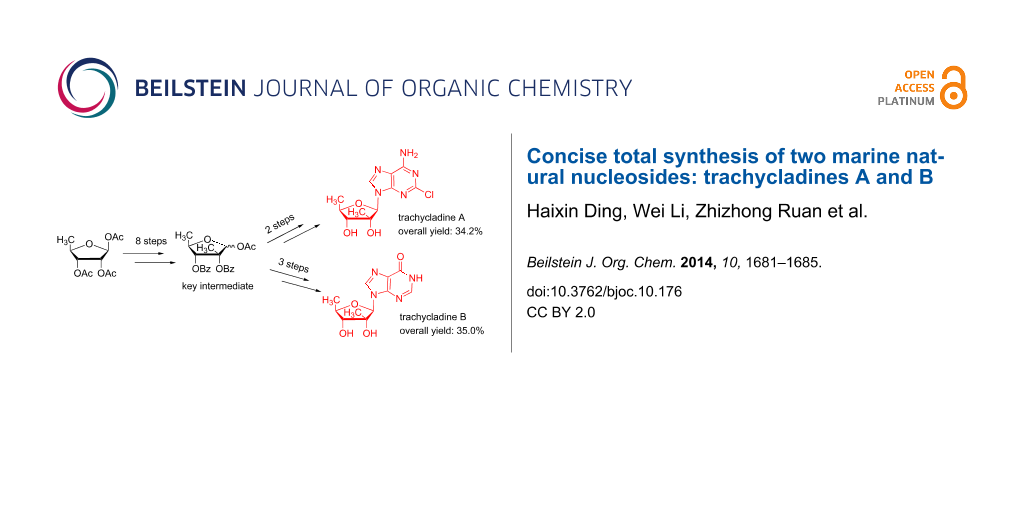
Abstract
We report the first total synthesis of trachycladines A (10 steps, 34.2% overall yield) and B (11 steps, 35.0% overall yield) by using 5-deoxy-1,2,3-tri-O-acetyl-β-D-ribofuranose as the starting material. The critical step was the SnCl4 assisted regio- and steroselective deprotection of perbenzylated 1-O-methyl-5-deoxyribofuranose. The enzyme adenylate deaminase (EC 3.5.4.6) was successfully applied to the chemoenzymatic synthesis of trachycladines B.

Graphical Abstract
Introduction
Marine organisms are a main source for the great diversity of naturally occurring nucleosides [1], which play an important role in medical research and pharmaceutical development, especially for the clinical treatment against malignant proliferation and infections by fungi, bacteria and viruses [2-7]. The initial discovery of marine nucleosides can be traced back to the identifications of spongothymidine and spongouridine in the early 1950s from the Caribbean sponge Tethyacrypta [8,9], which subsqeuently led to the commercialization of arabinofuranosylcytosine (Ara-C) [10], arabinofuranosyladenine (Ara-A) [11], and azidothymidine (AZT) [12] for clinical use.
As a key class of marine natural nucleosides, trachycladines A and B (Figure 1) hold an important position in drug development. Both trachycladines A and B contain a rare naturally occurring 2′-C-methyl-5′-deoxy-D-ribose sugar moiety. The biological activity of trachycladine B has not been reported yet due to its insufficient availability in natural resources. Trachycladines A, which was isolated from the same sponge natural product Trachycladus laevispirulifer (and also from the sponges Theonella cupola and Theonella sp) in 1995 [13,14], exhibits significant cytotoxicity in vitro against several human cell lines such as leukaemia (CCRF-CEM, IC50 0.4 µg/mL), colon tumour (HCT-116, IC50 0.9 µg/mL), and breast tumour cells (MCF-7, IC50 0.2 µg/mL) [13]. Furthermore, such 2′-C-branched ribonucleosides are also potential agonists for adenosine kinase receptors and play an important role in related drug discovery [15].
