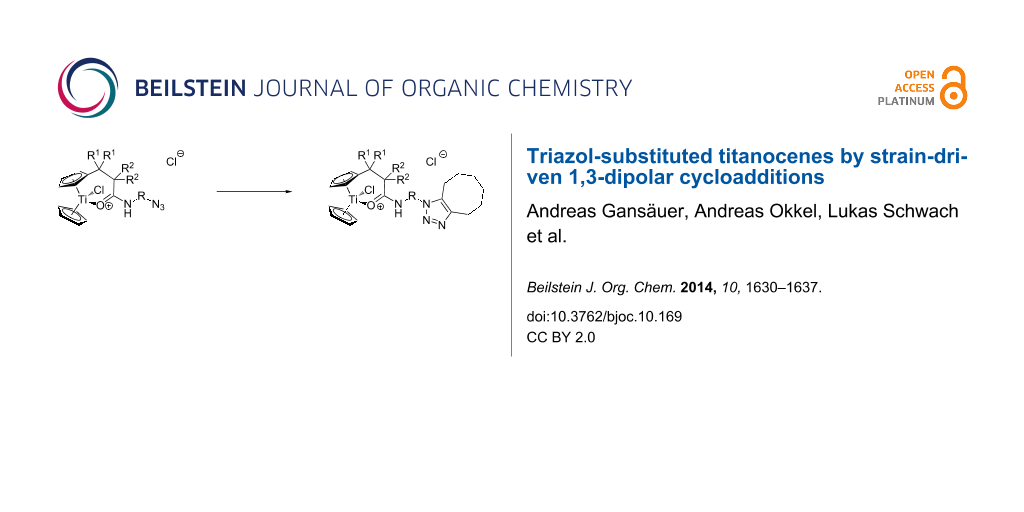
Abstract
An operationally simple, convenient, and mild strategy for the synthesis of triazole-substituted titanocenes via strain-driven 1,3-dipolar cycloadditions between azide-functionalized titanocenes and cyclooctyne has been developed. It features the first synthesis of titanocenes containing azide groups. These compounds constitute ‘second-generation’ functionalized titanocene building blocks for further synthetic elaboration. Our synthesis is modular and large numbers of the complexes can in principle be prepared in short periods of time. Some of the triazole-substituted titanocenes display high cyctotoxic activity against BJAB cells. Comparison of the most active complexes allows the identification of structural features essential for biological activity.

Graphical Abstract
Introduction
Group 4 metallocenes and derivatives of Cp2TiCl2, in particular, continue to be in the focus of contemporary research as a promising class of cytotoxic compounds [1-10], as efficient reagents and catalysts [11-16], as organometallic gelators [17-20], and in their own right [21,22]. In order to further investigate and improve these properties it is mandatory to access as yet unexplored functional titanocenes. This is most easily achieved with modular, efficient, and general strategies for the synthesis of these complexes. Classical approaches with metalation at the end of the sequence usually do not meet these requirements. This is because introduction of functional groups is difficult due to the nucleophilicity of the cyclopentadienyl anions before metalation and the electrophilicity of titanium after metalation [21,22].
We have devised a conceptually different approach addressing these issues. It relies on the use of carboxylate-containing titanocene building blocks [23-25]. From these compounds the corresponding acid chlorides can be prepared by addition of SOCl2. The acid chloride group is more electrophilic than the [TiCl2] fragment and therefore many titanocenes containing ligands with pending amide, ester, and ketone groups can be prepared with classical organic acylation reactions (Friedel–Crafts reaction, esterification, amide synthesis, Scheme 1). Some of these complexes have been used as organometallic gelators [19,20], as a novel class of cytostatic compounds [26], and catalysts for unusual radical cyclizations [27-31]. The ketone and amide substituted catalysts are cationic due to the intramolecular coordination of the carbonyl group [30,31]. This feature is essential for the cytostatic and catalytic activity. Moreover, in the amide complexes hydrogen bonding of the N–H bond to chloride or [ZnCl4]2− is a crucial structural feature [31]. The gelation ability of the neutral ester-substituted titanocenes critically depends on the steric demand of the substituents on the cyclopentadienyl ligands. The carboxylates are valuable complexes for mediating highly chemoselective Barbier type allylations [32,33].
![[1860-5397-10-169-i1]](/bjoc/content/inline/1860-5397-10-169-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Modular titanocene synthesis via acylation reactions [24].
Scheme 1: Modular titanocene synthesis via acylation reactions [24].
These findings demonstrate that the properties of our functional titanocenes critically depend on both the direct environment of the Ti center and the periphery of the complex. Therefore, it is desirable to develop novel entries to titanocenes with even higher structural and functional diversity to improve the observed functions. An especially attractive approach is the use of already functionalized building blocks as starting materials in diversity oriented synthesis that increases the molecular complexity. Any synthetic methodology used in this context must take into account the sensitivity of the titanocene towards nucleophiles.
We decided to address these issues by employing cationic amide-substituted titanocenes as building blocks and strain-driven 1,3-dipolar cycloadditions [34-40] as synthetic methodology for the preparation of such ‘second-generation’ functional titanocenes. This line of action seemed especially appealing for two reasons. First, the cationic amide titanocenes have already displayed interesting activity and therefore serve as our lead structures. Second, the strain-driven 1,3-dipolar cycloadditions have evolved as extremely mild reactions for the functionalization of complex molecules. Since no metal complexes are required to catalyze the 1,3-dipolar cycloaddition [41-43], the reaction can be even used for the functionalization of biomolecules in living systems and has therefore been called bioorthogonal [34,35].
