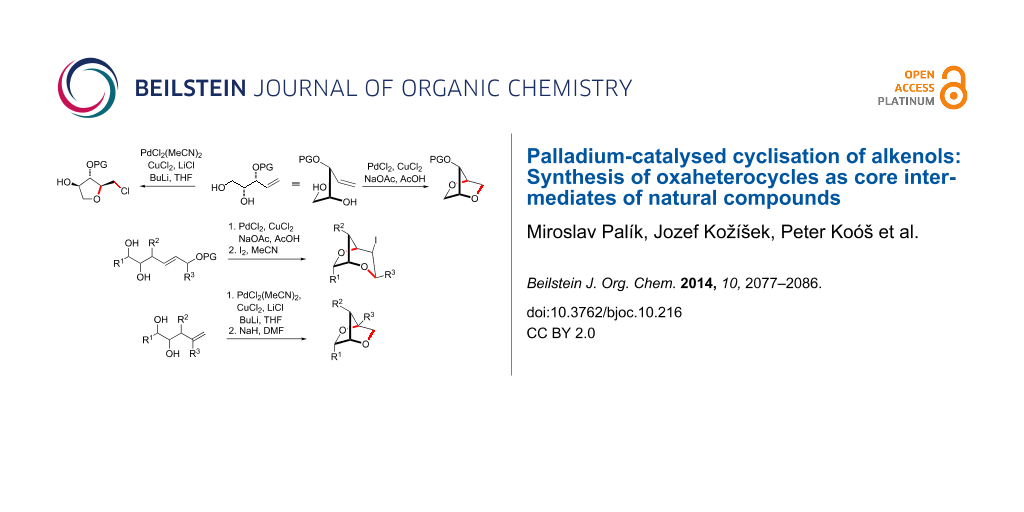
Abstract
The study of Pd-catalysed cyclisation reactions of alkenols using different catalytic systems is reported. These transformations affect the stereoselective construction of mono- and/or bicyclic oxaheterocyclic derivatives depending on a starting alkenol. The substrate scope and proposed mechanism of Pd-catalysed cyclisation reactions are also discussed. Moreover, the diastereoselective Pd-catalysed cyclisation of appropriate alkenols to tetrahydrofurans and subsequent cyclisation provided properly substituted 2,5-dioxabicyclo[2.2.1]heptane and 2,6-dioxabicyclo[3.2.1]octane, respectively. Such bicyclic ring subunits are found in many natural products including ocellenynes and aurovertines.

Graphical Abstract
Introduction
Oxaheterocycles of various sizes are found in many different biologically active compounds. Particularly, substituted tetrahydrofuran units are present in a large branch of natural products that display interesting biological properties, such as goniofufurone 1 [1], goniothalesdiol 2 [2], varitriol 3 [3], erythroskyrine 4 [4,5], ocellenynes 5 [6,7], sorangicin A 6 [8], aurovertins 7 [9-12] and epicitreoviridinol 8 [13] (Figure 1).
![[1860-5397-10-216-1]](/bjoc/content/figures/1860-5397-10-216-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of naturally occurring tetrahydrofurans.
Figure 1: Examples of naturally occurring tetrahydrofurans.
Over the last decades, an enormous work has been devoted to find an efficient stereoselective route to variously substituted tetrahydrofurans [14,15]. Among many described transformations, the metal-catalysed carboetherification reactions [16-18] and intramolecular oxycarbonylations of alkenes [19-21] are of particular importance. Although, many of these synthetic routes have showed their potential, there is still an area for improving the scope and stereocontrol of the new synthetic construction of substituted tetrahydrofurans.
Recently, we have described a novel type of PdCl2/CuCl2-catalysed bicyclisation reaction of α-O-benzyl-protected sugar-derived alkenitols A, that provided 2,5-dioxabicyclo[2.2.1]heptanes B with high 1,4-threo-selectivity (Scheme 1) [22,23]. In this process, the terminal carbon–carbon double bond is bis-O-functionalised with two hydroxy groups by sequential intramolecular–intramolecular reaction.
![[1860-5397-10-216-i1]](/bjoc/content/inline/1860-5397-10-216-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: PdCl2/CuCl2-catalysed bicyclisation of unsaturated polyols [22].
Scheme 1: PdCl2/CuCl2-catalysed bicyclisation of unsaturated polyols [22].
Based on our continuous interest in the palladium-catalysed cyclisation reactions and their applications in natural product syntheses [24,25], we have decided to explore the substrate scope and the limitations of this transformation. With this aim, we detailed the synthesis of a number of alkene alcohols, and described different catalytic systems in the Pd-catalysed cyclisation reaction. Additionally, we have also outlined the synthetic approach to substituted 2,5-dioxabicyclo[2.2.1]heptane and 2,6-dioxabicyclo[3.2.1]octane rings. Such substituted bicyclic rings are of further interest as they are the core substructures in a number of marine derived metabolites, including ocellenynes and sorangicin A.
Results and Discussion
Synthesis of starting materials
Palladium-catalysed cyclisations are substrate selective reactions. In most cases, the literature known cyclisation using similar substrate (with even small change in its substructure) can lead in different product formation. Although, there are several known rules-reactions (β-hydride elimination, η3-complex formation...) which are applicable to predict the behaviour of the used substrate in the Pd-catalysed reaction, there are cases where the results of such reactions still remain on experimental findings.
While the cyclisation reactions of alkenols have been relatively well described in the literature, only less attention was given to the reactions of unsaturated polyols. However, such cyclisations can provide a variety of products which are useful intermediates in many natural product syntheses. With this aim, we have designed syntheses of several substrates bearing different double bonds and substituents to cover certain possibilities for Pd-catalysed cyclisation screening.
At first, easily accessible C5-alkenitols (Figure 2) were chosen as simplest suitable substrates for screening the optimal reaction conditions of the previously described bicyclisation reaction. Thus, the known triols erythro-9 [26] and threo-10 [27] were prepared from divinylcarbinol using asymmetric epoxidation [28,29]. Diastereomeric mixtures of 3-O-benzyl 11, 3-O-silyl-protected 12 and fully unprotected triol 13 was prepared starting from 1,2-O-isopropylidene-D-glyceraldehyde using described procedures [30].
