Abstract
The near-unlimited availability of CO2 has stimulated a growing research effort in creating value-added products from this greenhouse gas. This paper presents the trends on the most important methods used in the electrochemical synthesis of carboxylic acids from carbon dioxide. An overview is given of different substrate groups which form carboxylic acids upon CO2 fixation, including mechanistic considerations. While most work focuses on the electrocarboxylation of substrates with sacrificial anodes, this review considers the possibilities and challenges of implementing other synthetic methodologies. In view of potential industrial application, the choice of reactor setup, electrode type and reaction pathway has a large influence on the sustainability and efficiency of the process.
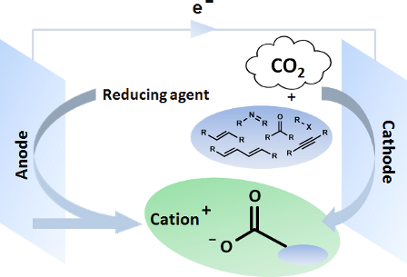
Graphical Abstract
Introduction
Carbon dioxide recycling
Implementing sustainable, resource-efficient chemical processes to meet the world’s growing demand for energy and chemicals is one of today’s major challenges. Depletion of fossil resources and the ongoing increase of atmospheric carbon dioxide levels urge to investigate alternative pathways to close the carbon cycle. The use of carbon dioxide as chemical feedstock is a logical strategy for this purpose, creating economical benefit from its capture. At the moment, only a very minor fraction (<1%) of anthropogenic CO2 emissions is actually used [1]. As an end product of combustion, CO2 has a high thermodynamic stability (ΔGf° = –396 kJ/mol), often demanding for an energy intensive activation. The hydrogenation of CO2 to methane for example is an industrial process which requires high temperatures and pressures to activate CO2 [2,3]. Efficient chemical incorporation of CO2 is limited to rather reactive substrates, like epoxides [4-6] and amines [7-9] to produce cyclic carbonates and carbamates, respectively, and even then, elevated reaction temperatures and/or complex catalyst systems are sometimes required. Urea is the main industrial product for which CO2 is applied as a C1 building block in a reaction with ammonia, still requiring pressures around 200 bar to drive the equilibrium to acceptable yields [10,11]. Another important end product of CO2 are inorganic carbonates, like CaCO3, produced by fast reaction with metal hydroxides [12]. Carboxylic acids are an interesting class of products, as important intermediates in the synthesis of polymers and pharmaceuticals. Hydroxybenzoic acids are among the few chemicals that are industrially produced from CO2, via the Kolbe–Schmidt reaction at high temperatures and CO2 pressures [13-15]. Sodium phenolate is selectively converted to salicylic acid, a precursor of Aspirin, while potassium phenolate exclusively yields p-hydroxybenzoic acid, used in polyester synthesis (Scheme 1) [16-20].
![[1860-5397-10-260-i1]](/bjoc/content/inline/1860-5397-10-260-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of salicylic acid and p-hydroxybenzoic acid via Kolbe–Schmidt reaction [16-20].
Scheme 1: Synthesis of salicylic acid and p-hydroxybenzoic acid via Kolbe–Schmidt reaction [16-20].
In order to generate carboxylic acids from CO2 under relatively mild conditions, reactive organometallic nucleophiles such as Grignard reagents can be used, generating a large amount of waste [21-24]. Electroreduction of CO2 can be a worthy alternative for these dangerous energy-intensive processes, replacing toxic or hazardous reducing agents by clean electrons. In this case, the high thermodynamic stability of CO2 is by-passed by a simple one-electron reduction at an electrode, leading to in situ generation of reactive intermediates. Often, room temperature conditions are sufficient, considering that the energy of the electrons is determined by the applied voltage [25]. Since the electroreduction takes place on a cathode surface, the need for complex homogeneous organometallic catalysts is minimized. Furthermore, electricity will be increasingly of renewable origin in the future, making organic electrosynthesis a promising technology for environmentally friendly chemical processes [26]. The electroreduction of CO2 can be applied for the synthesis of fuels like formic acid [27], methanol [28] or methane [29] via two-, six- and eight-electron reductions, respectively (Scheme 2). This way electric energy from periodic sustainable origin, like solar or wind energy, can be stored [30].
![[1860-5397-10-260-i2]](/bjoc/content/inline/1860-5397-10-260-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Electroreduction of carbon dioxide to formic acid, methanol or methane.
Scheme 2: Electroreduction of carbon dioxide to formic acid, methanol or methane.
Review
In this review, the focus will be on another approach, in which CO2 is fixed in organic chemicals by means of an energy-efficient reduction process to produce valuable carboxylic acids. This methodology requires only one or two electrons per CO2 molecule, as shown in Scheme 3 for olefins, with the C–C bond formation highlighted in bold.
![[1860-5397-10-260-i3]](/bjoc/content/inline/1860-5397-10-260-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Electrochemical fixation of CO2 in olefins.
Scheme 3: Electrochemical fixation of CO2 in olefins.
Industrial organic electrosynthesis
The chemical industry is devoting increasing research efforts to the field of organic electrosynthesis [31]. An extended series of electro-organic processes have already been implemented on an industrial scale, like for example the electrohydrodimerisation of acrylonitrile (Scheme 4) [32], or the production of p-methoxybenzaldehyde [33].
![[1860-5397-10-260-i4]](/bjoc/content/inline/1860-5397-10-260-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Electrohydrodimerisation of acrylonitrile to adiponitrile [32].
Scheme 4: Electrohydrodimerisation of acrylonitrile to adiponitrile [32].
Another interesting industrial process is the simultaneous production of phthalide and tert-butylbenzaldehyde dimethylacetal from dimethyl phthalate and tert-butyltoluene, respectively (Scheme 5). After separation, by distillation and precipitation, both products can be used in the production of pesticides [34]. This is an example of a paired electrosynthesis, in which the anodic and cathodic reactions simultaneously form compounds that are valuable. This way a combined electrochemical yield, i.e., the fraction of supplied current going to the desired reaction, is achieved, reducing energy consumption and reaction time. This methodology is very environmentally friendly since there is no generation of toxic wastes, electrical current is used more efficiently and a high atom economy is achievable [35]. The latter being the fraction of the molecular mass of all reactants which is transferred to the desired product(s). In Scheme 5, the total atom efficiency is 100% and the electrochemical yield reaches 180% [34].
![[1860-5397-10-260-i5]](/bjoc/content/inline/1860-5397-10-260-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Parallel paired electrosynthesis of phthalide and tert-butylbenzaldehyde dimethylacetal [34].
Scheme 5: Parallel paired electrosynthesis of phthalide and tert-butylbenzaldehyde dimethylacetal [34].
Moreover, numerous pilot scale processes have been demonstrated like the electrohydrodimerization of formaldehyde to ethylene glycol [36] or the production of glyoxylic acid [37]. The most important reasons for this raised interest are the higher energy efficiency compared to traditional thermochemical processes, the use of less expensive starting materials, less aggressive reaction conditions, fewer processing steps and the discovery of unique synthesis routes [31].