To the best of our knowledge, no examples of the Ti-containing substrates for our strategy, i.e., azide-functionalized titanocenes, have been reported in the literature. One aspect of our study is to establish if such complexes are stable and readily available in high yield. It should be noted that only a single example of a Cu-catalyzed 1,3-dipolar cycloaddition with an alkyne-functionalized titanocene has been described [44]. Therefore, the properties of triazol-substituted titanocenes, the products of the 1,3-dipolar cycloaddition, are also largely unexplored.
Results and Discussion
Synthesis of the titanocenes
Preparation of the starting materials
We started our investigation with the preparation of azide-substituted cationic titanocenes. To this end, the titanocene carboxylates 1–3 shown in Figure 1 were employed as substrates because their substitution pattern should allow a first simple assessment of structure–activity relationships.
![[1860-5397-10-169-1]](/bjoc/content/figures/1860-5397-10-169-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Carboxylates employed as titanocene starting materials for azide-substituted complexes.
Figure 1: Carboxylates employed as titanocene starting materials for azide-substituted complexes.
The compounds A–D shown in Figure 2 were used as amino-substituted azides. They are readily obtained from the corresponding diazides through a Staudinger reaction (see Supporting Information File 1 for details) [45,46]. As for the carboxylates the different tether lengths and substitution patterns of the arene allow to study the effect of substitution on the activity of the complexes. The ether tether in B serves as a model for PEG.
![[1860-5397-10-169-2]](/bjoc/content/figures/1860-5397-10-169-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Azides employed in this study and conditions for their synthesis.
Figure 2: Azides employed in this study and conditions for their synthesis.
The titanocene carboxylates 1–3 were transformed into the corresponding acid chlorides and then reacted with amino azides A–D in the presence of NaH without purification of the acid chlorides. Typical results are summarized in Table 1.
Table 1: Synthesis of cationic titanocenes containing azides (yield over two steps, see Supporting Information File 1 for details).
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-169-i2.svg?max-width=637&scale=1.0)
|
||
| substrates | product | yield/[%] |
|---|---|---|
| 1, A |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-169-i3.svg?max-width=637&scale=1.0)
4 |
78 |
| 1, B |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-169-i4.svg?max-width=637&scale=1.0)
5 |
51 |
| 1, C |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-169-i5.svg?max-width=637&scale=1.0)
6 |
89 |
| 2, A |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-169-i6.svg?max-width=637&scale=1.0)
7 |
89 |
| 2, C |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-169-i7.svg?max-width=637&scale=1.0)
8 |
63 |
| 2, D |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-169-i8.svg?max-width=637&scale=1.0)
9 |
71 |
| 3, B |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-169-i9.svg?max-width=637&scale=1.0)
10 |
51 |
| 3, D |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-169-i10.svg?max-width=637&scale=1.0)
11 |
31 |
Gratifyingly, the acylation reactions proceed without problems and the azide-functionalized titanocenes can generally be obtained in good yields after 16 h. The somewhat lower yields obtained with carboxylate 3 are probably due to an increased bulkiness of the ligand’s substituents. It should be noted that polyether groups can be readily incorporated into cationic titanocenes. This suggests that the cationic titanocenes can be readily immobilized by covalent binding to PEG.
In general, our results clearly demonstrate that the azide group is compatible with cationic titanocenes. Moreover, it is obvious that large libraries of such titanocenes can be accessed from our carboxylates in short periods of time.
Strain-driven 1,3-dipolar cycloadditions
With the new titanocene building blocks in hand we turned our attention to their further functionalization through the strain-driven 1,3-dipolar cycloaddition with cyclooctyne. The original conditions of Wittig [36], the reaction of cyclooctyne with phenyl azide, and the numerous applications pioneered by Bertozzi suggest that the reaction proceeds under mild conditions [34,35,37-40]. Therefore, we simply mixed the titanocenes and cyclooctyne in CH2Cl2 at room temperature. The concentration of the substrates was intentionally kept low (0.1 M) to avoid a too intense evolution of heat. Typical examples of the reaction are summarized in Table 2.
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-169-i11.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-169-i12.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-169-i13.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-169-i14.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-169-i15.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-169-i16.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-169-i17.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-169-i18.svg?max-width=637&scale=1.0)
The results demonstrate that the strain-driven 1,3-dipolar cycloaddition is a convenient and very mild route to triazole-functionalized cationic titanocenes. The yields between 75% and 92% are satisfying. It should be noted that the polyether-substituted complexes 13 and 17 are obtained in high yield. This opens further interesting perspectives for the immobilization of titanocene complexes.
Cytotoxicity studies
One of the pertinent features of titanocenes is their cytotoxicity [1-5]. Therefore, we investigated this particular property of our novel complexes. To guarantee comparability with a previous study [26] we discuss our results of the lymphoma cell line BJAB. Cell surface transmembrane receptor CD95, through which apoptosis can be induced, is expressed by BJAB cells. Cell death can be induced in these cells both by the extrinsic and the intrinsic apoptosis-signalling pathway [47-49]. Therefore, BJAB cells are well-suited for studying the induction of apoptosis by our cationic titanocenes [50-53]. It is logical to study apoptosis induction, expressed as AC50 values, instead of nonspecific cytotoxicity, which is usually reported as LC50 values, because cytotoxic drugs operate by specific induction of apoptosis. So we determined the AC50 values of our titanocenes, i.e., the concentrations causing specific apoptosis in 50% of lymphoma cells, counting all cells with membrane damage.