![[1860-5397-10-216-2]](/bjoc/content/figures/1860-5397-10-216-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The preparation of substrates 20–23 having a symmetrically disubstituted C–C double bond is depicted in Scheme 2. The synthesis started from known threose 15 [31] followed by a common synthetic sequence comprising the olefination reaction and the hydrolysis of the acetonide protecting group. Thus, Horner–Wadsworth–Emmons olefination of aldehyde 15 furnished the corresponding separable mixture of Z and E alkenes 16 and 17. In the case of utilising stabilised phosphorane ylides, the Wittig reaction provided only Z alkenes 18 and 19. Following acidic hydrolysis provided α-O-benzyl substrates 20–23 in good yields. Additionally, the synthesis of substrate 30 with 1,1-disubstituted C–C double bond was accomplished in 3 steps. The addition of methylmagnesium chloride to previously prepared threose 15, followed by Dess–Martin oxidation of the secondary alcohol gave methylketone 29. Subsequent Wittig olefination using (methylidene)triphenylphosphorane ylide and final hydrolysis of the acetonide provided the desired C5-substrate 30.
![[1860-5397-10-216-i2]](/bjoc/content/inline/1860-5397-10-216-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of alkenols 20-23 and 30. Reagents and conditions: a) lit. [31] (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C to rt, 2 h; b) Phosphonium salt, BuLi, THF, 0 °C to rt, overnight, MPLC; c) lit. [32] (EtO)2POCH2CO2Et, NaH, THF, 0 °C to rt, 3 h; d) 60% AcOH, 60 °C, 3 h; e) MeMgCl, Et2O, 0 °C to rt, 1 h; f) Dess–Martin periodinane, CH2Cl2, 0 °C, 1 h. L-DET = L-dimethyl tartrate.
Scheme 2: Synthesis of alkenols 20-23 and 30. Reagents and conditions: a) lit. [31] (COCl)2, DMSO, Et3N, CH2Cl2, ...
The synthesis of substrates 24–28 bearing an allylic hydroxy group is pictured in Scheme 3. At first, the ester group of previously prepared intermediate E-17 was reduced using DIBAL-H [32] providing the known allylic alcohol in very good yield. Following protection of the primary alcohol yielded fully protected alkene-tetraol and subsequent chemoselective removal of the acetonide protecting group in one pot led to substrates 24–26. The synthesis of substrate 28 bearing a tertiary allylic alcohol was performed in a two-step sequence. The addition of methyllithium to ester Z-17 and following deprotection of the corresponding alcohol 27 with aqueous acetic acid afforded tetraol 28 in good yield.
![[1860-5397-10-216-i3]](/bjoc/content/inline/1860-5397-10-216-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of alkenols 24–26 and 28. Reagents and conditions: a) lit. [32] DIBAL-H, CH2Cl2; b) TBDPSCl, imidazole, CH2Cl2, rt, overnight; c) NaH, THF then MeI, rt, overnight; d) FeCl3·6H2O, CHCl3, rt, 1 h (for 25); e) 60% AcOH, 60 °C, 3 h; f) MeLi, Et2O, −78 °C, 1 h. TBDPSCl = tert-butyldiphenylsilyl chloride.
Scheme 3: Synthesis of alkenols 24–26 and 28. Reagents and conditions: a) lit. [32] DIBAL-H, CH2Cl2; b) TBDPSCl, ...
Substrates syn-diols 33–35 (not bearing an α-O-protected group) were prepared in 2 steps starting from the aldehyde 31 using the Yamamoto’s [33] sequential O-nitrosoaldol and Grignard addition process using different reagents (Scheme 4).
![[1860-5397-10-216-i4]](/bjoc/content/inline/1860-5397-10-216-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of substrates 33–35, 37. Reagents and conditions: a) lit. [33] L-proline (0.25 equiv), 2-nitrosotoluene, CHCl3, −18 °C; b) RMgCl, CeCl3·2LiCl, THF, −78 °C to rt, overnight; c) acetone, PTSA, 3 h, rt; d) OsO4 (0.01 equiv), NMO (2 equiv), pyridine, 5 d, rt; e) NaIO4, MeOH/H2O, 3 h, rt; f) NaH, (EtO)2POCH2CO2Et, THF, −20 °C to rt, 15 min; g) DIBAL-H, CH2Cl2, −30 °C to −10 °C, 45 min; h) Ac2O, pyridine, CH2Cl2, 3 h, rt; i) AcOH/H2O, 3 h, 60 °C. PTSA = p-toluenesulfonic acid, NMO = N-methylmorpholine N-oxide, DIBAL = diisobutylaluminium hydride.
Scheme 4: Synthesis of substrates 33–35, 37. Reagents and conditions: a) lit. [33] L-proline (0.25 equiv), 2-nitr...
Thus, L-proline-catalysed oxidation of 31 with 2-nitrosotoluene gave the optically pure O-selective nitrosoaldol product 32, which underwent a reaction with the corresponding Grignard reagent in the presence of CeCl3·2LiCl providing diols 33–35 in good overall yields and high diastereoselectivity (d.r. >20:1).
Additionally, allylic acetate 37 was obtained starting from 33 in a seven-step sequence in 30% overall yield. The acetonisation of hydroxy groups of the previously prepared diol 33, followed by OsO4 dihydroxylation of the C–C double bond provided the corresponding diol in good yield. The resulting vicinal diol was then cleaved by sodium periodate to the corresponding aldehyde, which was immediately subjected to a Horner–Wadsworth–Emmons olefination using diethyl carbethoxyethylidenephopsphonate. Reduction of the resultant ester with DIBAL-H in dichloromethane afforded partially protected triol 36 in 39% yield over five steps. Finally, acetylation of the primary hydroxy group and subsequent removal of the acetonide provided the target compound 37 in good yield (77%).
Synthesis of the similar substrate rac-42 having two conjugated double bonds is shown in Scheme 5. The synthesis started from the known acetate 38, which was obtained by acetylation of commercially available non-3-ene-1-ol [34]. Epoxidation of acetate 38 with MCPBA in dichloromethane and subsequent acidic epoxide hydrolysis produced the syn-diol rac-39. The following protection of diol rac-39 as its acetonide and the primary hydroxy group deprotection using sodium methoxide afforded alcohol rac-40 in good yield (43% over 5 steps). Next, Swern oxidation of the primary hydroxy group provided the aldehyde, which was then transformed to diene derivative rac-41 by Wadsforth–Emmons olefination using diethyl allylphosphonate. Final deprotection of the hydroxy groups furnished rac-42 in 63% yield.