Despite numerous publications and patents in the field of CO2 electroreduction, no industrial processes are known in which CO2 is electrochemically incorporated in organic chemicals producing carboxylic acids. This review will give an overview of various types of electrocarboxylation procedures bearing in mind the requirements for future industrial application. Besides the identification of a profitable market for the product, minimized process costs are the major requirement for potential large scale implementation. Some parameters that influence these costs are current efficiency, reactor design, electrode material and reactant costs.
Electrocarboxylation setups
All electrochemical processes involve an anodic and cathodic reaction in order to close the electron cycle. Electrons supplied at the cathode must emerge from an oxidation reaction occurring at the anode (Scheme 6). The electrochemical system should be optimized to prevent unfavorable interference between both reactions, since such conflicts cause electric current to be lost and lead to a decrease in faradaic efficiency. Electrocarboxylation, the electrochemical fixation of carbon dioxide in organic chemicals, involves the electroreduction of carbon dioxide and/or an organic substrate. For olefins, alkynes, carbonyl compounds, imines and organic halides, this leads to the formation of carboxylate anions. A counter cation is required in order to obtain a stable reaction product and an anodic reaction is necessary to complete the electron cycle. The anodic generation of this counter cation has been a challenging and important point of discussion for many years, as will become clear further in this review. An overview of different possible setups for obtaining carboxylic acids is given in Scheme 6.
![[1860-5397-10-260-i6]](/bjoc/content/inline/1860-5397-10-260-i6.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Overview of electrocarboxylation setups using (a) a sacrificial anode, (b) an inert anode, generating protons, with a cation exchange membrane (CEM) and (c) an inert anode, releasing tetraalkylammonium cations.
Scheme 6: Overview of electrocarboxylation setups using (a) a sacrificial anode, (b) an inert anode, generati...
Electrocarboxylation reactions can either be conducted with a sacrificial anode, like magnesium or aluminum, or with an inert anode, like platinum or carbon. Most research has been focused on the fixation of CO2 using sacrificial anodes [38-40] (Scheme 6a). The higher oxidation potential of sacrificial anodes compared to that of the other reaction species makes this setup readily compatible with a simple undivided electrolysis cell, without a membrane separating the catholyte from the anolyte. This way high current densities can be obtained at relatively low potentials leading to minimized energy consumption. The absence of unwanted anodic reactions allows maintaining high current efficiencies without real difficulty. Furthermore, this counter electrode reaction delivers metal cations (Mg2+, Al3+), which rapidly are coordinated by the carboxylate anions formed at the cathode. Finally, the corresponding metal carboxylates can be precipitated from organic solvents allowing easy product isolation. Alongside all the benefits that are associated with these sacrificial anodes, the gradual consumption of the anode material is a major drawback for industrial applications. Not only is it rather expensive to consume such large amounts of metal; it also strongly hinders the implementation of a continuous process. Additionally, in order to obtain the free acids, an acid hydrolysis step is required, complicating product purification, and generating a significant amount of waste. The potential industrial use of electrochemical CO2 fixation with sacrificial anodes should be found in fine chemical applications, preferably when the carboxylate salt can be used as such.
Considerable efforts have been made in investigating other electrocarboxylation systems. Replacing the sacrificial anode with a stable anode brings along several challenges. First of all, a counter electrode reaction must be identified, delivering counter cations to balance the charge of the carboxylate anions. The anodic reactant should be more easily oxidized than the other species present in the reaction mixture. The possible side product formed in this anodic reaction should either have no effect on the system, or be a useful reactant for the cathodic reaction. The direct formation of free carboxylic acids is a very interesting approach in this respect, minimizing the amount of process steps and waste (Scheme 6b). Protons produced at the anode, however, can have a detrimental effect on the electrocarboxylation efficiency, through cathodic formation of hydrogen, formic acid and other side products [41,42]. Hydrogen formation can be limited by usage of cathode materials with high hydrogen overvoltage like lead and mercury, or more environmentally friendly tantalum and zinc [43]. In order to minimize other side reactions a cation exchange membrane (CEM) is necessary, allowing different conditions in both compartments, giving a controlled supply of protons to the catholyte. Besides the implementation and maintenance costs of such a membrane, it also causes an elevated ohmic resistance between the electrodes decreasing the energy efficiency of the process. Moreover, most membranes have difficulty operating in organic solvents and under high pressure conditions, limiting operational conditions [44]. The anodic oxidation of more reduction stable tetraalkylammonium salts is another approach compatible with non-sacrificial anodes (Scheme 6c). Here, the released tetraalkylammonium cations function as counter ions for the cathodically formed carboxylate anions. If the corresponding oxidation products are not harmful for the cathodic reaction a simple undivided cell can be envisaged. However, this can also be considered as a sacrificial process, since the tetraalkylammonium salts are consumed during the reaction. But more importantly, in contrast to the use of a dissolving anode, this method allows a more efficient implementation of a continuous process.
The first reports on electrocarboxylation date back to the early 1960s with a patent of Loveland, demonstrating the dicarboxylation of 1,3-butadiene in a two-compartment cell with a mercury cathode and a platinum anode, giving a mixture of mono- and dicarboxylic acids. CO2 bubbling through a catholyte solution of 1 wt % water in DMF, saturated with butadiene, yielded up to 50% of 3-hexenedioic acid [45], a result which unfortunately was difficult to reproduce [42,46]. At that time, various substrates, like olefins, alkynes and aromatic ketones, have been electrocarboxylated in these divided cells [47]. Shortly after, sacrificial anodes made the use of a diaphragm obsolete, giving satisfactory product yields and current efficiencies in an undivided cell [38]. From this point on most of the research in the field of CO2 fixation was focused on this type of setup.
Electrocarboxylation of conjugated dienes
The electrochemical fixation of carbon dioxide in 1,3-butadiene has been extensively investigated because of the importance of adipic acid for the polymer industry. 1,3-Butadiene is widely available, not only from steam cracking, but increasingly from dehydrogenation of linear butenes. Dicarboxylation of 1,3-butadiene yields a mixture of 3-hexene-1,6-dioic acid isomers, which are only one hydrogenation step removed from adipic acid, a monomer of nylon [41,42,44,48,49]. The general mechanism of CO2 fixation in conjugated dienes is illustrated in Scheme 7. It is remarkable that only one electron is used per CO2 molecule which is incorporated, making this a very energy efficient approach. There are two possible pathways to reach the monocarboxylate radical anion intermediate: one in which first CO2 is reduced to a reactive CO2•− radical anion, and another one in which a radical anion is formed from the alkene. It has been illustrated that both pathways may be operative at the same time [49,50]. Which mechanism is favored depends amongst other parameters mainly on the diene type [49-51], CO2 pressure [50] and cathode material [50].
![[1860-5397-10-260-i7]](/bjoc/content/inline/1860-5397-10-260-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: General mechanism of the electrochemical dicarboxylation of conjugated dienes [49].