![[1860-5397-10-176-1]](/bjoc/content/figures/1860-5397-10-176-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of trachycladine A and B.
Figure 1: Structures of trachycladine A and B.
To facilitate the discovery of lead compounds as anticancer reagents from marine nucleosides [16-18], the total synthesis of trachycladines A and trachycladines B are reported herein allowing to assemble their unique chemical structure for the first time.
Results and Discussion
Until now, there is only one publication about the total synthesis of analogues of trachycladines A and B. In 2005, Enders et al. reported the first asymmetric synthesis of 4′-epi-trachycladines A and B in 14 steps. They used 2,2-dimethyl-1,3-dioxan-5-one as a starting material and employed the SAMP-/RAMP-hydrazone methodology to obtain an overall yield of 18–21% [19,20]. Due to the formation of the opposite configuration at C-4′ and a lengthy synthetic route this strategy is not suitable for the total synthesis of trachycladine A and B.
From a synthetic point of view, the target nucleosides could be synthesized from either 1,2,3,5-tetra-O-benzoyl-2-C-β-methyl-D-ribofuranose (5) (route A) or 5-deoxy-1,2,3-tri-O-acetyl-β-D-ribofuranose (6) (route B). The corresponding retrosynthetic analysis is shown in Figure 2.
![[1860-5397-10-176-2]](/bjoc/content/figures/1860-5397-10-176-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis of trachycladines A and B.
Figure 2: Retrosynthetic analysis of trachycladines A and B.
In synthetic route (A), nucleoside 3 could be synthesized by using 1,2,3,5-tetra-O-benzoyl-2-C-methyl-D-ribofuranose (5) as a carbohydrate acceptor by a Vorbrüggen glycosylation with the corresponding silylated nucleobases and a Lewis acid as a catalyst. As the key intermediate for the preparation of the anti-HCV nucleoside valopicitabine (NM-283) [21,22] and related nucleosides, there are suitable methodologies for the synthesis of carbohydrate 5 [23]. After the selective deoxygenation of the C-5′ hydroxy group of nucleosides 3, trachycladine A and B could be afforded. According to the preliminary results from our lab (unpublished results), the deoxygenation procedure of the C-5′ hydroxy group was accompanied by several undesired side reactions.
Then we turned to synthetic route (B), which utilizes carbohydrate 4 as a Vorbrüggen glycosylation donor. Firstly, without the deoxygenation of the C-5′ hydroxy group at the late synthetic stage, carbohydrate 4 is a versatile intermediate for the diversity-oriented synthesis of the related 2′-C-β-methyl-5′-deoxyribonucleosides. As the key intermediate for the preparation of the antitumor drug capecitabine [24,25], 5-deoxy-1,2,3-tri-O-acetyl-β-D-ribofuranose (6) is commercially available with a cheap market price. In addition, the regioselective cleavage of the 2-O-benzyl protection group of perbenzylated 1-O-methyl ribofuranoside by using SnCl4 has been widely used for the synthesis of 2-O-substituted nucleosides [26-30]. To the best of our knowledge, an application for 5-deoxyribofuranoside is not published. This step would be vital for the introduction of the corresponding 2′-C-β-methyl group in our synthetic strategy.
Our synthetic route to 5-deoxy-1-O-acetyl-2,3-di-O-benzoyl-2-C-β-methyl-D-ribofuranose (4) is shown in Scheme 1. Treatment of 5-deoxy-1,2,3-tri-O-acetyl-β-D-ribofuranose (6) with catalytic NaOMe in MeOH gave 5-deoxy-D-ribose in quantitative yield. Without further work-up, an excess of freshly prepared Dowex-50 H+ resin was added to the mixture until pH 5 was reached. After stirred overnight, 5-deoxy-1-O-methylribofuranose 7 was obtained as a mixture of anomers (α:β = 2:3, 93% for two steps). Then, the other two hydroxy groups were protected as 2,4-dichlorobenzyl ether. Compound 8 was isolated in 95% yield after reacting compound 7 with NaH and 2,4-dichlorobenzyl chloride in anhydrous DMF. 2,4-Dichlorobenzyl chloride was used because it is less irritant than the usually used benzyl bromide.