![[1860-5397-10-216-i5]](/bjoc/content/inline/1860-5397-10-216-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of rac-42. Reagents and conditions: a) MCPBA, CH2Cl2, 0 °C to rt, 45 min; b) TFA, H2O, THF, 60 °C overnight, c) acetone, PTSA, 3 h, rt; d) NaOMe, MeOH, 48 h, rt, 43% over 5 steps; e) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C to rt, 2 h; f) diethyl allylphosphonate, BuLi, THF, 0 °C to rt, overnight, 30% over 2 steps; g) 60% AcOH, 60 °C, 3 h, 63%. MCPBA = m-chloroperbenzoic acid, TFA = trifluoacetic acid, PTSA = p-toluenesulfonic acid.
Scheme 5: Synthesis of rac-42. Reagents and conditions: a) MCPBA, CH2Cl2, 0 °C to rt, 45 min; b) TFA, H2O, TH...
In addition, enantiomerically pure substrate 43 was synthetised from D-glucose in 11 steps according to a protocol of Szewczyk [35] (Figure 3).
![[1860-5397-10-216-3]](/bjoc/content/figures/1860-5397-10-216-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Pd-catalysed cyclisations of unsaturated polyols
Prepared substrates were then subjected to the Pd-catalysed transformation under several reaction conditions. At first, we tried the reaction conditions which were recently developed for the bicyclisation of α-O-benzyl-protected polyols bearing a terminal alkene moiety [22]. Thus, the reactions incorporating the PdII–Pd0 catalytic cycle (Scheme 6) were carried out using PdCl2 (0.1 equiv) as a catalyst, CuCl2 (3 equiv) as a reoxidant, NaOAc (3 equiv) as a buffer in AcOH at room temperature (Table 1, Method A, see Supporting Information File 1 for full experimental data).
Table 1: Pd-Catalysed cyclisations of unsaturated polyols.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-216-i8.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate | Reaction conditionsa | Product(s) | Yield (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-216-i9.svg?max-width=637&scale=1.0)
11 |
Method A |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-216-i10.svg?max-width=637&scale=1.0)
44 |
79 [23] |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-216-i11.svg?max-width=637&scale=1.0)
12 |
Method A
Method C |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-216-i12.svg?max-width=637&scale=1.0)
45 |
63
40 |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-216-i13.svg?max-width=637&scale=1.0)
13 |
Method A | Complex mixture | |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-216-i14.svg?max-width=637&scale=1.0)
33 |
Method A
Method B |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-216-i15.svg?max-width=637&scale=1.0)
51 + 52 |
15 (51), 25 (52)
65 (52) |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-216-i16.svg?max-width=637&scale=1.0)
E-21 |
Method A |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-216-i17.svg?max-width=637&scale=1.0)
46 |
30 |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-216-i18.svg?max-width=637&scale=1.0)
24–26 |
Method A |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-216-i19.svg?max-width=637&scale=1.0)
47 + 48 |
66 (47/48, 5:3) |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-216-i20.svg?max-width=637&scale=1.0)
28 |
Method A |
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-216-i21.svg?max-width=637&scale=1.0)
49 + 50 |
54 (49/50, 5:3) |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-216-i22.svg?max-width=637&scale=1.0)
37 |
Method A
Method B Pd(PPh3)4b |
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-216-i23.svg?max-width=637&scale=1.0)
56 + 57 |
70 (56/57, 1:3)
69 (56/57, 1:3) 84 (56/57, 1:3) |
| 9 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-216-i24.svg?max-width=637&scale=1.0)
35 |
Method A |
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-216-i25.svg?max-width=637&scale=1.0)
53 |
33 |
| 10 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-216-i26.svg?max-width=637&scale=1.0)
30 |
Method A
Method B |
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-216-i27.svg?max-width=637&scale=1.0)
54 + 55 |
38 (54), 35 (55)
70 (55) |
| 11 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-216-i28.svg?max-width=637&scale=1.0)
threo-9 |
Method B |
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-216-i29.svg?max-width=637&scale=1.0)
58 |
78 |
aMethod A: PdCl2 (0.1 equiv), CuCl2 (3 equiv), NaOAc (3 equiv), AcOH, rt.; method B: PdCl2(MeCN)2 (0.1 equiv), BuLi (2 equiv), CuCl2 (3 equiv), LiCl (3 equiv), THF, rt; method C: Pd(OAc)2 (0.1 equiv), PhI(OAc)2 (2 equiv), Me4N+Cl− (1 equiv), NaOAc (1 equiv), AcOH, rt. bLit. [36-38] Pd(PPh3)4 (0.1 equiv), THF, rt.
It is clear, that the chemoselectivity of the cyclisation reaction directly correlates to the nature of the C=C bond of the substrate. Also, the configuration and the position of participating substituents have immense influence on the reaction output and the obtained results showed that the cyclisation reactions can progress through four different transformation pathways, yielding various types of products (I, II, III and IV).
Under these conditions (method A) the reactions of the simplest alkenols having a terminal alkene moiety and those with an α-O-protected allylic system, i.e., 11 [23] (Table 1, entry 1) and 12 (Table 1, entry 2) provided corresponding bicycles of the type I. Likewise, the alkenediol 33 without α-allylic hydroxy group provided bicyclic product 51, however, as a minor product in only 15% yield along with the furan compound 52 (Table 1, entry 4). The furan derivative 52 in this reaction was probably formed through a monocyclisation, followed by β-H−-elimination and aromatisation. In the case of substrate 13 having an unprotected α-hydroxy group, the reaction provided only a mixture of unidentified products (Table 1, entry 3). These results are consistent with previous observations and it is evident that a protection of the α-allylic hydroxy function is required for a successful bicyclisation reaction.
Next, we have also examined the compatibility of substrates having a symmetrically disubstituted C=C bond in the cyclisation reactions. Unfortunately, butadienes rac-42 and 43 underwent uncontrollable transformations under these conditions providing a complex mixture of products. Similarly, the transformations of substrates 20, Z-21, 22 and 34 failed, while the reaction of E-21 (Table 1, entry 5) afforded surprisingly tetrahydrofuran derivative 46 as a product of a Wacker-type cyclisation.
Interestingly, the reactions of alkenols having an additional allylic OR group provided only products of type II. Thus, diastereomeric mixtures of vinyltetrahydrofurans 47, 48 (Table 1, entry 6), 49, 50 (Table 1, entry 7) and 56, 57 (Table 1, entry 8) were formed starting from alkenols 24–26, 28 and 37. Formation of these products (type II) in this type of PdII-catalysed cyclisation [21,39-43] possibly involved an intramolecular Wacker-type reaction to form PdII-intermediate E and subsequent regeneration of the PdII-catalyst via cleavage of the C–OR bond (Scheme 6).