Scheme 7: General mechanism of the electrochemical dicarboxylation of conjugated dienes [49].
The different reactor setups used for the electrocarboxylation of 1,3-butadiene and the effect of the counter cation are illustrated in Table 1.
Table 1: Electrocarboxylation of 1,3-butadiene in different setups.a
| Entry | Anode | Reducing agent | CEMb |
Solvent
(catholyte – anolyte) |
C5:C6:C10c | ηd (%) (Yield (%)) | Ref. |
|---|---|---|---|---|---|---|---|
| 1e | Pt | H2O | Yes | CH3CN – H2Of | 67:0:33 | 18 (11) | [41] |
| 2g | Pt | dry MgO | Yes | CH3CN – CH3CN | 26:58:16 | 31 (21) | [41] |
| 3g | Pth | H2 | No | CH3CN | 0:0:0 | 0 (0) | [42] |
| 4g | Pth | H2 + dry MgO | No | CH3CN | 75:25:0 | 3.7 (–) | [42] |
| 5g,i | Pth | H2O | Yes | DMF – H2Of | –:–:– | 3.4 (–) | [42] |
| 6g | Pt | NH3 | No | CH3CN | 33:54:13 | 5.8 (–) | [42] |
| 7j | Pt | (TEA)2oxalate + TEA formatek | No | CH3CN | 36:52:12 | 39 (98) | [44] |
| 8l | Mg | Anode | No | DMF | 2:98:0 | – (81) | [48] |
| 9m | Al | Anode | No | DMF | 0:100:0 | 42 (84) | [49] |
aReactions were performed in conditions presented in corresponding references; bcation exchange membrane; cC5:C6:C10 = 3-pentenoic acid:3-hexenedioic acid:3,7-decadienedioic acid; dtotal current efficiency; emercury cathode; f1 wt % H2SO4 solution; glead cathode; hplatinum hydrogen gas-diffusion electrode; iNH3 present in catholyte; jcarbon felt cathode; kTEA = tetraethylammonium; ltantalum cathode with 2,4,4-trimethyl-1,5,9-triazacyclododecene nickel(II) tetrafluoroborate mediator; mnickel cathode.
The different anodic reactions that have been used for the electrocarboxylation of 1,3-butadiene (Table 1) are illustrated in Scheme 8. They are divided into three categories, with (a) the sacrificial anode dissolution, (b) the proton forming reactions and (c) the oxidations evolving other free cations.
![[1860-5397-10-260-i8]](/bjoc/content/inline/1860-5397-10-260-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reported anodic reactions for the electrocarboxylation of 1,3-butadiene.
Scheme 8: Reported anodic reactions for the electrocarboxylation of 1,3-butadiene.
In case a stable platinum anode is used, a C5:C6:C10 product mixture is formed, containing isomers of 3-pentenoic acid (C5), 3-hexenedioic acid (C6) and 3,7-decadienedioic acid (C10) (Table 1, entries 1–7). The product distribution is among other things influenced by the water and proton content in the reaction system. In a divided cell, in which the anolyte consists of 1% H2SO4 in water, no C6 product is formed (Table 1, entry 1). Only in an anhydrous catholyte and anolyte, applying MgO in the anolyte as reducing agent, the carboxylation becomes more selective for C6, reaching appreciable current efficiencies (Table 1, entry 2). Furthermore, the presence of water in the catholyte decreases the total current efficiency, due to the formation of formic acid [41,42]. It must be noted that, when using an aqueous and organic solvent in anolyte and catholyte, respectively, the transfer of hydrated protons through the membrane will cause water to enter the catholyte. Protons in turn can also favor formic acid generation, but on top of that, they promote the C5 and C10 formation [41]. It must be noted that the presence of formic acid illustrates the existence of mechanism I (Scheme 7). When working in an undivided cell and using protons as sole counter cation through hydrogen oxidation in anhydrous conditions, only formic acid is produced (Table 1, entry 3) [42]. The latter stresses the effect of a cation exchange membrane in providing a controlled proton supply to the catholyte, minimizing cathodic formation of formic acid. In these electrosynthesis setups, in which protons are generated at the anode, cathodes with high hydrogen overpotential, like mercury and lead, are necessary to minimize current loss through hydrogen gas formation [41,42].
The counter ion seems to have a significant influence on the fate of the cathodically formed CO2•− radical anion. For carboxylation of a carbon skeleton to occur, this radical anion needs to react via the radical centered at its carbon atom. Grinberg et al. claim that metal or ammonium cations can cause a reversible migration of the negative charge between the carbon and oxygen atoms, allowing the CO2•− radical anion to react with its C-centered radical [42]. When this CO2•− radical anion abstracts a proton from the solvent, proton migration from oxygen to carbon results in formation of a strong covalent CH bond, yielding a formyloxy radical, which is further reduced to a formate anion (Table 1, entry 3). In general terms, one can conclude that the electrocarboxylation of 1,3-butadiene is not efficiently performed in aqueous or protic media. MgO has been considered as an alternative cation source (Table 1, entry 4), but its solubility in organic solvents is low. Using ammonia as proton scavenger, with formation of ammonium counter cations, allows electrocarboxylation of 1,3-butadiene, and can increase the selectivity for the C6 product (Table 1, entries 5 and 6). The faradaic efficiencies, however, are rather low, hence, there has been a search to find alternative reducing agents.
Tetraethylammonium oxalate and formate salts appeared to be very promising for this purpose, fulfilling both the role of electrolyte and reducing agent. Tetraethylammonium cations have high reduction stability, while still possessing good ion pairing properties. The degree of delocalization of the positive charge is large enough to prevent cathodic reduction and small enough to allow a quick and stable interaction with the cathodically formed carboxylate anions. Furthermore, oxalate and formate are easily oxidized at a Pt anode, gradually releasing the tetraethylammonium cations. The combination of both salts in acetonitrile gives near quantitative yields of the C5:C6:C10 product mixture, with C6 as the main product (Table 1, entry 7) [44]. The anodic reaction produces CO2 which can directly be used as reactant at the cathode (Scheme 8), sustaining the atom economy of the process. This way, however, there is no net incorporation of gaseous CO2 into the organic substrate, but only net conversion of more energetic oxalate and formate. On top of that, it is important to realize that both the MgO and the tetraalkylammonium salts can also be considered as sacrificial reducing agents. Their advantage over sacrificial anodes, however, is an easier implementation in a continuous process. The anodic oxidation of formate generates one CO2 molecule and one proton, giving a controlled supply of protons to the cathode (Scheme 8). Conducting the electrocarboxylation with tetraethylammonium oxalate, without formate salts, increases the amount of C6 compared to C5 and C10 [44]. The use of acetonitrile as solvent is ideal when working in an undivided cell, thanks to its adequate oxidation stability and high dielectric constant. Anhydrous conditions and the easier oxidation of oxalate and formate, at a Pt anode, compared to butadiene and the cathodically generated carboxylates, make the use of a membrane redundant, enabling the use of a simple undivided cell [44]. Moreover, the use of membranes in non-aqueous or aprotic environments is unsatisfactory as they become poorly conducting [44]. This procedure was patented and extended to various other substrates, like activated olefins, imines, carbonyl and halogen compounds, and other anions like an azide, which forms inert N2 upon oxidation [52].