![[1860-5397-10-176-i1]](/bjoc/content/inline/1860-5397-10-176-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 5-deoxy-1-O-acetyl-2,3-di-O-benzoyl-2-C-β-methyl-D-ribofuranose (4). Reagents and conditions: (a) i. CH3ONa (cat.), CH3OH, 0 °C; ii. Dowex-50 H+ resin, pH 5, rt, overall 93% yield in two steps; (b) NaH, 2,4-dichlorobenzyl chloride, DMF, 0 °C, 95%; (c) SnCl4, DCM, 0 °C, 87%; (d) Dess–Martin oxidation, DCM, reflux, 92%; (e) CH3MgBr, ether, −10 °C, 87%; (f) H2, 20% Pd(OH)2/C, Et3N, THF/EtOAc 1:1, rt, 91%; (g) Benzoyl chloride, DMAP (2 equiv), DCM, rt, 92%; (h) Ac2O/AcOH 1:1, H2SO4, rt, 84%.
Scheme 1: Synthesis of 5-deoxy-1-O-acetyl-2,3-di-O-benzoyl-2-C-β-methyl-D-ribofuranose (4). Reagents and cond...
Regioselective cleavage of the 2-O-(2,4-dichlorobenzyl) group was attempted with anhydrous SnCl4 in DCM by the method reported by Brown [31,32] and our group [16]. To our delight, this approach can afford 3-O-(2,4-dichlorobenzyl)-1-O-methyl-α-D-ribofuranose (9). After extensive optimization, compound 9 was obtained as an α-anomer only in 87% yield with exclusive regio- and stereoselectivity (Scheme 1). The anomeric hydrogen atom of 9 shows a characteristic doublet with a coupling constant of J = 4.8 Hz at 4.87 ppm. The structure of 9 was further unambiguously confirmed by X-ray diffraction analysis (Figure 3). This is the first report on the usage of SnCl4 as a regio- and stereoselective deprotection reagent in perbenzylated 5-deoxy-1-O-methylribofuranose. Based on these findings SnCl4 may prove useful for the regio- and stereoselective cleavage of the 2-O-benzyl group at perbenzylated 1-O-methylribofuranose analogues.
![[1860-5397-10-176-3]](/bjoc/content/figures/1860-5397-10-176-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The X-ray crystal structural of 1-O-methyl-3-O-(2,4-dichlorobenzyl)-5-deoxy-α-D-ribofuranose (9).
Figure 3: The X-ray crystal structural of 1-O-methyl-3-O-(2,4-dichlorobenzyl)-5-deoxy-α-D-ribofuranose (9).
Dess–Martin oxidation of compound 9 provided ketone 10 in 92% yield. Next, the addition reaction with CH3MgBr in dry ether gave 11 exclusively with the requisite β-methyl configuration at C-2. NOE experiments showed correct correlations of H-1, H-3, 5-CH3 and 2-β-methyl. Then, the remaining 3-(O-2,4-dichlorobenzyl) group was removed under hydrogenation with 20% Pd(OH)2/C as a catalyst to give 12 in 91% yield. As the anomerization of the 1-O-α-methyl glycoside may occur under acidic conditions, the structural characterization may be much more complex. The addition of triethylamine is necessary to neutralize the possibly formed hydrogen chloride due to the reduction of the aromatic chloro substitutes during hydrogenation. The protection of all free hydroxy groups of 12 as benzoates afforded 13 in 92% yield. Due to the steric environment of 2-OH the benzoylation with benzoyl chloride required an extended reaction time (48 h) and 2 equiv DMAP as base and catalyst. At last, the 1-O-methyl group was transformed to 1-O-acetate glycosylation acceptor 4 as an anomeric mixture (α:β = 2:3) in 84% yield. Therefore, the key intermediate 4 was synthesized from commercial available 5-deoxy-1,2,3-tri-O-acetyl-β-D-ribofuranose (6) in 8 steps in 43.2% overall yield.