![[1860-5397-10-216-i6]](/bjoc/content/inline/1860-5397-10-216-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Suggested mechanisms for PdII–Pd0, PdII–PdIV and PdII-chloro/cyclisation of unsaturated polyols.
Scheme 6: Suggested mechanisms for PdII–Pd0, PdII–PdIV and PdII-chloro/cyclisation of unsaturated polyols.
Surprisingly, the reaction of 1,1-disubstituted alkenes 30, 35 (Table 1, entries 10 and 9) provided only chlorinated tetrahydropyran (type III) and tetrahydrofuran (type IV) derivatives. The chloroderivatives III and IV were probably formed from σ-alkyl PdII-complexes F and G by reductive elimination of Pd0 (Scheme 6). Additionally, the X-ray analysis [44] of 53 confirmed the absolute configuration and structure of the six-membered heterocycle (Figure 4).
![[1860-5397-10-216-4]](/bjoc/content/figures/1860-5397-10-216-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: An ORTEP [44] view of crystal and molecular structure of 53.
Figure 4: An ORTEP [44] view of crystal and molecular structure of 53.
Recently, Wolfe reported a tetrahydrofuran-forming reaction via Pd-catalysed carboetherification [45-49] under strong basic conditions in the presence of a phosphine ligand. In order to enhance the ligand affinity of the hydroxy group in the formation of σ-palladiumII-complex D (Scheme 6), we have decided to adopt the described conditions and to examine the transformation of alkenols in the presence of PdCl2(MeCN)2, butyllithium and LiCl (method B). Unfortunately, these experiments in most cases did not afford any cyclisation products and reactions of non-terminal olefinic substrates 20–23, 34, rac-42 and 43 provided only a complex mixture of inseparable products. However, terminal olefins 30 and threo-9 underwent chlorocyclisation most probably due to the presence of an excess of chloride anions (Table 1, entries 10 and 11). Interestingly, this chlorocyclisation reaction proceeded with high trans-diastereoselectivity, which is in accordance with Wolfe´s TS model [17,18]. In both cases, only 2,3-trans diastereomers 55 and 58 were isolated in good yields. In addition, this reaction represents a new synthetic access to the 3-hydroxy-2,3-trans-tetrahydrofuran skeleton and is complementary to the known X+-mediated cyclisation methodology producing exclusively 2,3-cis-diastereomer [14,15,50,51].
Based on the published findings, we have also examined the cyclisation reactions incorporating the PdII–PdIV catalytic cycle [52,53] (Scheme 6). The experiments were carried out using Pd(OAc)2 salt as a catalyst, PhI(OAc)2 as reoxidant, AcONa and Me4N+Cl− as buffer in AcOH (method C). Unfortunately, all reactions and their modifications (temperature, solvents: AcOH, AcOH–H2O, NMP, DMF, MeOH, THF, Et2O, DCM, CHCl3) did not provide cyclisation products and only complex mixtures of β-H−-elimination and consequential products were observed. Only one exception to previously unpleasant findings was a reaction of O-silyl-protected triol 12, which provided the bicycle 45 but only in a decreased yield of 40% (Table 1, entry 2).
To show the usefulness of such cyclisation products, we have investigated the possibility of employing prepared tetrahydrofuran derivatives bearing suitable moieties in the next cyclisation step (Scheme 7).
![[1860-5397-10-216-i7]](/bjoc/content/inline/1860-5397-10-216-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Bicyclisation of 55–58. Reagents and conditions: a) NaH, DMF, 50 °C, 2 h; b) I2, CH3CN, rt, overnight.
Scheme 7: Bicyclisation of 55–58. Reagents and conditions: a) NaH, DMF, 50 °C, 2 h; b) I2, CH3CN, rt, overnig...
Gratifyingly, the chloromethyltetrahydrofurans 55 and 58 were both transformed into the bicyclic products 59 and 44 by treatment with sodium hydride in DMF. Also, this transformation step has approved the relative configurations of substituents on the tetrahydrofuran ring. Interestingly, an iodo-cyclisation reaction of a 1:3 diastereomeric mixture of vinyltetrahydrofurans 56 and 57 in acetonitrile provided only one corresponding product derived from 56. Thus, pure (4R,7R)-4,7-disubstituted 2,6-dioxabicyclo[3.2.1]octane 60 was isolated in 52% yield. The trans arrangement of the substituents at C4/C7 in the product of the 6-endo-trig cyclisation was determined by means of 1H NMR and NOE interactions.
In conclusion, we have also shown the possibility to construct interesting bicyclic intermediates in a 2 step sequence combining the PdII-catalysed cyclisation [36-40] or Pd0-allylic substitution [42,43] of alkenols having an allylic OR group and additional halocyclisation.
Conclusion
In summary, we have developed the syntheses of several unsaturated alcohols. The chiral alkenols 20–28, 34–37 and 43 represent useful C5–C12 chain building blocks.
The stereoselective PdII–CuII-catalysed cyclisation [22] and its substrate scope has been investigated. The bicyclisation reaction appears to be applicable only to terminal olefinic substrates, while the reaction of alkenols bearing nonterminal and/or disubstituted olefins did not provide bicyclisation products. Moreover, alkenes having both an allylic OR group and a hydroxylated tether underwent intramolecular Wacker-type cyclisation affording corresponding vinyltetrahydrofurans, which constitute useful intermediates for the synthesis of naturally occurring tetrahydrofuran derivatives.
We have also explored the Pd-cyclisation of unsaturated polyols in the presence of a strong base or a high oxidation state palladium catalyst. The PdII–PdIV-catalysed transformation toward the bicyclisation product proceeded only on the O-silyl-protected triol 12. The PdII-cyclisation of terminal olefinic substrates in the presence of BuLi and LiCl provided selectively 5-exo-trig cyclisation products with excellent 2,3-trans diastereoselectivity.
Finally, we have also proposed a synthetic access to the dioxaheptane core of natural C15 acetogenins and dioxaoctane, a substructure of the macrolide-polyether antibiotic sorangicin A and aurovertins. Thus, Pd-catalysed cyclisation of appropriate alkenols to tetrahydrofurans and subsequent iodo-cyclisation yielded properly substituted 2,5-dioxabicyclo[2.2.1]heptane and 2,6-dioxabicyclo[3.2.1]octane, respectively with defined stereochemistry and excellent diastereoselectivity. The further synthetic studies toward ocellenynes are currently underway.