Eventually the electrocarboxylation of 1,3-butadiene was also conducted with sacrificial anodes, resulting in a high selectivity for the C6 product (Table 1, entries 8 and 9) [48,49]. Working under anhydrous conditions allows using another range of cathode materials, such as nickel, without being limited by the hydrogen overvoltage. The cathode metal has a large effect on the product distribution, acting as a catalyst [44,49]. Secondly, the absence of a membrane allowed the use of high CO2 pressures, pushing the selectivity towards the C6 product, completely eliminating C10 formation [49]. The above-mentioned benefits of sacrificial anode systems have lead to the electrocarboxylation of various other conjugated dienes [49-51,53,54]. In order to increase the selectivity for the C6 product, dissolved nickel and iron redox mediators have been used [48,53,54]. The need for such organometallic complexes could be eliminated by direct electrocarboxylation on nickel or stainless steel cathode surfaces [49,50]. Substrates with an internal conjugated system appear to be less reactive towards CO2 fixation, due to steric hindrance and the presence of electron donating substituents [49,51]. However, working at atmospheric CO2 pressures and at lower current densities allows effectively performing the double carboxylation of internal conjugated double bonds in open chains. This way, conjugated linoleic acids could be dicarboxylated with a yield approaching 80% at current efficiencies of over 50%, opening the reactant scope to other renewable dienes [50]. The occurrence of mechanism I (Scheme 7) was illustrated via the formation of oxalic acid, the CO2 dimerization product [50]. Additional insight in the carboxylation mechanism was gained by comparing the reactivity of 1,3-cyclohexadiene with a mixture of 2,4-hexadiene isomers. The fixed cyclic conformation of 1,3-cyclohexadiene increases its reactivity towards CO2 fixation, explained by a higher adsorption strength on the cathode surface. This suggests that dienes undergo carboxylation according to mechanism II while adsorbed on the surface, combined with mechanism I (Scheme 7). Moreover it was shown that diene configuration has a strong stereoelectronic effect on the rate of the dicarboxylation, with the Z,Z-configuration being the most reactive one [50].
Electrocarboxylation of olefins and alkynes
Many reports have been published on the electrocarboxylation of olefins and alkynes [55-74]. Most research has been done using a setup with a sacrificial magnesium or aluminum anode. The general mechanism of alkyne electrocarboxylation to a 1,4-dicarboxylated product is shown in Scheme 9. It is highly similar to the mechanism for electrocarboxylation of olefins. In these reactions a high selectivity for dicarboxylation can be achieved.
![[1860-5397-10-260-i9]](/bjoc/content/inline/1860-5397-10-260-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: General mechanism for electrocarboxylation of alkynes.
Scheme 9: General mechanism for electrocarboxylation of alkynes.
Alkynes and olefins react according to similar pathways, although a separate mechanism has been proposed for selective monocarboxylation of alkynes using nickel mediators, usually with bipyridine or N,N,N′,N′,N′′-pentamethyldiethylenetriamine ligands [55-60]. These organometallic complexes have also proven their value in increasing the carboxylation efficiency of alkenes, without including a specific selectivity for the monocarboxylic acid [56]. Monocarboxylation can readily occur as a side reaction when a small amount of protons are present in the reaction mixture, or through a proton/hydrogen radical abstraction from the reaction medium. The triple bond of alkynes is more active towards carboxylation than the olefin double bond [56]; furthermore, terminal alkynes are more reactive than internal alkynes [56-58], both leading to highly selective CO2 fixation. The selectivity towards the dicarboxylation product can be significantly increased by working at higher CO2 pressures [61,66], although an optimum must be found to minimize oxalic acid formation at high CO2 pressures by electrodimerization of CO2 [72]. Under rigorously anhydrous conditions, the dicarboxylation product of alkynes, shown in Scheme 9, can be transformed to a maleic anhydride [61,62]. Alkynes are rather reactive as such; olefins can be rendered more reactive by introduction of electron withdrawing groups [68-71,73,74]. Thus ethyl cinnamate was electrocarboxylated in 78% yield to give a mixture of mono- and dicarboxylated product (Scheme 10) [70]. Activated olefins like dimethyl maleate and acrylonitrile have also been reacted in a setup with a stable anode utilizing tetraethylammonium oxalate, formate or azide salts as the reductant [52].
![[1860-5397-10-260-i10]](/bjoc/content/inline/1860-5397-10-260-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Electrocarboxylation of ethyl cinnamate [70].
Scheme 10: Electrocarboxylation of ethyl cinnamate [70].
Electrocarboxylation of ketones, aldehydes and imines
As is the case for conjugated dienes, olefins and alkynes, two possible pathways exist for electrocarboxylation of carbonyl and imine compounds. One starts with CO2 reduction; another starts with reduction of the substrate (Scheme 11). The second route is considered as the predominant one [75]. In case the carbonyl or imine compound is reduced first, the negative charge may reside either on the carbon or on the heteroatom. This results in a first CO2 fixation on the carbon or on the heteroatom, depending on the electron-withdrawing/electron-donating properties of the substituents R1 and R2. In both cases, a second electron reduction in presence of CO2 yields a carboxylate intermediate with an additional carbonate or carbamate group. The latter is converted to the corresponding α-hydroxy acid or to an α-amino acid after acid hydrolysis in the product work-up [75-77].
![[1860-5397-10-260-i11]](/bjoc/content/inline/1860-5397-10-260-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: General electrocarboxylation mechanism for carbonyl compounds (Y = O) and imines (Y = NH) [75-77].
Scheme 11: General electrocarboxylation mechanism for carbonyl compounds (Y = O) and imines (Y = NH) [75-77].
An alternative mechanism has been proposed for aliphatic aldehydes, in which not α-hydroxy acids are formed but in which CO2 is incorporated on the α-carbon according to Scheme 12 [78]. Here, the reduced aldehyde abstracts a proton from the α-carbon of an unreacted aldehyde.
![[1860-5397-10-260-i12]](/bjoc/content/inline/1860-5397-10-260-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Electrocarboxylation mechanism of butyraldehyde proposed by Doherty [78].
Scheme 12: Electrocarboxylation mechanism of butyraldehyde proposed by Doherty [78].
