiller, F.; Krska, R.; Adam, G.; Fröhlich, J. Tetrahedron Lett. 2013, 54, 3290–3293. doi:10.1016/j.tetlet.2013.04.059 |
| 36. | Ingram, L. J.; Desoky, A.; Ali, A. M.; Taylor, S. D. J. Org. Chem. 2009, 74, 6479–6485. doi:10.1021/jo9014112 |
| 37. | Liu, Y.; Lien, I.-F. F.; Ruttgaizer, S.; Dove, P.; Taylor, S. D. Org. Lett. 2004, 6, 209–212. doi:10.1021/ol036157o |
| 31. | Wegmann, B.; Schmidt, R. R. J. Carbohydr. Chem. 1987, 6, 357–375. doi:10.1080/07328308708057926 |
| 32. | Damager, I.; Olsen, C. E.; Møller, B. L.; Motawia, M. S. Carbohydr. Res. 1999, 320, 19–30. doi:10.1016/S0008-6215(99)00131-7 |
| 33. | Motawia, M. S.; Olsen, C. E.; Enevoldsen, K.; Marcussen, J.; Møller, B. L. Carbohydr. Res. 1995, 277, 109–123. doi:10.1016/0008-6215(95)00203-6 |
| 34. | Motawia, M. S.; Olsen, C. E.; Møller, B. L.; Marcussen, J. Carbohydr. Res. 1994, 252, 69–84. doi:10.1016/0008-6215(94)90006-X |
| 35. | Green, T. W.; Wuts, P. G. W. Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: New York, 1999; pp 269–270. doi:10.1002/0471220574 |
| 30. | Mydock, L. K.; Demchenko, A. V. Org. Biomol. Chem. 2010, 8, 497–510. doi:10.1039/b916088d |
| 28. | Muñoz, L.; Castro, J. L.; Cardelle, M.; Castedo, L.; Riguera, R. Phytochemistry 1989, 28, 83–85. doi:10.1016/0031-9422(89)85014-9 |
| 29. | Mikula, H.; Hametner, C.; Fröhlich, J. Synth. Commun. 2013, 43, 1939–1946. doi:10.1080/00397911.2012.681827 |
© 2014 Mikula et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)