Supporting Information
| Supporting Information File 1: Mechanisms, general information, experimental procedures and spectroscopic data for all new compounds. | ||
| Format: PDF | Size: 696.9 KB | Download |
| Supporting Information File 2: 1H NMR and 13C NMR spectra of selected compounds. | ||
| Format: PDF | Size: 3.9 MB | Download |
| Supporting Information File 3: X-ray crystal structure analysis of 53. | ||
| Format: PDF | Size: 574.5 KB | Download |
References
-
Gracza, T.; Jäger, V. Synlett 1992, 191–193. doi:10.1055/s-1992-21309
Return to citation in text: [1] -
Cao, S.-G.; Wu, X.-H.; Sim, K.-Y.; Tan, B. K. H.; Pereira, J. T.; Goh, S.-H. Tetrahedron 1998, 54, 2143–2148. doi:10.1016/S0040-4020(97)10422-7
Return to citation in text: [1] -
Malmstrøm, J.; Christophersen, C.; Barrero, A. F.; Oltra, J. E.; Justicia, J.; Rosales, A. J. Nat. Prod. 2002, 65, 364–367. doi:10.1021/np0103214
Return to citation in text: [1] -
Howard, B. H.; Raistrick, H. Biochem. J. 1949, 44, 227–233.
Return to citation in text: [1] -
Beutler, J. A.; Hilton, B. D.; Clark, P.; Tempesta, M. S.; Corley, D. G. J. Nat. Prod. 1988, 51, 562–566. doi:10.1021/np50057a018
Return to citation in text: [1] -
Schulte, G. R.; Chung, M. C. H.; Scheuer, P. J. J. Org. Chem. 1981, 46, 3870–3873. doi:10.1021/jo00332a022
Return to citation in text: [1] -
Wright, A. D.; König, G. M.; de Nys, R.; Sticher, O. J. Nat. Prod. 1993, 56, 394–401. doi:10.1021/np50093a012
Return to citation in text: [1] -
Jansen, R.; Wray, V.; Irschik, H.; Reichenbach, H.; Höfle, G. Tetrahedron Lett. 1985, 26, 6031–6034. doi:10.1016/S0040-4039(00)95117-7
Return to citation in text: [1] -
Baldwin, C. L.; Weaver, L. C.; Brooker, R. M.; Jacobsen, T. N.; Osborne, C. E., Jr.; Nash, H. A. Lloydia 1964, 27, 88–95.
Return to citation in text: [1] -
Osselton, M. D.; Baum, H.; Beechey, R. B. Biochem. Soc. Trans. 1974, 200–202.
Return to citation in text: [1] -
Mulheirn, L. J.; Beechey, R. B.; Leworthy, D. P.; Osselton, M. D. J. Chem. Soc., Chem. Commun. 1974, 874–876. doi:10.1039/C39740000874
Return to citation in text: [1] -
Wang, F.; Luo, D.-Q.; Liu, J.-K. J. Antibiot. 2005, 58, 412–415. doi:10.1038/ja.2005.53
Return to citation in text: [1] -
Lai, S.; Matsunaga, K.; Shizuri, Y.; Yamamura, S. Tetrahedron Lett. 1990, 31, 5503–5506. doi:10.1016/S0040-4039(00)97883-3
Return to citation in text: [1] -
Cardillo, G.; Orena, M. Tetrahedron 1990, 46, 3321–3408. doi:10.1016/S0040-4020(01)81510-6
Return to citation in text: [1] [2] -
Cardillo, G.; Orena, M. Formation of C–O bonds by cyclization onto olefinic double bonds forming lactones and ethers. In Houben-Weyl, 4th ed.; Helmchen, G.; Hoffmann, R.; Mulzer, J., Eds.; Thieme: Stuttgart, 1995; Vol. E21e, pp 4698–4817.
Return to citation in text: [1] [2] -
Wolfe, J. P. Synthesis of saturated heterocycles via metal-catalyzed alkene carboamination or carboalkoxylation reactions. In Synthesis of Heterocycles via Metal-Catalyzed Reactions that Generate One or More Carbon-Heteroatom Bonds; Wolfe, J. P., Ed.; Topics in Heterocyclic Chemistry, Vol. 32; Springer: Berlin, 2013; pp 1–38.
Return to citation in text: [1] -
Wolfe, J. P. Eur. J. Org. Chem. 2007, 571–582. doi:10.1002/ejoc.200600767
Return to citation in text: [1] [2] -
Wolfe, J. P. Synlett 2008, 2913–2937. doi:10.1055/s-0028-1087339
Return to citation in text: [1] [2] -
Gracza, T. Intramolecular oxycarbonylation in stereoselective synthesis. In Stereoselective Synthesis of Drugs and Natural Products; Andrushko, V.; Andrushko, N., Eds.; Wiley: Hoboken, New Jersey, 2013; Vol. 1, pp 421–440. doi:10.1002/9781118596784.ssd015
Return to citation in text: [1] -
Doháňošová, J.; Gracza, T. Molecules 2013, 18, 6173–6192. doi:10.3390/molecules18066173
Return to citation in text: [1] -
Muzart, J. J. Mol. Catal. A: Chem. 2010, 319, 1–29. doi:10.1016/j.molcata.2009.12.001
Return to citation in text: [1] [2] -
Babjak, M.; Remeň, L.; Szolcsányi, P.; Zálupský, P.; Mikloš, D.; Gracza, T. J. Organomet. Chem. 2006, 691, 928–940. doi:10.1016/j.jorganchem.2005.10.036
Return to citation in text: [1] [2] [3] [4] -
Babjak, M.; Remeň, L.; Karlubíková, O.; Gracza, T. Synlett 2005, 1609–1611. doi:10.1055/s-2005-869840
Return to citation in text: [1] [2] [3] -
Palík, M.; Karlubíková, O.; Lásiková, A.; Kožíšek, J.; Gracza, T. Eur. J. Org. Chem. 2009, 709–715. doi:10.1002/ejoc.200801070
Return to citation in text: [1] -
Palík, M.; Karlubíková, O.; Lackovičová, D.; Lásiková, A.; Gracza, T. Tetrahedron 2010, 66, 5244–5249. doi:10.1016/j.tet.2010.04.074
Return to citation in text: [1] -
Bravo, F.; Kassou, M.; Castillón, S. Tetrahedron Lett. 1999, 40, 1187–1190. doi:10.1016/S0040-4039(98)02561-1
Return to citation in text: [1] -
Fürstner, A.; Jumbam, D.; Teslic, J.; Weidmann, H. J. Org. Chem. 1991, 56, 2213–2217. doi:10.1021/jo00006a047
Return to citation in text: [1] -
Koppenhoefer, B.; Walser, M.; Schröter, D.; Häfele, B.; Jäger, V. Tetrahedron 1987, 43, 2059–2064. doi:10.1016/S0040-4020(01)86787-9
Return to citation in text: [1] -
Jäger, V.; Schröter, D.; Koppenhoefer, B. Tetrahedron 1991, 47, 2195–2210. doi:10.1016/S0040-4020(01)96130-7
Return to citation in text: [1] -
Babjak, M.; Zálupský, P.; Gracza, T. ARKIVOC 2005, No. v, 45–57.