The electrocarboxylation of ketones was first described by Wawzonek, converting benzophenone and acetophenone to benzylic acid and 2-hydroxy-2-phenylpropionic acid, respectively [79]. This offers an electrochemical route for several commercially relevant α-aryl propionic acids, used as non-steroidal anti-inflammatory drugs (NSAIDs) [80]. Therefore, the electrocarboxylation of aromatic ketones with sacrificial anodes has been extensively investigated [75-77,81-98]. Some researchers focused on replacing toxic and volatile organic solvents with ionic liquids [81-83]. Their negligible vapor pressure, large electrochemical window, good intrinsic conductivity and high CO2 solubility make them interesting solvents for electrochemical CO2 valorization [81-83,99-101]. Under similar conditions, ketones are carboxylated with higher selectivity for the α-hydroxy acid compared to aldehydes, and especially compared to aliphatic aldehydes, like acetaldehyde, which give very poor yields [84]. Since carbonyl compounds have the tendency to accept an electron more easily than CO2, the reaction mixture contains a lot of carbonyl radical anions, which can form vicinal diol dimers (pinacols) as side product [75,76,83]. The ratio of CO2 to substrate is of great importance in obtaining a high selectivity for the α-hydroxy acid products. Working at high CO2 pressures and low carbonyl concentrations gives the highest faradaic efficiencies, minimizing pinacol formation. The presence of protons drastically increases the amount of dimerization product and favors the hydrogenation of the carbonyl group to the alcohol [75,76,82,91]. The cathode material again plays an important role in the electrocarboxylation of carbonyl compounds. Toxic lead cathodes [85] and expensive platinum cathodes [87] can easily be replaced by better performing stainless steel [90] and nickel [75,89]. Concerning the reactivity of carbonyl compounds, it has been demonstrated that the carboxylation rate of benzophenones is decreased by electron donating substituents [90]. The pharmaceutical value of the electrocarboxylation of aromatic ketones to NSAIDs has lead to a significant number of patents using a sacrificial anode [92-95]. Since enantioselectivity is crucial for anti-inflammatory drugs, research has also been performed on the enantioselective electrocarboxylation of aromatic ketones, using chiral alkaloids [96-98].
The most important semi-industrial scale electrocarboxylation processes are related to the synthesis of these NSAIDs. The α-hydroxy acid is an intermediate, still requiring a chemical hydrogenation to obtain the desired product. 2-Acetyl-6-methoxynaphthalene (AMN) can be converted to hydroxynaproxen (HN), a precursor of naproxen (Scheme 13) [85,86]. Although yields up to 90% were obtained in a 1 L flow reactor, the switch to a 75 L system was accompanied by leaks and instrument problems resulting in a low yield (58%) and current efficiency (30%) [86].
![[1860-5397-10-260-i13]](/bjoc/content/inline/1860-5397-10-260-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Electrocarboxylation of AMN to HN using a sacrificial aluminum anode [86].
Scheme 13: Electrocarboxylation of AMN to HN using a sacrificial aluminum anode [86].
Besides carbonyl compounds, imines have also shown value as substrates for electrocarboxylation, namely in the synthesis of non-natural amino acids [102-108]. A semi-industrial setup was designed for the electrocarboxylation of benzalaniline (Scheme 14). Scale up was done in a filter press type cell with flow distribution, which is commercially available. The electrodes are pressed together with a PTFE coated glass fibre net between them. This way, the inter-electrode gap remains constant during consumption of the anode. In a 2 L solution with 200 g of reactant, a product yield of 85% and a current efficiency of 80% were obtained [105].
![[1860-5397-10-260-i14]](/bjoc/content/inline/1860-5397-10-260-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Electrocarboxylation of benzalaniline using a sacrificial aluminum anode [105].
Scheme 14: Electrocarboxylation of benzalaniline using a sacrificial aluminum anode [105].
The electrocarboxylation of aromatic ketones was also conducted with stable electrodes as shown in Scheme 15 [94,95]. In these patents p-isobutylacetophenone is carboxylated to hydroxyibuprofen, which is readily hydrogenolyzed to ibuprofen. A nafion membrane is used, allowing a selective passage of protons and tetraalkylammonium cations from the anolyte to the catholyte. Cyclohexene is added to the anolyte to scavenge the anodically formed bromine. A current efficiency of up to 90% was reached with a copper cathode and graphite anode. The method appeared also suitable for the synthesis of other NSAIDs like naproxen, cicloprofen, isoprofen, flurbiprofen, fenoprofen and carprofen.
![[1860-5397-10-260-i15]](/bjoc/content/inline/1860-5397-10-260-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Electrocarboxylation of p-isobutylacetophenone with stable electrodes [94,95].
Scheme 15: Electrocarboxylation of p-isobutylacetophenone with stable electrodes [94,95].
The electrocarboxylation of aliphatic aldehydes was also patented, namely for the production of 2-hydroxy-4-methylmercaptobutyric acid (MHA) by electrochemical carboxylation of 3-methylmercaptopropionaldehyde (MMP), with both a sacrificial anode [109] and a stable anode [110,111] (Scheme 16). The system, using the stable electrodes, can be extended to aldehydes, ketones and imines.
![[1860-5397-10-260-i16]](/bjoc/content/inline/1860-5397-10-260-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Electrochemical carboxylation of MMP to MHA [110,111].
Scheme 16: Electrochemical carboxylation of MMP to MHA [110,111].
MHA is an industrial scale feed additive, which is conventionally prepared using cyanides. In the proposed setup for the electrocatalytic preparation of MHA, a boron-doped diamond coated permeable cathode and Pt coated permeable anode rest directly on the cation exchange membrane to decrease ohmic resistance by the membrane, minimizing the required voltage. Hydrogen, supplied to the anolyte gives protons, which gradually enter the catholyte to form the free carboxylic acids. Both anolyte and catholyte are based on DMF. Unfortunately, no yields or current efficiencies higher than 30% were obtained [111].
There are also examples in which aromatic ketones and imines are electrocarboxylated in an undivided cell with stable anode. CO2 fixation in acetophenone was done in good yields in an undivided cell using a quaternary ammonium oxalate as an electrolyte and a sacrificial reducing agent [52,94,95,112,113]. Benzalanilines were carboxylated electrochemically with 79% faradaic yield, using an oxalate electrolyte [52].
Electrocarboxylation of organic halides
Numerous reports have been published on the electrocarboxylation of organic halides [114-146]. In a first step, a one electron reduction causes a halide anion to dissociate, forming a reactive radical. The latter undergoes a second reduction in the presence of CO2, yielding a monocarboxylate anion (Scheme 17) [122,124-126,128].
![[1860-5397-10-260-i17]](/bjoc/content/inline/1860-5397-10-260-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: General mechanism for electrocarboxylation of alkyl halides [122,124-126,128].
Scheme 17: General mechanism for electrocarboxylation of alkyl halides [122,124-126,128].
The first reactions reported were conducted in a divided cell, giving only moderate yields [114,115]. A drastic increase in efficiency was obtained by employing sacrificial anodes [116], especially magnesium anodes [117,118]. The cathode material is again of great importance, with silver and platinum giving the highest carboxylation selectivity [119-125]. Redox mediators allow working at a less negative cathodic potential, which results in a better energy efficiency and a more selective CO2 fixation [128]. The most common redox mediators are organometallic nickel [129-131], palladium [132] and cobalt complexes [133-137].