Unfortunately, the Vorbrüggen glycosylation of carbohydrate 4 with bis(trimethylsilyl)hypoxanthine with TMSOTf as a catalyst afforded a mixture of inseparable N9 and N7 isomers with 40% yield. To further improve the yield and add to the molecular diversity, N6-benzoyladenine 17 was used as a base to give nucleoside 14 in 87% yield (Scheme 2). All benzoyl protecting groups were removed simultaneously by using ammonia saturated methanol in a sealed press tube to give 15 in 96% yield. In the next transformation to trachycladine B the classic procedure with sodium nitrite in acetic acid proved to be inefficient as it gave complicated mixtures. Therefore, we applied an alternative method.
![[1860-5397-10-176-i2]](/bjoc/content/inline/1860-5397-10-176-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of trachycladine B (2). Reagents and conditions: (a) i. N,O-Bis(trimethylsilyl)acetamide (BSA), MeCN; ii. TMSOTf, MeCN, 87%; (b) NH3 sat. methanol, 96%; (c) AMPDA, phosphate buffer at pH 5.6 and 40 °C, 97%.
Scheme 2: Synthesis of trachycladine B (2). Reagents and conditions: (a) i. N,O-Bis(trimethylsilyl)acetamide ...
In the past decades, adenosine deaminase (adenosine aminohydrolase, ADA, EC 3.5.4.4) and adenylate deaminase (5′-adenylic acid deaminase, AMP deaminase, AMPDA, EC 3.5.4.6) have been widely used in enzymatic hydrolysis [33-36]. In our experiment, ADA failed to give trachycladine A due to its lack of the 5′-hydroxy group, which plays a crucial role for the activity of the enzyme [35]. AMPDA is commercially available as a practical lyophilate powder and was widely used in the food industry for the production of inosine 5′-phosphate. AMPDA is a more versatile biocatalyst compared to ADA, since it can convert a larger number of adenosine derivatives into the corresponding inosine derivatives.
AMPDA was used as a biocatalyst in the enzymatic hydrolysis of adenosine 15. The reaction proceeded smoothly in the presence of AMPDA in phosphate buffer at pH 5.6 and 40 °C with 3% DMSO as a co-solvent. Adenosine 15 was enzymatically deaminated to give trachycladine B in almost quantitative yield. The reaction is a novel instance of the broad substrate tolerance of AMPDA and extends its usage in nucleoside chemistry. Therefore, trachycladine B was synthesized in 35.0% overall yield from 5-deoxy-1,2,3-tri-O-acetyl-β-D-ribofuranose (6).
Next, the coupling of carbohydrate 4 with 2,6-dichloropurine (18) afforded nucleoside 16 in 86% yield with DBU as a base and TMSOTf as a Lewis acid (Scheme 3). After heated in a pressure tube with ammonia saturated methanol, the benzoyl protecting groups were removed and the chlorine atom at position C6 was substituted at the same time to afford trachycladine A in 92% yield. Therefore, trachycladine A was synthesized in 34.2% overall yield from 5-deoxy-1,2,3-tri-O-acetyl-β-D-ribofuranose (6).
![[1860-5397-10-176-i3]](/bjoc/content/inline/1860-5397-10-176-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of trachycladine A (1). Reagents and conditions: (a) DBU, TMSOTf, CH3CN, 86%; (b) NH3 sat. methanol, sealed press tube, 92%.
Scheme 3: Synthesis of trachycladine A (1). Reagents and conditions: (a) DBU, TMSOTf, CH3CN, 86%; (b) NH3 sat...