The azide-substituted complexes showed no significant apoptosis induction (AC50 > 100 µM). Introduction of the triazole ring through 1,3-dipolar cycloaddition markedly changes the activity of our titanocenes as a function of the substitution pattern. The most active complexes are highlighted in Figure 3.
![[1860-5397-10-169-3]](/bjoc/content/figures/1860-5397-10-169-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Most active titanocenes of this study and their AC50 values.
Figure 3: Most active titanocenes of this study and their AC50 values.
Gratifyingly, 18, together with a ketone-substituted titanocene, displays the highest activity against the BJAB cell line of our cationic carbonyl-substituted titanocenes. Comparison of the three most active complexes also allows the identification of structural features essential for cytotoxic activity. First, a bulky substitution of the cyclopentadienyl ligand is favorable. Second, positioning of the triazol in close proximity – ortho-substitution in 16 leads to a lower AC50 value than para-substitution in 15 – of the metal center enhances the biological activity.
Conclusion
In summary, we have devised an operationally simple, convenient, and mild strategy for the synthesis of triazole substituted titanocenes via strain-driven 1,3-dipolar cycloadditions between azide-functionalized titanocenes and cyclooctyne. It features the first synthesis of titanocenes containing azide groups. These compounds constitute functionalized ‘second-generation’ titanocene building blocks for further synthetic elaboration. Our synthesis is modular and large numbers of the complexes can in principle be prepared in short periods of time. Some of the triazole-substituted titanocenes display high cyctotoxic activity against BJAB cells. Comparison of the most active complexes allows the identification of structural features essential for biological activity.
Future studies will concentrate on the preparation of neutral and of enantiomerically pure triazole-substituted titanocenes, also via metal catalyzed cycloadditions, for further increasing the biological activity of the complexes and for applications in enantioselective [54,55] cyclizations [56-58].
Supporting Information
| Supporting Information File 1: Experimental procedures and compound characterization, cytotoxicity studies. | ||
| Format: PDF | Size: 487.5 KB | Download |
References
-
Buettner, K. M.; Valentine, A. M. Chem. Rev. 2012, 112, 1863–1881. doi:10.1021/cr1002886
Return to citation in text: [1] [2] -
Hartinger, C. G.; Metzler-Nolte, N.; Dyson, P. J. Organometallics 2012, 31, 5677–5685. doi:10.1021/om300373t
Return to citation in text: [1] [2] -
Tshuva, E. Y.; Ashenhurst, J. A. Eur. J. Inorg. Chem. 2009, 2203–2218. doi:10.1002/ejic.200900198
Return to citation in text: [1] [2] -
Strohfeldt, K.; Tacke, M. Chem. Soc. Rev. 2008, 37, 1174–1187. doi:10.1039/b707310k
Return to citation in text: [1] [2] -
Köpf-Maier, P.; Köpf, H. Chem. Rev. 1987, 87, 1137–1152. doi:10.1021/cr00081a012
Return to citation in text: [1] [2] -
Sirignano, E.; Saturnino, C.; Botta, A.; Sinicropi, M. S.; Caruso, A.; Pisano, A.; Lappano, R.; Maggiolini, M.; Longo, P. Bioorg. Med. Chem. Lett. 2013, 23, 3458–3462. doi:10.1016/j.bmcl.2013.03.059
Return to citation in text: [1] -
Schur, J.; Manna, C. M.; Deally, A.; Köster, R. W.; Tacke, M.; Tshuva, E. Y.; Ott, I. Chem. Commun. 2013, 49, 4785–4787. doi:10.1039/c3cc38604j
Return to citation in text: [1] -
Kater, L.; Claffey, J.; Hogan, M.; Jesse, P.; Kater, B.; Strauss, S.; Tacke, M.; Prokop, A. Toxicol. in Vitro 2012, 26, 119–124. doi:10.1016/j.tiv.2011.09.010
Return to citation in text: [1] -
González-Pantoja, J. F.; Stern, M.; Jarzecki, A. A.; Royo, E.; Robles-Escajeda, E.; Varela-Ramírez, A.; Aguilera, R. J.; Contel, M. Inorg. Chem. 2011, 50, 11099–11110. doi:10.1021/ic201647h