Return to citation in text: [1] -
Sánches-Sancho, F.; Valverde, S.; Herradón, B. Tetrahedron: Asymmetry 1996, 7, 3209–3246. doi:10.1016/0957-4166(96)00424-7
Return to citation in text: [1] [2] -
Chandrasekhar, S.; Parida, B. B.; Rambabu, C. J. Org. Chem. 2008, 73, 7826–7828. doi:10.1021/jo801377s
Return to citation in text: [1] [2] [3] -
Jiao, P.; Kawasaki, M.; Yamamoto, H. Angew. Chem., Int. Ed. 2009, 48, 3333–3336. doi:10.1002/anie.200900682
Return to citation in text: [1] [2] -
Prat, I.; Font, D.; Company, A.; Junge, K.; Ribas, X.; Beller, M.; Costas, M. Adv. Synth. Catal. 2013, 355, 947–956. doi:10.1002/adsc.201200938
Return to citation in text: [1] -
Jarosz, S.; Skóra, S.; Szewczyk, K. Tetrahedron: Asymmetry 2000, 11, 1997–2006. doi:10.1016/S0957-4166(00)00135-X
Return to citation in text: [1] -
Trost, B. M.; Tenaglia, A. Tetrahedron Lett. 1988, 29, 2927–2930. doi:10.1016/0040-4039(88)85049-4
Return to citation in text: [1] [2] -
Passiniemi, M.; Koskinen, A. M. P. Tetrahedron Lett. 2008, 49, 980–983. doi:10.1016/j.tetlet.2007.12.014
Return to citation in text: [1] [2] -
Roulland, E. Angew. Chem., Int. Ed. 2008, 47, 3762–3765. doi:10.1002/anie.200800585
Return to citation in text: [1] [2] -
Uenishi, J.; Ohmi, M.; Ueda, A. Tetrahedron: Asymmetry 2005, 16, 1299–1303. doi:10.1016/j.tetasy.2005.02.006
Return to citation in text: [1] [2] -
Uenishi, J.; Ohmi, M. Angew. Chem., Int. Ed. 2005, 44, 2756–2760. doi:10.1002/anie.200500029
Return to citation in text: [1] [2] -
Kawai, N.; Lagrange, J.-M.; Ohmi, M.; Uenishi, J. J. Org. Chem. 2006, 71, 4530–4537. doi:10.1021/jo060415o
Return to citation in text: [1] -
Kawai, N.; Lagrange, J.-M.; Uenishi, J. Eur. J. Org. Chem. 2007, 2808–2814. doi:10.1002/ejoc.200601103
Return to citation in text: [1] [2] -
Kawai, N.; Hande, S. M.; Uenishi, J. Tetrahedron 2007, 63, 9049–9056. doi:10.1016/j.tet.2007.06.081
Return to citation in text: [1] [2] -
CCDC 963459 (for compound 53) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif
Return to citation in text: [1] [2] -
Wolfe, J. P.; Rossi, M. A. J. Am. Chem. Soc. 2004, 126, 1620–1621. doi:10.1021/ja0394838
Return to citation in text: [1] -
Hay, M. B.; Hardin, A. R.; Wolfe, J. P. J. Org. Chem. 2005, 70, 3099–3107. doi:10.1021/jo050022+
Return to citation in text: [1] -
Hay, M. B.; Wolfe, J. P. J. Am. Chem. Soc. 2005, 127, 16468–16476. doi:10.1021/ja054754v
Return to citation in text: [1] -
Hay, M. B.; Wolfe, J. P. Tetrahedron Lett. 2006, 47, 2793–2796. doi:10.1016/j.tetlet.2006.02.066
Return to citation in text: [1] -
Ward, A. F.; Wolfe, J. P. Org. Lett. 2010, 12, 1268–1271. doi:10.1021/ol1001472
Return to citation in text: [1] -
Chamberlin, A. R.; Mulholland, R. L., Jr.; Kahn, S. D.; Hehre, W. J. J. Am. Chem. Soc. 1987, 109, 672–677. doi:10.1021/ja00237a006
Return to citation in text: [1] -
Kahn, S. D.; Hehre, W. J. J. Am. Chem. Soc. 1987, 109, 666–671. doi:10.1021/ja00237a005
Return to citation in text: [1] -
Muñiz, K. Angew. Chem., Int. Ed. 2009, 48, 9412–9423. doi:10.1002/anie.200903671
Return to citation in text: [1] -
Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Chem. Soc. Rev. 2010, 39, 712–733. doi:10.1039/b809912j
Return to citation in text: [1]
| 22. | Babjak, M.; Remeň, L.; Szolcsányi, P.; Zálupský, P.; Mikloš, D.; Gracza, T. J. Organomet. Chem. 2006, 691, 928–940. doi:10.1016/j.jorganchem.2005.10.036 |
| 23. | Babjak, M.; Remeň, L.; Karlubíková, O.; Gracza, T. Synlett 2005, 1609–1611. doi:10.1055/s-2005-869840 |
| 36. | Trost, B. M.; Tenaglia, A. Tetrahedron Lett. 1988, 29, 2927–2930. doi:10.1016/0040-4039(88)85049-4 |
| 37. | Passiniemi, M.; Koskinen, A. M. P. Tetrahedron Lett. 2008, 49, 980–983. doi:10.1016/j.tetlet.2007.12.014 |
| 38. | Roulland, E. Angew. Chem., Int. Ed. 2008, 47, 3762–3765. doi:10.1002/anie.200800585 |
| 6. | Schulte, G. R.; Chung, M. C. H.; Scheuer, P. J. J. Org. Chem. 1981, 46, 3870–3873. doi:10.1021/jo00332a022 |
| 7. | Wright, A. D.; König, G. M.; de Nys, R.; Sticher, O. J. Nat. Prod. 1993, 56, 394–401. doi:10.1021/np50093a012 |
| 26. | Bravo, F.; Kassou, M.; Castillón, S. Tetrahedron Lett. 1999, 40, 1187–1190. doi:10.1016/S0040-4039(98)02561-1 |