Conclusion
In conclusion, the total synthesis of trachycladine A and B have been successfully accomplished for the first time with 34.8% and 35.6% overall yield, respectively. All spectral data of synthetic trachycladine A and B are in accordance with those of the natural products. We would like to particularly emphasize three aspects of this work : (1) this is the first report using SnCl4 as a regio- and stereoselective deprotection reagent in perbenzylated 1-O-methyl-5-deoxyribofuranosides, (2) adenylate deaminase (EC 3.5.4.6) facilitated chemoenzymatic hydrolysis was used for the synthesis of trachycladine B, and (3) the reported carbohydrate 4 (1-O-acetyl-2,3-O-dibenzoyl-2-β-C-methyl-5-deoxy-D-ribofuranose) can be used as a common intermediate for the synthesis of versatile 5′-deoxy-2′-C-branched nucleosides.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, characterization data of compounds, and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 2.3 MB | Download |
| Supporting Information File 2: X-ray crystal structural of compound 9. | ||
| Format: CIF | Size: 11.2 KB | Download |
References
-
Bhakuni, D. S.; Rawat, D. S. Bioactive marine natural products; Co-published by Springer with Anamaya Publishers: New Delhi, New York, 2005.
Return to citation in text: [1] -
Md, S. H.; Fareed, S.; Ansari, S.; Khan, M. S. Indian J. Mar. Sci. 2012, 41, 27–39.
Return to citation in text: [1] -
Vo, T.-S.; Ngo, D.-H.; Ta, Q. V.; Kim, S.-K. Eur. J. Pharm. Sci. 2011, 44, 11–20. doi:10.1016/j.ejps.2011.07.005
Return to citation in text: [1] -
Sagar, S.; Kaur, M.; Minneman, K. P. Mar. Drugs 2010, 8, 2619–2638. doi:10.3390/md8102619
Return to citation in text: [1] -
Newman, D. J.; Cragg, G. M. Curr. Med. Chem. 2004, 11, 1693–1713. doi:10.2174/0929867043364982
Return to citation in text: [1] -
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2004, 67, 1216–1238. doi:10.1021/np040031y
Return to citation in text: [1] -
Müller, W. E. G.; Grebenjuk, V. A.; Pennec, G. L.; Schröder, H.-C.; Brümmer, F.; Hentschel, U.; Müller, I. M.; Breter, H.-J. Mar. Biotechnol. 2004, 6, 105–117. doi:10.1007/s10126-002-0098-6
Return to citation in text: [1] -
Bergmann, W.; Stempien, M. F., Jr. J. Org. Chem. 1957, 22, 1575–1577. doi:10.1021/jo01363a009
Return to citation in text: [1] -
Bergmann, W.; Feeney, R. J. J. Org. Chem. 1951, 16, 981–987. doi:10.1021/jo01146a023
Return to citation in text: [1] -
Kern, W.; Estey, E. H. Cancer 2006, 107, 116–124. doi:10.1002/cncr.21543
Return to citation in text: [1] -
Becher, H. J. Inn. Med. 1986, 13, 237–245.