Return to citation in text: [1] -
Gao, L. M.; Vera, J. L.; Matt, J.; Meléndez, E. J. Biol. Inorg. Chem. 2010, 15, 851–859. doi:10.1007/s00775-010-0649-7
Return to citation in text: [1] -
Carter, M. B.; Schiøtt, B.; Gutiérrez, A.; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 11667–11670. doi:10.1021/ja00105a006
Return to citation in text: [1] -
Pohlki, F.; Doye, S. Angew. Chem., Int. Ed. 2001, 40, 2305–2308. doi:10.1002/1521-3773(20010618)40:12<2305::AID-ANIE2305>3.0.CO;2-7
Return to citation in text: [1] -
Cuerva, J. M.; Justicia, J.; Oller-López, J. L.; Oltra, J. E. Top. Curr. Chem. 2006, 264, 63–91. doi:10.1007/128_025
Return to citation in text: [1] -
Gansäuer, A.; Justicia, J.; Fan, C.-A.; Worgull, D.; Piestert, F. Top. Curr. Chem. 2007, 279, 25–52. doi:10.1007/128_2007_130
Return to citation in text: [1] -
Gansäuer, A.; Fleckhaus, A.; Lafont, M. A.; Okkel, A.; Kotsis, K.; Anoop, A.; Neese, F. J. Am. Chem. Soc. 2009, 131, 16989–16999. doi:10.1021/ja907817y
Return to citation in text: [1] -
Streuff, J.; Feurer, M.; Bichovski, P.; Frey, G.; Gellrich, U. Angew. Chem., Int. Ed. 2012, 51, 8661–8664. doi:10.1002/anie.201204469
Return to citation in text: [1] -
Dastidar, P. Chem. Soc. Rev. 2008, 37, 2699–2715. doi:10.1039/b807346e
Return to citation in text: [1] -
Piepenbrok, M.-O. M.; Lloyd, G. O.; Clarke, N.; Steed, J. W. Chem. Rev. 2010, 110, 1960–2004. doi:10.1021/cr9003067
Return to citation in text: [1] -
Klawonn, T.; Gansäuer, A.; Winkler, I.; Lauterbach, T.; Franke, D.; Nolte, R. J. M.; Feiters, M. C.; Börner, H.; Hentschel, J.; Dötz, K. H. Chem. Commun. 2007, 1894–1895. doi:10.1039/b701565h
Return to citation in text: [1] [2] -
Gansäuer, A.; Winkler, I.; Klawonn, T.; Nolte, R. J. M.; Feiters, M. C.; Börner, H. G.; Hentschel, J.; Dötz, K. H. Organometallics 2009, 28, 1377–1382. doi:10.1021/om801022c
Return to citation in text: [1] [2] -
Halterman, R. L. Chem. Rev. 1992, 92, 965–994. doi:10.1021/cr00013a011
Return to citation in text: [1] [2] -
Qian, Y.; Huang, J.; Bala, M. D.; Lian, B.; Zhang, H.; Zhang, H. Chem. Rev. 2003, 103, 2633–2690. doi:10.1021/cr020002x
Return to citation in text: [1] [2] -
Gansäuer, A.; Franke, D.; Lauterbach, T.; Nieger, M. J. Am. Chem. Soc. 2005, 127, 11622–11623. doi:10.1021/ja054185r
Return to citation in text: [1] -
Gansäuer, A.; Winkler, I.; Worgull, D.; Franke, D.; Lauterbach, T.; Okkel, A.; Nieger, M. Organometallics 2008, 27, 5699–5707. doi:10.1021/om800700c
Return to citation in text: [1] [2] -
Gansäuer, A.; Okkel, A.; Worgull, D.; Schnakenburg, G. Organometallics 2010, 29, 3227–3230. doi:10.1021/om100245r
Return to citation in text: [1] -
Gansäuer, A.; Winkler, I.; Worgull, D.; Lauterbach, T.; Franke, D.; Selig, A.; Wagner, L.; Prokop, A. Chem.–Eur. J. 2008, 14, 4160–4163. doi:10.1002/chem.200800407
Return to citation in text: [1] [2] -
Gansäuer, A.; Lauterbach, T.; Geich-Gimbel, D. Chem.–Eur. J. 2004, 10, 4983–4990. doi:10.1002/chem.200400685
Return to citation in text: [1] -
Friedrich, J.; Dolg, M.; Gansäuer, A.; Geich-Gimbel, D.; Lauterbach, T. J. Am. Chem. Soc. 2005, 127, 7071–7077. doi:10.1021/ja050268w
Return to citation in text: [1] -
Friedrich, J.; Walczak, K.; Dolg, M.; Piestert, F.; Lauterbach, T.; Worgull, D.; Gansäuer, A. J. Am. Chem. Soc. 2008, 130, 1788–1796. doi:10.1021/ja077596b
Return to citation in text: [1] -
Gansäuer, A.; Worgull, D.; Knebel, K.; Huth, I.; Schnakenburg, G. Angew. Chem., Int. Ed. 2009, 48, 8882–8885. doi:10.1002/anie.200904428
Return to citation in text: [1] [2] -
Gansäuer, A.; Knebel, K.; Kube, C.; van Gastel, M.; Cangönül, A.; Daasbjerg, K.; Hangele, T.; Hülsen, M.; Dolg, M.; Friedrich, J. Chem.–Eur. J. 2012, 18, 2591–2599. doi:10.1002/chem.201102959