Similar to what was mentioned above for aromatic ketones, benzylic chlorides can also be converted to 2-arylpropionic acids (Scheme 18), with applications in the pharmaceutical industry, mainly as NSAIDs. Here too, some articles described the use of ionic liquids as solvent for electrocarboxylation reactions, in order to increase the safety and efficiency of the process [120,122].
![[1860-5397-10-260-i18]](/bjoc/content/inline/1860-5397-10-260-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Electrocarboxylation of benzylic chlorides as synthesis route for NSAIDs.
Scheme 18: Electrocarboxylation of benzylic chlorides as synthesis route for NSAIDs.
The industrial potential of this reaction has been assessed in several pilot applications, for example, using a setup with a sacrificial anode. In a 400 L reactor, a narrow and constant interelectrode gap was maintained in time and space, avoiding an ohmic drop during consumption of the anode material. A polyethylene grid is placed between the electrodes, allowing the anode to press down on the cathode by its own weight. A good agitation is obtained by pumping the reaction mixture through the cell [138]. Unfortunately, industrial production was not developed because of difficulties in the purification of the product, arising from the presence of impurities generated by the degradation of the solvent [139]. Another pilot scale experiment, conducted for the production of NSAIDs, uses a stable graphite anode in an undivided cell. The anodic reaction is the oxidation of lithium oxalate, giving yields up to 85% of 2-phenylpropionic acid [140]. The same group also investigated a setup in which a metal powder, like zinc, is oxidized at the anode, giving similar results [141].
The electrocarboxylation of organic halides can also be considered as an alternative dechlorination pathway for chlorobenzenes [122] and polychloromethanes [142]. While the synthesis of halogenated reagents is rather hazardous, the electrocarboxylation of organic halides is an interesting method to revalue waste products, like for example carbon tetrachloride, a toxic liquid, causing ozone depletion. In an undivided cell and acetonitrile as solvent, tri- and dichloroacetic acid are formed with current efficiencies between 50 and 60%. The exact anodic reaction(s), however, are not really specified. Oxidation of chloride, which itself originates in the CCl4 reactant, likely results in partial chlorination of the acetonitrile solvent, releasing protons which in turn cause the formation of chloroform by attacking the cathodically formed carbanions [143].
An undivided electrosynthesis setup with stable anode can also be used for CO2 fixation in other aliphatic halides. The anodic oxidation of tetraethylammonium oxalate is used in the electrocarboxylation of 1-bromo-2-methylpentane, which is almost quantitatively converted into 3-methylhexanoic acid [52]. The electrocarboxylation of 1,4-dibromo-2-butene is another reaction for which a stable anode and an undivided cell were proposed. The goal here is to form 3-hexenedioic acid, a precursor of adipic acid, although very poor yields and current efficiencies were obtained. Besides 1,4-dibromo-2-butene, 1,3-butadiene is added in the one-compartment cell to capture the bromine generated at the anode, forming the reactant (Scheme 19). The low yields are caused by debromodimerization and oligomerization [144].
![[1860-5397-10-260-i19]](/bjoc/content/inline/1860-5397-10-260-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Electrocarboxylation of 1,4-dibromo-2-butene [144].
Scheme 19: Electrocarboxylation of 1,4-dibromo-2-butene [144].
A significant effort has been devoted to efficiently produce cyanoacetic acid through CO2 fixation in chloroacetonitrile, as an alternative for the hazardous synthesis via alkali metal cyanides [137,145,146]. Derivatives of cyanoacetic acid are precious starting materials in pharmaceutical and agrochemical synthesis [137]. When the anodic reaction is the oxidation of a halide, lower current efficiencies can be attributed to a successive oxidation and reduction of respectively halides and halonium species. Therefore, the use of a membrane or glass frit can be interesting to minimize this effect. However, in a divided cell, the electrocarboxylation of chloroacetonitrile to cyanoacetic acid still appeared to give higher current efficiencies when using a sacrificial anode [146]. The major downside in carboxylating organic halides is the release of halides in the system, which can moreover induce a significant number of side reactions in a non-sacrificial setup, and which is in any case disadvantageous for the atom economy.
Innovative electrocarboxylation of other substrates
Besides the electrocarboxylation of chloroacetonitrile, CO2 can also be incorporated electrocatalytically in acetonitrile itself. Such reaction was successfully conducted in a two-compartment cell divided by a medium porosity glass frit (Scheme 20). This is an interesting alternative for the conventional synthesis of cyanoacetic acid, which is carried out by the reaction of chloroacetic acid and alkaline cyanides [147-149].
![[1860-5397-10-260-i20]](/bjoc/content/inline/1860-5397-10-260-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Convergent paired electrosynthesis of cyanoacetic acid, with X− = F4B−, ClO4−, HSO4−, Cl−, Br− [147].
Scheme 20: Convergent paired electrosynthesis of cyanoacetic acid, with X− = F4B−, ClO4−, HSO4−, Cl−, Br− [147].
The electrocarboxylation of acetonitrile to cyanoacetic acid is an example of a convergent paired electrosynthesis, meaning that two different substrates undergo either oxidation or reduction to afford products that react among themselves to generate a single product. Cyanomethyl radicals are formed by anodic oxidation of the supporting electrolyte anion followed by hydrogen radical abstraction from the acetonitrile solvent. These cyanomethyl radicals are then coupled to the CO2•− radical anion, forming cyanoacetic acid after protonation. The authors claim that the product is solely formed in the anolyte after CO2•− transport from the catholyte to the anolyte, although transport from cyanomethyl radicals to the catholyte is not excluded. However, product formation through cathodic reduction of acetonitrile is ruled out properly, since no cyanoacetic acid was formed when using a cation-exchange membrane. Since most of the product is present in the anolyte, current yields are rather low (24%). Moreover, the electrolyte anion and the cyanoacetic acid product have similar oxidation potentials. On top of that, some halogenation of the solvent to chloroacetonitrile was observed as a side reaction [147]. This reaction setup was also tested with propionitrile, butyronitrile, benzyl chloride and toluene in the anolyte compartment. Adjacent functional groups weaken C–H bonds, yielding relatively stable radicals, in turn resulting in selective CO2 fixation [150].
Another patented system uses electrogenerated bases to deprotonate a weakly acidic hydrocarbon group forming anions which are carboxylated in the presence of CO2. Meanwhile, proton scavengers remove protons released from the anodic regeneration of the base precursors as shown in Scheme 21 [151].
![[1860-5397-10-260-i21]](/bjoc/content/inline/1860-5397-10-260-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: General scheme of carboxylation of weak acidic hydrocarbons with electrogenerated bases. RH: weakly acidic hydrocarbon; BP: base precursor; EGB−: electrogenerated base; EGBH: conjugate acid of electrogenerated base [151].
Scheme 21: General scheme of carboxylation of weak acidic hydrocarbons with electrogenerated bases. RH: weakly...
The electrogenerated bases are redox mediators, used as catalysts in the carboxylation process. The net reaction can be written as follows:
RH + CO2 + scavenger → RCOO− + innocuous scavenger reaction product.