Return to citation in text: [1] -
Sachs, M. K. Arch. Intern. Med. 1992, 152, 485–501. doi:10.1001/archinte.1992.00400150019005
Return to citation in text: [1] -
Searle, P. A.; Molinski, T. F. J. Org. Chem. 1995, 60, 4296–4298. doi:10.1021/jo00118a059
Return to citation in text: [1] [2] -
Ichiba, T.; Nakao, Y.; Scheuer, P. J.; Sata, N. U.; Kelly-Borges, M. Tetrahedron Lett. 1995, 36, 3977–3980. doi:10.1016/0040-4039(95)00692-6
Return to citation in text: [1] -
Li, N.-S.; Lu, J.; Piccirilli, J. A. Org. Prep. Proced. Int. 2010, 42, 191–283. doi:10.1080/00304948.2010.483657
Return to citation in text: [1] -
Song, Y.; Ding, H.; Dou, Y.; Yang, R.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis 2011, 1442–1446. doi:10.1055/s-0030-1259975
Return to citation in text: [1] [2] -
Sun, J.; Dou, Y.; Ding, H.; Yang, R.; Sun, Q.; Xiao, Q. Mar. Drugs 2012, 10, 881–889. doi:10.3390/md10040881
Return to citation in text: [1] -
Dou, Y.-H.; Ding, H.-X.; Yang, R.-C.; Li, W.; Xiao, Q. Chin. Chem. Lett. 2013, 24, 379–382. doi:10.1016/j.cclet.2013.03.014
Return to citation in text: [1] -
Enders, D.; Breuer, I.; Drosdow, E. Synthesis 2005, 3239–3244. doi:10.1055/s-2005-918439
Return to citation in text: [1] -
Enders, D.; Nühring, A.; Runsink, J.; Raabe, G. Synthesis 2001, 1406–1414. doi:10.1055/s-2001-15236
Return to citation in text: [1] -
Toniutto, P.; Fabris, C.; Bitetto, D.; Fornasiere, E.; Rapetti, R.; Pirisi, M. Curr. Opin. Invest. Drugs 2007, 8, 150–158.
Return to citation in text: [1] -
Bichko, V.; Lallos, L.; Soubasakos, M.; LaColla, M.; Tausek, M. M.; Gillum, J.; Standring, D. N. J. Hepatol. 2007, 46 (Suppl. 1), S163–S164. doi:10.1016/S0168-8278(07)62025-X
Return to citation in text: [1] -
Harry-O'kuru, R. E.; Smith, J. M.; Wolfe, M. S. J. Org. Chem. 1997, 62, 1754–1759. doi:10.1021/jo961893+
Return to citation in text: [1] -
McGavin, J. K.; Goa, K. L. Drugs 2001, 61, 2309–2326. doi:10.2165/00003495-200161150-00015
Return to citation in text: [1] -
Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H.; Kuruma, I.; Horii, I.; Ishitsuka, H. Bioorg. Med. Chem. 2000, 8, 1697–1706. doi:10.1016/S0968-0896(00)00087-0
Return to citation in text: [1] -
Li, N.-S.; Piccirilli, J. A. J. Org. Chem. 2004, 69, 4751–4759. doi:10.1021/jo0495337
Return to citation in text: [1] -
Jeannot, F.; Gosselin, G.; Mathé, C. Org. Biomol. Chem. 2003, 1, 2096–2102. doi:10.1039/b303093h
Return to citation in text: [1] -
Martin, P. Helv. Chim. Acta 1995, 78, 486–504. doi:10.1002/hlca.19950780219
Return to citation in text: [1] -
Hori, H.; Nishida, Y.; Ohrui, H.; Meguro, H. J. Org. Chem. 1989, 54, 1346–1353. doi:10.1021/jo00267a022
Return to citation in text: [1] -
Martin, O. R.; Kurz, K. G.; Rao, S. P. J. Org. Chem. 1987, 52, 2922–2925. doi:10.1021/jo00389a049