Return to citation in text: [1] [2] [3] -
Jiménez, T.; Morcillo, S. P.; Martín-Lasanta, A.; Collado-Sanz, D.; Cárdenas, D. J.; Gansäuer, A.; Justicia, J.; Cuerva, J. M. Chem.–Eur. J. 2012, 18, 12825–12833. doi:10.1002/chem.201201534
Return to citation in text: [1] -
Morcillo, S. P.; Martinez-Peragón, Á.; Jakoby, V.; Mota, A. J.; Kube, C.; Justicia, J.; Cuerva, J. M.; Gansäuer, A. Chem. Commun. 2014, 50, 2211–2213. doi:10.1039/c3cc49230c
Return to citation in text: [1] -
Sletten, E. M.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. doi:10.1002/anie.200900942
Return to citation in text: [1] [2] [3] -
Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. doi:10.1039/b901970g
Return to citation in text: [1] [2] [3] -
Wittig, G.; Krebs, A. Chem. Ber. 1961, 94, 3260–3275. doi:10.1002/cber.19610941213
Return to citation in text: [1] [2] -
Agard, N. J.; Prescher, J. A.; Bertozzi, C. R. J. Am. Chem. Soc. 2004, 126, 15046–15047. doi:10.1021/ja044996f
Return to citation in text: [1] [2] -
Gordon, C. G.; Mackey, J. L.; Jewett, J. C.; Sletten, E. M.; Houk, K. N.; Bertozzi, C. R. J. Am. Chem. Soc. 2012, 134, 9199–9208. doi:10.1021/ja3000936
Return to citation in text: [1] [2] -
Hudak, J. E.; Barfield, R. M.; de Hart, G. W.; Grob, P.; Nogales, E.; Bertozzi, C. R.; Rabuka, D. Angew. Chem., Int. Ed. 2012, 51, 4161–4165. doi:10.1002/anie.201108130
Return to citation in text: [1] [2] -
de Almeida, G.; Sletten, E. M.; Nakamura, H.; Palaniappan, K. K.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2012, 51, 2443–2447. doi:10.1002/anie.201106325
Return to citation in text: [1] [2] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Return to citation in text: [1] -
Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s
Return to citation in text: [1] -
Zagerman, J.; Deally, A.; Metzler-Nolte, N.; Müller-Bunz, H.; Wallis, D.; Tacke, M. Polyhedron 2011, 30, 2387–2390. doi:10.1016/j.poly.2011.05.036
Return to citation in text: [1] -
Golobov, Y. M.; Kasukhin, L. F. Tetrahedron 1992, 48, 1353–1406. doi:10.1016/S0040-4020(01)92229-X
Return to citation in text: [1] -
Köhn, M.; Breinbauer, R. Angew. Chem., Int. Ed. 2004, 43, 3106–3116. doi:10.1002/anie.200401744
Return to citation in text: [1] -
Bantel, H.; Engels, I. H.; Völter, W.; Schulze-Osthoff, K.; Wesselborg, S. Cancer Res. 1999, 59, 2083–2090.
Return to citation in text: [1] -
Klener, P., Jr.; Anděra, L.; Klener, P.; Nečas, E.; Živný, J. Folia Biol. (Prague, Czech Repub.) 2006, 52, 34–44.
Return to citation in text: [1] -
Meiler, J.; Schuler, M. Curr. Drug Targets 2006, 7, 1361–1369. doi:10.2174/138945006778559175
Return to citation in text: [1] -
Wieder, T.; Prokop, A.; Bagci, B.; Essmann, F.; Bernicke, D.; Schulze-Osthoff, K.; Dörken, B.; Schmalz, H.-G.; Daniel, P. T.; Henze, G. Leukemia 2001, 15, 1735–1742. doi:10.1038/sj.leu.2402284
Return to citation in text: [1] -
Diller, R. A.; Riepl, H. M.; Rose, O.; Frias, C.; Henze, G.; Prokop, A. Planta Med. 2007, 73, 755–761. doi:10.1055/s-2007-981545
Return to citation in text: [1] -
Prokop, A.; Wrasidlo, W.; Lode, H.; Herold, R.; Lang, F.; Henze, G.; Dörken, B.; Wieder, T.; Daniel, P. T. Oncogene 2003, 22, 9107–9120. doi:10.1038/sj.onc.1207196
Return to citation in text: [1] -
Rudner, J.; Lepple-Wienhues, A.; Budach, W.; Berschauer, J.; Friedrich, B.; Wesselborg, S.; Schulze-Osthoff, K.; Belka, C. J. Cell Sci. 2001, 114, 4161–4172.
Return to citation in text: [1] -
Gansäuer, A.; Fan, C.-A.; Keller, F.; Karbaum, P. Chem.–Eur. J. 2007, 13, 8084–8090. doi:10.1002/chem.200701021
Return to citation in text: [1] -
Gansäuer, A.; Shi, L.; Otte, M. J. Am. Chem. Soc. 2010, 132, 11858–11859. doi:10.1021/ja105023y
Return to citation in text: [1] -
Gansäuer, A.; Pierobon, M. Synlett 2000, 1357–1359. doi:10.1055/s-2000-7133