| 14. | Cardillo, G.; Orena, M. Tetrahedron 1990, 46, 3321–3408. doi:10.1016/S0040-4020(01)81510-6 |
| 15. | Cardillo, G.; Orena, M. Formation of C–O bonds by cyclization onto olefinic double bonds forming lactones and ethers. In Houben-Weyl, 4th ed.; Helmchen, G.; Hoffmann, R.; Mulzer, J., Eds.; Thieme: Stuttgart, 1995; Vol. E21e, pp 4698–4817. |
| 50. | Chamberlin, A. R.; Mulholland, R. L., Jr.; Kahn, S. D.; Hehre, W. J. J. Am. Chem. Soc. 1987, 109, 672–677. doi:10.1021/ja00237a006 |
| 51. | Kahn, S. D.; Hehre, W. J. J. Am. Chem. Soc. 1987, 109, 666–671. doi:10.1021/ja00237a005 |
| 4. | Howard, B. H.; Raistrick, H. Biochem. J. 1949, 44, 227–233. |
| 5. | Beutler, J. A.; Hilton, B. D.; Clark, P.; Tempesta, M. S.; Corley, D. G. J. Nat. Prod. 1988, 51, 562–566. doi:10.1021/np50057a018 |
| 27. | Fürstner, A.; Jumbam, D.; Teslic, J.; Weidmann, H. J. Org. Chem. 1991, 56, 2213–2217. doi:10.1021/jo00006a047 |
| 52. | Muñiz, K. Angew. Chem., Int. Ed. 2009, 48, 9412–9423. doi:10.1002/anie.200903671 |
| 53. | Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Chem. Soc. Rev. 2010, 39, 712–733. doi:10.1039/b809912j |
| 3. | Malmstrøm, J.; Christophersen, C.; Barrero, A. F.; Oltra, J. E.; Justicia, J.; Rosales, A. J. Nat. Prod. 2002, 65, 364–367. doi:10.1021/np0103214 |
| 22. | Babjak, M.; Remeň, L.; Szolcsányi, P.; Zálupský, P.; Mikloš, D.; Gracza, T. J. Organomet. Chem. 2006, 691, 928–940. doi:10.1016/j.jorganchem.2005.10.036 |
| 45. | Wolfe, J. P.; Rossi, M. A. J. Am. Chem. Soc. 2004, 126, 1620–1621. doi:10.1021/ja0394838 |
| 46. | Hay, M. B.; Hardin, A. R.; Wolfe, J. P. J. Org. Chem. 2005, 70, 3099–3107. doi:10.1021/jo050022+ |
| 47. | Hay, M. B.; Wolfe, J. P. J. Am. Chem. Soc. 2005, 127, 16468–16476. doi:10.1021/ja054754v |
| 48. | Hay, M. B.; Wolfe, J. P. Tetrahedron Lett. 2006, 47, 2793–2796. doi:10.1016/j.tetlet.2006.02.066 |
| 49. | Ward, A. F.; Wolfe, J. P. Org. Lett. 2010, 12, 1268–1271. doi:10.1021/ol1001472 |
| 2. | Cao, S.-G.; Wu, X.-H.; Sim, K.-Y.; Tan, B. K. H.; Pereira, J. T.; Goh, S.-H. Tetrahedron 1998, 54, 2143–2148. doi:10.1016/S0040-4020(97)10422-7 |
| 24. | Palík, M.; Karlubíková, O.; Lásiková, A.; Kožíšek, J.; Gracza, T. Eur. J. Org. Chem. 2009, 709–715. doi:10.1002/ejoc.200801070 |
| 25. | Palík, M.; Karlubíková, O.; Lackovičová, D.; Lásiková, A.; Gracza, T. Tetrahedron 2010, 66, 5244–5249. doi:10.1016/j.tet.2010.04.074 |
| 17. | Wolfe, J. P. Eur. J. Org. Chem. 2007, 571–582. doi:10.1002/ejoc.200600767 |
| 18. | Wolfe, J. P. Synlett 2008, 2913–2937. doi:10.1055/s-0028-1087339 |
| 14. | Cardillo, G.; Orena, M. Tetrahedron 1990, 46, 3321–3408. doi:10.1016/S0040-4020(01)81510-6 |
| 15. | Cardillo, G.; Orena, M. Formation of C–O bonds by cyclization onto olefinic double bonds forming lactones and ethers. In Houben-Weyl, 4th ed.; Helmchen, G.; Hoffmann, R.; Mulzer, J., Eds.; Thieme: Stuttgart, 1995; Vol. E21e, pp 4698–4817. |
| 19. | Gracza, T. Intramolecular oxycarbonylation in stereoselective synthesis. In Stereoselective Synthesis of Drugs and Natural Products; Andrushko, V.; Andrushko, N., Eds.; Wiley: Hoboken, New Jersey, 2013; Vol. 1, pp 421–440. doi:10.1002/9781118596784.ssd015 |
| 20. | Doháňošová, J.; Gracza, T. Molecules 2013, 18, 6173–6192. doi:10.3390/molecules18066173 |
| 21. | Muzart, J. J. Mol. Catal. A: Chem. 2010, 319, 1–29. doi:10.1016/j.molcata.2009.12.001 |
| 44. | CCDC 963459 (for compound 53) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif |
| 13. | Lai, S.; Matsunaga, K.; Shizuri, Y.; Yamamura, S. Tetrahedron Lett. 1990, 31, 5503–5506. doi:10.1016/S0040-4039(00)97883-3 |
| 22. | Babjak, M.; Remeň, L.; Szolcsányi, P.; Zálupský, P.; Mikloš, D.; Gracza, T. J. Organomet. Chem. 2006, 691, 928–940. doi:10.1016/j.jorganchem.2005.10.036 |
| 23. | Babjak, M.; Remeň, L.; Karlubíková, O.; Gracza, T. Synlett 2005, 1609–1611. doi:10.1055/s-2005-869840 |
| 44. | CCDC 963459 (for compound 53) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif |
| 9. | Baldwin, C. L.; Weaver, L. C.; Brooker, R. M.; Jacobsen, T. N.; Osborne, C. E., Jr.; Nash, H. A. Lloydia 1964, 27, 88–95. |