Return to citation in text: [1] -
Lou, C.; Xiao, Q.; Tailor, R. R.; Ben Gaied, N.; Gale, N.; Light, M. E.; Fox, K. R.; Brown, T. Med. Chem. Commun. 2011, 2, 550–558. doi:10.1039/c1md00068c
Return to citation in text: [1] -
Lou, C.; Xiao, Q.; Brennan, L.; Light, M. E.; Vergara-Irigaray, N.; Atkinson, E. M.; Holden-Dye, L. M.; Fox, K. R.; Brown, T. Bioorg. Med. Chem. 2010, 18, 6389–6397. doi:10.1016/j.bmc.2010.07.005
Return to citation in text: [1] -
Ferrero, M.; Gotor, V. Monatsh. Chem. 2000, 131, 585–616. doi:10.1007/s007060070088
Return to citation in text: [1] -
Ferrero, M.; Gotor, V. Chem. Rev. 2000, 100, 4319–4348. doi:10.1021/cr000446y
Return to citation in text: [1] -
Santaniello, E.; Ciuffreda, P.; Alessandrini, L. Synthesis 2005, 509–526. doi:10.1055/s-2005-861821
Return to citation in text: [1] [2] -
Gupta, M.; Nair, V. Collect. Czech. Chem. Commun. 2006, 71, 769–787. doi:10.1135/cccc20060769
Return to citation in text: [1]
| 35. | Santaniello, E.; Ciuffreda, P.; Alessandrini, L. Synthesis 2005, 509–526. doi:10.1055/s-2005-861821 |
| 16. | Song, Y.; Ding, H.; Dou, Y.; Yang, R.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis 2011, 1442–1446. doi:10.1055/s-0030-1259975 |
| 33. | Ferrero, M.; Gotor, V. Monatsh. Chem. 2000, 131, 585–616. doi:10.1007/s007060070088 |
| 34. | Ferrero, M.; Gotor, V. Chem. Rev. 2000, 100, 4319–4348. doi:10.1021/cr000446y |
| 35. | Santaniello, E.; Ciuffreda, P.; Alessandrini, L. Synthesis 2005, 509–526. doi:10.1055/s-2005-861821 |
| 36. | Gupta, M.; Nair, V. Collect. Czech. Chem. Commun. 2006, 71, 769–787. doi:10.1135/cccc20060769 |
| 1. | Bhakuni, D. S.; Rawat, D. S. Bioactive marine natural products; Co-published by Springer with Anamaya Publishers: New Delhi, New York, 2005. |
| 26. | Li, N.-S.; Piccirilli, J. A. J. Org. Chem. 2004, 69, 4751–4759. doi:10.1021/jo0495337 |
| 27. | Jeannot, F.; Gosselin, G.; Mathé, C. Org. Biomol. Chem. 2003, 1, 2096–2102. doi:10.1039/b303093h |
| 28. | Martin, P. Helv. Chim. Acta 1995, 78, 486–504. doi:10.1002/hlca.19950780219 |
| 29. | Hori, H.; Nishida, Y.; Ohrui, H.; Meguro, H. J. Org. Chem. 1989, 54, 1346–1353. doi:10.1021/jo00267a022 |
| 30. | Martin, O. R.; Kurz, K. G.; Rao, S. P. J. Org. Chem. 1987, 52, 2922–2925. doi:10.1021/jo00389a049 |
| 31. | Lou, C.; Xiao, Q.; Tailor, R. R.; Ben Gaied, N.; Gale, N.; Light, M. E.; Fox, K. R.; Brown, T. Med. Chem. Commun. 2011, 2, 550–558. doi:10.1039/c1md00068c |
| 32. | Lou, C.; Xiao, Q.; Brennan, L.; Light, M. E.; Vergara-Irigaray, N.; Atkinson, E. M.; Holden-Dye, L. M.; Fox, K. R.; Brown, T. Bioorg. Med. Chem. 2010, 18, 6389–6397. doi:10.1016/j.bmc.2010.07.005 |
| 8. | Bergmann, W.; Stempien, M. F., Jr. J. Org. Chem. 1957, 22, 1575–1577. doi:10.1021/jo01363a009 |