Return to citation in text: [1] -
Gansäuer, A.; Pierobon, M.; Bluhm, H. Synthesis 2001, 2500–2520. doi:10.1055/s-2001-18713
Return to citation in text: [1] -
Barrero, A. F.; Quilez del Moral, J. F.; Sánchez, E. M.; Arteaga, J. F. Eur. J. Org. Chem. 2006, 1627–1641. doi:10.1002/ejoc.200500849
Return to citation in text: [1]
| 1. | Buettner, K. M.; Valentine, A. M. Chem. Rev. 2012, 112, 1863–1881. doi:10.1021/cr1002886 |
| 2. | Hartinger, C. G.; Metzler-Nolte, N.; Dyson, P. J. Organometallics 2012, 31, 5677–5685. doi:10.1021/om300373t |
| 3. | Tshuva, E. Y.; Ashenhurst, J. A. Eur. J. Inorg. Chem. 2009, 2203–2218. doi:10.1002/ejic.200900198 |
| 4. | Strohfeldt, K.; Tacke, M. Chem. Soc. Rev. 2008, 37, 1174–1187. doi:10.1039/b707310k |
| 5. | Köpf-Maier, P.; Köpf, H. Chem. Rev. 1987, 87, 1137–1152. doi:10.1021/cr00081a012 |
| 6. | Sirignano, E.; Saturnino, C.; Botta, A.; Sinicropi, M. S.; Caruso, A.; Pisano, A.; Lappano, R.; Maggiolini, M.; Longo, P. Bioorg. Med. Chem. Lett. 2013, 23, 3458–3462. doi:10.1016/j.bmcl.2013.03.059 |
| 7. | Schur, J.; Manna, C. M.; Deally, A.; Köster, R. W.; Tacke, M.; Tshuva, E. Y.; Ott, I. Chem. Commun. 2013, 49, 4785–4787. doi:10.1039/c3cc38604j |
| 8. | Kater, L.; Claffey, J.; Hogan, M.; Jesse, P.; Kater, B.; Strauss, S.; Tacke, M.; Prokop, A. Toxicol. in Vitro 2012, 26, 119–124. doi:10.1016/j.tiv.2011.09.010 |
| 9. | González-Pantoja, J. F.; Stern, M.; Jarzecki, A. A.; Royo, E.; Robles-Escajeda, E.; Varela-Ramírez, A.; Aguilera, R. J.; Contel, M. Inorg. Chem. 2011, 50, 11099–11110. doi:10.1021/ic201647h |
| 10. | Gao, L. M.; Vera, J. L.; Matt, J.; Meléndez, E. J. Biol. Inorg. Chem. 2010, 15, 851–859. doi:10.1007/s00775-010-0649-7 |
| 21. | Halterman, R. L. Chem. Rev. 1992, 92, 965–994. doi:10.1021/cr00013a011 |
| 22. | Qian, Y.; Huang, J.; Bala, M. D.; Lian, B.; Zhang, H.; Zhang, H. Chem. Rev. 2003, 103, 2633–2690. doi:10.1021/cr020002x |
| 41. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 42. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 43. | Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s |
| 21. | Halterman, R. L. Chem. Rev. 1992, 92, 965–994. doi:10.1021/cr00013a011 |
| 22. | Qian, Y.; Huang, J.; Bala, M. D.; Lian, B.; Zhang, H.; Zhang, H. Chem. Rev. 2003, 103, 2633–2690. doi:10.1021/cr020002x |
| 34. | Sletten, E. M.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. doi:10.1002/anie.200900942 |
| 35. | Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. doi:10.1039/b901970g |
| 17. | Dastidar, P. Chem. Soc. Rev. 2008, 37, 2699–2715. doi:10.1039/b807346e |
| 18. | Piepenbrok, M.-O. M.; Lloyd, G. O.; Clarke, N.; Steed, J. W. Chem. Rev. 2010, 110, 1960–2004. doi:10.1021/cr9003067 |
| 19. | Klawonn, T.; Gansäuer, A.; Winkler, I.; Lauterbach, T.; Franke, D.; Nolte, R. J. M.; Feiters, M. C.; Börner, H.; Hentschel, J.; Dötz, K. H. Chem. Commun. 2007, 1894–1895. doi:10.1039/b701565h |
| 20. | Gansäuer, A.; Winkler, I.; Klawonn, T.; Nolte, R. J. M.; Feiters, M. C.; Börner, H. G.; Hentschel, J.; Dötz, K. H. Organometallics 2009, 28, 1377–1382. doi:10.1021/om801022c |
| 24. | Gansäuer, A.; Winkler, I.; Worgull, D.; Franke, D.; Lauterbach, T.; Okkel, A.; Nieger, M. Organometallics 2008, 27, 5699–5707. doi:10.1021/om800700c |
| 11. | Carter, M. B.; Schiøtt, B.; Gutiérrez, A.; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 11667–11670. doi:10.1021/ja00105a006 |
| 12. | Pohlki, F.; Doye, S. Angew. Chem., Int. Ed. 2001, 40, 2305–2308. doi:10.1002/1521-3773(20010618)40:12<2305::AID-ANIE2305>3.0.CO;2-7 |
| 13. | Cuerva, J. M.; Justicia, J.; Oller-López, J. L.; Oltra, J. E. Top. Curr. Chem. 2006, 264, 63–91. doi:10.1007/128_025 |
| 14. | Gansäuer, A.; Justicia, J.; Fan, C.-A.; Worgull, D.; Piestert, F. Top. Curr. Chem. 2007, 279, 25–52. doi:10.1007/128_2007_130 |