| 10. | Osselton, M. D.; Baum, H.; Beechey, R. B. Biochem. Soc. Trans. 1974, 200–202. |
| 11. | Mulheirn, L. J.; Beechey, R. B.; Leworthy, D. P.; Osselton, M. D. J. Chem. Soc., Chem. Commun. 1974, 874–876. doi:10.1039/C39740000874 |
| 12. | Wang, F.; Luo, D.-Q.; Liu, J.-K. J. Antibiot. 2005, 58, 412–415. doi:10.1038/ja.2005.53 |
| 23. | Babjak, M.; Remeň, L.; Karlubíková, O.; Gracza, T. Synlett 2005, 1609–1611. doi:10.1055/s-2005-869840 |
| 8. | Jansen, R.; Wray, V.; Irschik, H.; Reichenbach, H.; Höfle, G. Tetrahedron Lett. 1985, 26, 6031–6034. doi:10.1016/S0040-4039(00)95117-7 |
| 16. | Wolfe, J. P. Synthesis of saturated heterocycles via metal-catalyzed alkene carboamination or carboalkoxylation reactions. In Synthesis of Heterocycles via Metal-Catalyzed Reactions that Generate One or More Carbon-Heteroatom Bonds; Wolfe, J. P., Ed.; Topics in Heterocyclic Chemistry, Vol. 32; Springer: Berlin, 2013; pp 1–38. |
| 17. | Wolfe, J. P. Eur. J. Org. Chem. 2007, 571–582. doi:10.1002/ejoc.200600767 |
| 18. | Wolfe, J. P. Synlett 2008, 2913–2937. doi:10.1055/s-0028-1087339 |
| 21. | Muzart, J. J. Mol. Catal. A: Chem. 2010, 319, 1–29. doi:10.1016/j.molcata.2009.12.001 |
| 39. | Uenishi, J.; Ohmi, M.; Ueda, A. Tetrahedron: Asymmetry 2005, 16, 1299–1303. doi:10.1016/j.tetasy.2005.02.006 |
| 40. | Uenishi, J.; Ohmi, M. Angew. Chem., Int. Ed. 2005, 44, 2756–2760. doi:10.1002/anie.200500029 |
| 41. | Kawai, N.; Lagrange, J.-M.; Ohmi, M.; Uenishi, J. J. Org. Chem. 2006, 71, 4530–4537. doi:10.1021/jo060415o |
| 42. | Kawai, N.; Lagrange, J.-M.; Uenishi, J. Eur. J. Org. Chem. 2007, 2808–2814. doi:10.1002/ejoc.200601103 |
| 43. | Kawai, N.; Hande, S. M.; Uenishi, J. Tetrahedron 2007, 63, 9049–9056. doi:10.1016/j.tet.2007.06.081 |
| 31. | Sánches-Sancho, F.; Valverde, S.; Herradón, B. Tetrahedron: Asymmetry 1996, 7, 3209–3246. doi:10.1016/0957-4166(96)00424-7 |
| 28. | Koppenhoefer, B.; Walser, M.; Schröter, D.; Häfele, B.; Jäger, V. Tetrahedron 1987, 43, 2059–2064. doi:10.1016/S0040-4020(01)86787-9 |
| 29. | Jäger, V.; Schröter, D.; Koppenhoefer, B. Tetrahedron 1991, 47, 2195–2210. doi:10.1016/S0040-4020(01)96130-7 |
| 36. | Trost, B. M.; Tenaglia, A. Tetrahedron Lett. 1988, 29, 2927–2930. doi:10.1016/0040-4039(88)85049-4 |
| 37. | Passiniemi, M.; Koskinen, A. M. P. Tetrahedron Lett. 2008, 49, 980–983. doi:10.1016/j.tetlet.2007.12.014 |
| 38. | Roulland, E. Angew. Chem., Int. Ed. 2008, 47, 3762–3765. doi:10.1002/anie.200800585 |
| 39. | Uenishi, J.; Ohmi, M.; Ueda, A. Tetrahedron: Asymmetry 2005, 16, 1299–1303. doi:10.1016/j.tetasy.2005.02.006 |
| 40. | Uenishi, J.; Ohmi, M. Angew. Chem., Int. Ed. 2005, 44, 2756–2760. doi:10.1002/anie.200500029 |
| 42. | Kawai, N.; Lagrange, J.-M.; Uenishi, J. Eur. J. Org. Chem. 2007, 2808–2814. doi:10.1002/ejoc.200601103 |
| 43. | Kawai, N.; Hande, S. M.; Uenishi, J. Tetrahedron 2007, 63, 9049–9056. doi:10.1016/j.tet.2007.06.081 |
| 22. | Babjak, M.; Remeň, L.; Szolcsányi, P.; Zálupský, P.; Mikloš, D.; Gracza, T. J. Organomet. Chem. 2006, 691, 928–940. doi:10.1016/j.jorganchem.2005.10.036 |
| 34. | Prat, I.; Font, D.; Company, A.; Junge, K.; Ribas, X.; Beller, M.; Costas, M. Adv. Synth. Catal. 2013, 355, 947–956. doi:10.1002/adsc.201200938 |
| 35. | Jarosz, S.; Skóra, S.; Szewczyk, K. Tetrahedron: Asymmetry 2000, 11, 1997–2006. doi:10.1016/S0957-4166(00)00135-X |
| 33. | Jiao, P.; Kawasaki, M.; Yamamoto, H. Angew. Chem., Int. Ed. 2009, 48, 3333–3336. doi:10.1002/anie.200900682 |
| 33. | Jiao, P.; Kawasaki, M.; Yamamoto, H. Angew. Chem., Int. Ed. 2009, 48, 3333–3336. doi:10.1002/anie.200900682 |
| 32. | Chandrasekhar, S.; Parida, B. B.; Rambabu, C. J. Org. Chem. 2008, 73, 7826–7828. doi:10.1021/jo801377s |
| 32. | Chandrasekhar, S.; Parida, B. B.; Rambabu, C. J. Org. Chem. 2008, 73, 7826–7828. doi:10.1021/jo801377s |
| 31. | Sánches-Sancho, F.; Valverde, S.; Herradón, B. Tetrahedron: Asymmetry 1996, 7, 3209–3246. doi:10.1016/0957-4166(96)00424-7 |
| 32. | Chandrasekhar, S.; Parida, B. B.; Rambabu, C. J. Org. Chem. 2008, 73, 7826–7828. doi:10.1021/jo801377s |
© 2014 Palík et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)