| 9. | Bergmann, W.; Feeney, R. J. J. Org. Chem. 1951, 16, 981–987. doi:10.1021/jo01146a023 |
| 23. | Harry-O'kuru, R. E.; Smith, J. M.; Wolfe, M. S. J. Org. Chem. 1997, 62, 1754–1759. doi:10.1021/jo961893+ |
| 2. | Md, S. H.; Fareed, S.; Ansari, S.; Khan, M. S. Indian J. Mar. Sci. 2012, 41, 27–39. |
| 3. | Vo, T.-S.; Ngo, D.-H.; Ta, Q. V.; Kim, S.-K. Eur. J. Pharm. Sci. 2011, 44, 11–20. doi:10.1016/j.ejps.2011.07.005 |
| 4. | Sagar, S.; Kaur, M.; Minneman, K. P. Mar. Drugs 2010, 8, 2619–2638. doi:10.3390/md8102619 |
| 5. | Newman, D. J.; Cragg, G. M. Curr. Med. Chem. 2004, 11, 1693–1713. doi:10.2174/0929867043364982 |
| 6. | Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2004, 67, 1216–1238. doi:10.1021/np040031y |
| 7. | Müller, W. E. G.; Grebenjuk, V. A.; Pennec, G. L.; Schröder, H.-C.; Brümmer, F.; Hentschel, U.; Müller, I. M.; Breter, H.-J. Mar. Biotechnol. 2004, 6, 105–117. doi:10.1007/s10126-002-0098-6 |
| 24. | McGavin, J. K.; Goa, K. L. Drugs 2001, 61, 2309–2326. doi:10.2165/00003495-200161150-00015 |
| 25. | Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H.; Kuruma, I.; Horii, I.; Ishitsuka, H. Bioorg. Med. Chem. 2000, 8, 1697–1706. doi:10.1016/S0968-0896(00)00087-0 |
| 15. | Li, N.-S.; Lu, J.; Piccirilli, J. A. Org. Prep. Proced. Int. 2010, 42, 191–283. doi:10.1080/00304948.2010.483657 |
| 19. | Enders, D.; Breuer, I.; Drosdow, E. Synthesis 2005, 3239–3244. doi:10.1055/s-2005-918439 |
| 20. | Enders, D.; Nühring, A.; Runsink, J.; Raabe, G. Synthesis 2001, 1406–1414. doi:10.1055/s-2001-15236 |
| 13. | Searle, P. A.; Molinski, T. F. J. Org. Chem. 1995, 60, 4296–4298. doi:10.1021/jo00118a059 |
| 21. | Toniutto, P.; Fabris, C.; Bitetto, D.; Fornasiere, E.; Rapetti, R.; Pirisi, M. Curr. Opin. Invest. Drugs 2007, 8, 150–158. |
| 22. | Bichko, V.; Lallos, L.; Soubasakos, M.; LaColla, M.; Tausek, M. M.; Gillum, J.; Standring, D. N. J. Hepatol. 2007, 46 (Suppl. 1), S163–S164. doi:10.1016/S0168-8278(07)62025-X |
| 13. | Searle, P. A.; Molinski, T. F. J. Org. Chem. 1995, 60, 4296–4298. doi:10.1021/jo00118a059 |
| 14. | Ichiba, T.; Nakao, Y.; Scheuer, P. J.; Sata, N. U.; Kelly-Borges, M. Tetrahedron Lett. 1995, 36, 3977–3980. doi:10.1016/0040-4039(95)00692-6 |
| 12. | Sachs, M. K. Arch. Intern. Med. 1992, 152, 485–501. doi:10.1001/archinte.1992.00400150019005 |
| 16. | Song, Y.; Ding, H.; Dou, Y.; Yang, R.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis 2011, 1442–1446. doi:10.1055/s-0030-1259975 |
| 17. | Sun, J.; Dou, Y.; Ding, H.; Yang, R.; Sun, Q.; Xiao, Q. Mar. Drugs 2012, 10, 881–889. doi:10.3390/md10040881 |
| 18. | Dou, Y.-H.; Ding, H.-X.; Yang, R.-C.; Li, W.; Xiao, Q. Chin. Chem. Lett. 2013, 24, 379–382. doi:10.1016/j.cclet.2013.03.014 |
© 2014 Ding et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)