The base precursor should be more easily electroreduced than the weakly acidic hydrocarbon group and carbon dioxide, and should not undergo a nucleophilic attack by either the hydrocarbon anion or the electrogenerated base. Therefore, the base precursor should be sterically hindered at or near the site(s) where reduction will occur. The electrogenerated base must be a strong enough Brønsted base to deprotonate the weakly acidic hydrocarbon group. Ethenetetracarboxylate tetraesters are typical base precursors, suited for the electrocarboxylation of N-alkyldiglycolimides (Scheme 22). This process provides a feasible route to methoxymethane-1,1,1’-tricarboxylate salts, which are excellent detergent builders. The reaction should be carried out in strictly anhydrous conditions, since water is a stronger acid than the weakly acidic hydrocarbons employed herein. Electrogenerated bromine is used to regenerate the base precursor. Via a radical bromination, followed by a nucleophilic elimination under alkaline conditions, the alkane is oxidized to the alkene base precursor. These alkaline conditions and the four electron withdrawing ethoxy carbonyl groups (R1), prevent further bromination of the acquired double bond. Sodium carbonate is a suitable proton scavenger, providing the required alkalinity and being a convenient source of sodium ions. Crown ethers are added to dissolve the alkali metal salts in the organic solvent system. After reaction, anolyte and catholyte are filtered and base precursor and conjugate acid of electrogenerated base must be transferred to the other compartment. In this divided cell a yield of 85% for product (A) could be obtained (Scheme 22). In an undivided setup however, lower yields and current efficiencies were observed [151].
![[1860-5397-10-260-i22]](/bjoc/content/inline/1860-5397-10-260-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Electrocarboxylation of N-methyldiglycolimide to methoxymethane-1,1,1’-tricarboxylate precursors. R1: ethoxycarbonyl [151].
Scheme 22: Electrocarboxylation of N-methyldiglycolimide to methoxymethane-1,1,1’-tricarboxylate precursors. R1...
Oxalic acid is another carboxylic acid which can be formed through electrocarboxylation, namely of CO2 itself. This complexing agent has applications in cleaning industry, dyeing processes and metallurgy [152]. Besides its easy synthesis under anhydrous conditions in a cell with sacrificial anode, it can also be produced in a stable electrode setup, with current efficiencies over 50% (Scheme 23) [153]. In the catholyte an organic solvent is used while the anolyte consists of an aqueous NaCl solution. The anodically formed chlorine gas is continuously removed from the anolyte. A cation exchange membrane allows the selective transport of sodium cations to the catholyte. The sodium oxalate that is produced precipitates from the solution. A downside of this setup is the gradual transfer of water from the aqueous anolyte to the organic catholyte, this way steadily lowering the selectivity of the process [153].
![[1860-5397-10-260-i23]](/bjoc/content/inline/1860-5397-10-260-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Electrochemical dimerization of CO2 with stable electrodes [153].
Scheme 23: Electrochemical dimerization of CO2 with stable electrodes [153].
Conclusion
Electrochemical reduction is an efficient approach to activate thermodynamically stable CO2 under relatively mild and safe conditions. Electrocarboxylation allows the production of valuable carboxylic acids, through incorporation of CO2 in a wide range of organic chemicals. This way, polymer building blocks are produced from conjugated dienes, NSAIDs can easily be obtained from aromatic ketones and benzylic halides, and various other interesting applications are possible. Despite the vast amount of papers and patents on this subject, no industrial applications have emerged yet; only a couple of pilot plant scale processes have been demonstrated. The sustainable and efficient formation of carboxylic acids from carbon dioxide presents many intriguing challenges. The choice of reactor setup, electrode type and reaction pathway, not only affects the implementation cost but also determines operational characteristics like process continuity, atom economy and current efficiency. The shortcomings illustrated in this review emphasize the need for more innovative pathways to invent even more efficient and sustainable electrocarboxylation reactions.
References
-
Proceedings of “CO2 Emission from Fuels Combustion: Highlights 2012”, October, 2012, Paris, France. http://www.iea.org/publications/freepublications/publication/CO2emissionfromfuelcombustionhighlightsMarch2013.pdf (accessed October 27, 2014).
Return to citation in text: [1] -
Hoekman, S. K.; Broch, A.; Robbins, C.; Purcell, R. Int. J. Greenhouse Gas Control 2010, 4, 44–50. doi:10.1016/j.ijggc.2009.09.012
Return to citation in text: [1] -
Wang, W.; Wang, S.; Ma, X.; Gong, J. Chem. Soc. Rev. 2011, 40, 3703–3727. doi:10.1039/c1cs15008a
Return to citation in text: [1] -
Ion, A.; Parvulescu, V.; Jacobs, P.; de Vos, D. Appl. Catal., A 2009, 363, 40–44. doi:10.1016/j.apcata.2009.04.036
Return to citation in text: [1] -
Darensbourg, D. J.; Horn, A., Jr.; Moncada, A. I. Green Chem. 2010, 12, 1376–1379. doi:10.1039/c0gc00136h
Return to citation in text: [1] -
Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N.; Gao, J.; Yin, Z.-S. Green Chem. 2012, 14, 519–527. doi:10.1039/c2gc16039k
Return to citation in text: [1] -
Tomishige, K.; Yasuda, H.; Yoshida, Y.; Nurunnabi, M.; Li, B.; Kunimori, K. Green Chem. 2004, 6, 206–214. doi:10.1039/b401215a
Return to citation in text: [1] -
Ion, A.; Parvulescu, V.; Jacobs, P.; De Vos, D. Green Chem. 2007, 9, 158–161. doi:10.1039/b612403h
Return to citation in text: [1] -
Ion, A.; Van Doorslaer, C.; Parvulescu, V.; Jacobs, P.; De Vos, D. Green Chem. 2008, 10, 111–116. doi:10.1039/b711197e
Return to citation in text: [1] -
Guyer, A. Process for the manufacture of urea. U.S. Patent 2,854,482, Sept 30, 1958.
Return to citation in text: [1] -
Krase, N. W.; Gaddy, V. L. Ind. Eng. Chem. 1922, 14, 611–615. doi:10.1021/ie50151a009
Return to citation in text: [1] -
Aresta, M.; Dibenedetto, A. Dalton Trans. 2007, 2975–2992. doi:10.1039/b700658f
Return to citation in text: [1] -
Moore, E. R.; McDonald, D. C.; Willner, J.; Briggs, R. L. Carbonation of alkali metal phenates. U.S. Patent 4,171,453, Oct 16, 1979.
Return to citation in text: [1] -
Kolbe, H. Ann. Chem. Pharm. 1860, 113, 125–127. doi:10.1002/jlac.18601130120
Return to citation in text: [1] -
Schmitt, R. J. Prakt. Chem. 1885, 31, 397–411. doi:10.1002/prac.18850310130
Return to citation in text: [1] -
Hunt, S. E.; Jones, J. I.; Lindsey, A. S.; Killoh, D. C.; Turner, H. S. J. Chem. Soc. 1958, 3152–3160. doi:10.1039/jr9580003152
Return to citation in text: [1] [2] -
Markovic, Z.; Engelbrecht, J. P.; Markovic, S. Z. Naturforsch. 2002, 57, 812–818.