| 15. | Gansäuer, A.; Fleckhaus, A.; Lafont, M. A.; Okkel, A.; Kotsis, K.; Anoop, A.; Neese, F. J. Am. Chem. Soc. 2009, 131, 16989–16999. doi:10.1021/ja907817y |
| 16. | Streuff, J.; Feurer, M.; Bichovski, P.; Frey, G.; Gellrich, U. Angew. Chem., Int. Ed. 2012, 51, 8661–8664. doi:10.1002/anie.201204469 |
| 34. | Sletten, E. M.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. doi:10.1002/anie.200900942 |
| 35. | Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. doi:10.1039/b901970g |
| 36. | Wittig, G.; Krebs, A. Chem. Ber. 1961, 94, 3260–3275. doi:10.1002/cber.19610941213 |
| 37. | Agard, N. J.; Prescher, J. A.; Bertozzi, C. R. J. Am. Chem. Soc. 2004, 126, 15046–15047. doi:10.1021/ja044996f |
| 38. | Gordon, C. G.; Mackey, J. L.; Jewett, J. C.; Sletten, E. M.; Houk, K. N.; Bertozzi, C. R. J. Am. Chem. Soc. 2012, 134, 9199–9208. doi:10.1021/ja3000936 |
| 39. | Hudak, J. E.; Barfield, R. M.; de Hart, G. W.; Grob, P.; Nogales, E.; Bertozzi, C. R.; Rabuka, D. Angew. Chem., Int. Ed. 2012, 51, 4161–4165. doi:10.1002/anie.201108130 |
| 40. | de Almeida, G.; Sletten, E. M.; Nakamura, H.; Palaniappan, K. K.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2012, 51, 2443–2447. doi:10.1002/anie.201106325 |
| 27. | Gansäuer, A.; Lauterbach, T.; Geich-Gimbel, D. Chem.–Eur. J. 2004, 10, 4983–4990. doi:10.1002/chem.200400685 |
| 28. | Friedrich, J.; Dolg, M.; Gansäuer, A.; Geich-Gimbel, D.; Lauterbach, T. J. Am. Chem. Soc. 2005, 127, 7071–7077. doi:10.1021/ja050268w |
| 29. | Friedrich, J.; Walczak, K.; Dolg, M.; Piestert, F.; Lauterbach, T.; Worgull, D.; Gansäuer, A. J. Am. Chem. Soc. 2008, 130, 1788–1796. doi:10.1021/ja077596b |
| 30. | Gansäuer, A.; Worgull, D.; Knebel, K.; Huth, I.; Schnakenburg, G. Angew. Chem., Int. Ed. 2009, 48, 8882–8885. doi:10.1002/anie.200904428 |
| 31. | Gansäuer, A.; Knebel, K.; Kube, C.; van Gastel, M.; Cangönül, A.; Daasbjerg, K.; Hangele, T.; Hülsen, M.; Dolg, M.; Friedrich, J. Chem.–Eur. J. 2012, 18, 2591–2599. doi:10.1002/chem.201102959 |
| 31. | Gansäuer, A.; Knebel, K.; Kube, C.; van Gastel, M.; Cangönül, A.; Daasbjerg, K.; Hangele, T.; Hülsen, M.; Dolg, M.; Friedrich, J. Chem.–Eur. J. 2012, 18, 2591–2599. doi:10.1002/chem.201102959 |
| 26. | Gansäuer, A.; Winkler, I.; Worgull, D.; Lauterbach, T.; Franke, D.; Selig, A.; Wagner, L.; Prokop, A. Chem.–Eur. J. 2008, 14, 4160–4163. doi:10.1002/chem.200800407 |
| 32. | Jiménez, T.; Morcillo, S. P.; Martín-Lasanta, A.; Collado-Sanz, D.; Cárdenas, D. J.; Gansäuer, A.; Justicia, J.; Cuerva, J. M. Chem.–Eur. J. 2012, 18, 12825–12833. doi:10.1002/chem.201201534 |
| 33. | Morcillo, S. P.; Martinez-Peragón, Á.; Jakoby, V.; Mota, A. J.; Kube, C.; Justicia, J.; Cuerva, J. M.; Gansäuer, A. Chem. Commun. 2014, 50, 2211–2213. doi:10.1039/c3cc49230c |
| 19. | Klawonn, T.; Gansäuer, A.; Winkler, I.; Lauterbach, T.; Franke, D.; Nolte, R. J. M.; Feiters, M. C.; Börner, H.; Hentschel, J.; Dötz, K. H. Chem. Commun. 2007, 1894–1895. doi:10.1039/b701565h |
| 20. | Gansäuer, A.; Winkler, I.; Klawonn, T.; Nolte, R. J. M.; Feiters, M. C.; Börner, H. G.; Hentschel, J.; Dötz, K. H. Organometallics 2009, 28, 1377–1382. doi:10.1021/om801022c |
| 23. | Gansäuer, A.; Franke, D.; Lauterbach, T.; Nieger, M. J. Am. Chem. Soc. 2005, 127, 11622–11623. doi:10.1021/ja054185r |
| 24. | Gansäuer, A.; Winkler, I.; Worgull, D.; Franke, D.; Lauterbach, T.; Okkel, A.; Nieger, M. Organometallics 2008, 27, 5699–5707. doi:10.1021/om800700c |
| 25. | Gansäuer, A.; Okkel, A.; Worgull, D.; Schnakenburg, G. Organometallics 2010, 29, 3227–3230. doi:10.1021/om100245r |
| 30. | Gansäuer, A.; Worgull, D.; Knebel, K.; Huth, I.; Schnakenburg, G. Angew. Chem., Int. Ed. 2009, 48, 8882–8885. doi:10.1002/anie.200904428 |
| 31. | Gansäuer, A.; Knebel, K.; Kube, C.; van Gastel, M.; Cangönül, A.; Daasbjerg, K.; Hangele, T.; Hülsen, M.; Dolg, M.; Friedrich, J. Chem.–Eur. J. 2012, 18, 2591–2599. doi:10.1002/chem.201102959 |
| 36. | Wittig, G.; Krebs, A. Chem. Ber. 1961, 94, 3260–3275. doi:10.1002/cber.19610941213 |
| 44. | Zagerman, J.; Deally, A.; Metzler-Nolte, N.; Müller-Bunz, H.; Wallis, D.; Tacke, M. Polyhedron 2011, 30, 2387–2390. doi:10.1016/j.poly.2011.05.036 |
| 45. | Golobov, Y. M.; Kasukhin, L. F. Tetrahedron 1992, 48, 1353–1406. doi:10.1016/S0040-4020(01)92229-X |
| 46. | Köhn, M.; Breinbauer, R. Angew. Chem., Int. Ed. 2004, 43, 3106–3116. doi:10.1002/anie.200401744 |
| 56. | Gansäuer, A.; Pierobon, M. Synlett 2000, 1357–1359. doi:10.1055/s-2000-7133 |
| 57. | Gansäuer, A.; Pierobon, M.; Bluhm, H. Synthesis 2001, 2500–2520. doi:10.1055/s-2001-18713 |
| 58. | Barrero, A. F.; Quilez del Moral, J. F.; Sánchez, E. M.; Arteaga, J. F. Eur. J. Org. Chem. 2006, 1627–1641. doi:10.1002/ejoc.200500849 |
| 50. | Wieder, T.; Prokop, A.; Bagci, B.; Essmann, F.; Bernicke, D.; Schulze-Osthoff, K.; Dörken, B.; Schmalz, H.-G.; Daniel, P. T.; Henze, G. Leukemia 2001, 15, 1735–1742. doi:10.1038/sj.leu.2402284 |
| 51. | Diller, R. A.; Riepl, H. M.; Rose, O.; Frias, C.; Henze, G.; Prokop, A. Planta Med. 2007, 73, 755–761. doi:10.1055/s-2007-981545 |
| 52. | Prokop, A.; Wrasidlo, W.; Lode, H.; Herold, R.; Lang, F.; Henze, G.; Dörken, B.; Wieder, T.; Daniel, P. T. Oncogene 2003, 22, 9107–9120. doi:10.1038/sj.onc.1207196 |
| 53. | Rudner, J.; Lepple-Wienhues, A.; Budach, W.; Berschauer, J.; Friedrich, B.; Wesselborg, S.; Schulze-Osthoff, K.; Belka, C. J. Cell Sci. 2001, 114, 4161–4172. |
| 54. | Gansäuer, A.; Fan, C.-A.; Keller, F.; Karbaum, P. Chem.–Eur. J. 2007, 13, 8084–8090. doi:10.1002/chem.200701021 |
| 55. | Gansäuer, A.; Shi, L.; Otte, M. J. Am. Chem. Soc. 2010, 132, 11858–11859. doi:10.1021/ja105023y |
| 26. | Gansäuer, A.; Winkler, I.; Worgull, D.; Lauterbach, T.; Franke, D.; Selig, A.; Wagner, L.; Prokop, A. Chem.–Eur. J. 2008, 14, 4160–4163. doi:10.1002/chem.200800407 |
| 47. | Bantel, H.; Engels, I. H.; Völter, W.; Schulze-Osthoff, K.; Wesselborg, S. Cancer Res. 1999, 59, 2083–2090. |
| 48. | Klener, P., Jr.; Anděra, L.; Klener, P.; Nečas, E.; Živný, J. Folia Biol. (Prague, Czech Repub.) 2006, 52, 34–44. |
| 49. | Meiler, J.; Schuler, M. Curr. Drug Targets 2006, 7, 1361–1369. doi:10.2174/138945006778559175 |
| 34. | Sletten, E. M.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. doi:10.1002/anie.200900942 |
| 35. | Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279. doi:10.1039/b901970g |
| 37. | Agard, N. J.; Prescher, J. A.; Bertozzi, C. R. J. Am. Chem. Soc. 2004, 126, 15046–15047. doi:10.1021/ja044996f |
| 38. | Gordon, C. G.; Mackey, J. L.; Jewett, J. C.; Sletten, E. M.; Houk, K. N.; Bertozzi, C. R. J. Am. Chem. Soc. 2012, 134, 9199–9208. doi:10.1021/ja3000936 |
| 39. | Hudak, J. E.; Barfield, R. M.; de Hart, G. W.; Grob, P.; Nogales, E.; Bertozzi, C. R.; Rabuka, D. Angew. Chem., Int. Ed. 2012, 51, 4161–4165. doi:10.1002/anie.201108130 |
| 40. | de Almeida, G.; Sletten, E. M.; Nakamura, H.; Palaniappan, K. K.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2012, 51, 2443–2447. doi:10.1002/anie.201106325 |
| 1. | Buettner, K. M.; Valentine, A. M. Chem. Rev. 2012, 112, 1863–1881. doi:10.1021/cr1002886 |
| 2. | Hartinger, C. G.; Metzler-Nolte, N.; Dyson, P. J. Organometallics 2012, 31, 5677–5685. doi:10.1021/om300373t |
| 3. | Tshuva, E. Y.; Ashenhurst, J. A. Eur. J. Inorg. Chem. 2009, 2203–2218. doi:10.1002/ejic.200900198 |
| 4. | Strohfeldt, K.; Tacke, M. Chem. Soc. Rev. 2008, 37, 1174–1187. doi:10.1039/b707310k |
| 5. | Köpf-Maier, P.; Köpf, H. Chem. Rev. 1987, 87, 1137–1152. doi:10.1021/cr00081a012 |
© 2014 Gansäuer et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)