Return to citation in text: [1] [2] -
Rahim, M. A.; Matsui, Y.; Matsuyama, T.; Kosugi, Y. Bull. Chem. Soc. Jpn. 2003, 76, 2191–2195. doi:10.1246/bcsj.76.2191
Return to citation in text: [1] [2] -
Kosugi, Y.; Imaoka, Y.; Gotoh, F.; Rahim, M. A.; Matsui, Y.; Sakanishi, S. Org. Biomol. Chem. 2003, 1, 817–821. doi:10.1039/b210793g
Return to citation in text: [1] [2] -
Marković, Z.; Marković, S.; Manojlović, N.; Predojević-Simović, J. J. Chem. Inf. Model. 2007, 47, 1520–1525. doi:10.1021/ci700068b
Return to citation in text: [1] [2] -
Finnegan, R. A.; Altschuld, J. W. J. Organomet. Chem. 1967, 9, 193–204. doi:10.1016/S0022-328X(00)83721-3
Return to citation in text: [1] -
Quirk, R. P.; Yin, J.; Fetters, L. J.; Kastrup, R. V. Macromolecules 1992, 25, 2262–2267. doi:10.1021/ma00034a030
Return to citation in text: [1] -
Ebert, G. W.; Juda, W. L.; Kosakowski, R. H.; Ma, B.; Dong, L.; Cummings, K. E.; Phelps, M. V. B.; Mostafa, A. E.; Luo, J. J. Org. Chem. 2005, 70, 4314–4317. doi:10.1021/jo047731s
Return to citation in text: [1] -
Correa, A.; Martín, R. Angew. Chem., Int. Ed. 2009, 48, 6201–6204. doi:10.1002/anie.200900667
Return to citation in text: [1] -
Jitaru, M. J. Univ. Chem. Technol. Metall. 2007, 42, 333–344.
Return to citation in text: [1] -
Schäfer, H. J. C. R. Chim. 2011, 14, 745–765. doi:10.1016/j.crci.2011.01.002
Return to citation in text: [1] -
Agarwal, A. S.; Zhai, Y.; Hill, D.; Sridhar, N. ChemSusChem 2011, 4, 1301–1310. doi:10.1002/cssc.201100220
Return to citation in text: [1] -
Ganesh, I. Renewable Sustainable Energy Rev. 2014, 31, 221–257. doi:10.1016/j.rser.2013.11.045
Return to citation in text: [1] -
Cook, R. L.; MacDuff, R. C.; Sammells, A. F. J. Electrochem. Soc. 1988, 135, 1320–1326. doi:10.1149/1.2095972
Return to citation in text: [1] -
Olah, G. A.; Prakash, G. K. S.; Goeppert, A. J. Am. Chem. Soc. 2011, 133, 12881–12898. doi:10.1021/ja202642y
Return to citation in text: [1] -
Sequeira, C. A. C.; Santos, D. M. F. J. Braz. Chem. Soc. 2009, 20, 387–406. doi:10.1590/S0103-50532009000300002
Return to citation in text: [1] [2] -
Danly, D. E. J. Electrochem. Soc. 1984, 131, 435C–442C. doi:10.1149/1.2115324
Return to citation in text: [1] [2] -
Matthews, M. A. Pure Appl. Chem. 2001, 73, 1305–1308. doi:10.1351/pac200173081305
Return to citation in text: [1] -
Pütter, H.; Hannebaum, H. Preparation of phthalides. U.S. Patent 6,063,256, May 16, 2000.
Return to citation in text: [1] [2] [3] -
Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d
Return to citation in text: [1] -
Doherty, A. P.; Christensen, P. A.; Hamnett, A.; Scott, K. J. Electroanal. Chem. 1995, 386, 39–44. doi:10.1016/0022-0728(94)03816-L
Return to citation in text: [1] -
Li, J.; Hu, X.; Su, Y.; Li, Q. Chem. Eng. Sci. 2007, 62, 6784–6793. doi:10.1016/j.ces.2007.02.021
Return to citation in text: [1] -
Silvestri, G.; Gambino, S.; Filardo, G. Acta Chem. Scand. 1991, 45, 987–992. doi:10.3891/acta.chem.scand.45-0987
Return to citation in text: [1] [2] -
Tokuda, M. J. Nat. Gas Chem. 2006, 15, 275–281. doi:10.1016/S1003-9953(07)60006-1
Return to citation in text: [1] -
Silvestri, G.; Scialdone, O. Recent Scientific and Technological Developments in Electrochemical Carboxylation Based on Carbon Dioxide. In Carbon dioxide as Chemical feedstock; Aresta, M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp 317–334. doi:10.1002/9783527629916.ch12
Return to citation in text: [1] -
van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Scott, K. Dev. Chem. Eng. Miner. Process. 1993, 1, 71–117. doi:10.1002/apj.5500010202
Return to citation in text: [1] -
Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Loveland, J. W. Electrolytic production of acyclic carboxylic acids from hydrocarbons. U.S. Patent 3,032,489, May 1, 1962.
Return to citation in text: [1] -
Neikam, W. C. Electrolytic preparation of carboxylic acids. U.S. Patent 3,344,045, Sept 26, 1967.
Return to citation in text: [1] -
Tyssee, D. A.; Wagenknecht, J. H.; Baizer, M. M.; Chruma, J. L. Tetrahedron Lett. 1972, 13, 4809–4812. doi:10.1016/S0040-4039(01)94435-1
Return to citation in text: [1] -
Bringmann, J.; Dinjus, E. Appl. Organomet. Chem. 2001, 15, 135–140. doi:10.1002/1099-0739(200102)15:2<135::AID-AOC108>3.0.CO;2-L
Return to citation in text: [1] [2] [3] [4] -
Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. RSC Adv. 2013, 3, 4634–4642. doi:10.1039/c3ra00129f
Return to citation in text: [1] [2] [3] -
van Tilborg, W. J. M.; Smit, C. J.; Engels, R. A process for the electroreductive preparation of organic compounds. Eur. Patent 0,028,430, May 13, 1981.
Return to citation in text: [1] [2] [3] [4] [5] -
Ballivet-Tkatchenko, D.; Folest, J.-C.; Tanji, J. Appl. Organomet. Chem. 2000, 14, 847–849. doi:10.1002/1099-0739(200012)14:12<847::AID-AOC78>3.0.CO;2-7
Return to citation in text: [1] [2] -
Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. Tetrahedron 1992, 48, 5235–5248. doi:10.1016/S0040-4020(01)89021-9
Return to citation in text: [1] [2] -
Labbé, E.; Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 353, C51–C56. doi:10.1016/0022-328X(88)80330-9
Return to citation in text: [1] [2] -
Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 352, 239–246. doi:10.1016/0022-328X(88)83038-9
Return to citation in text: [1] [2] [3] [4] [5] -
Duñach, E.; Dérien, S.; Périchon, J. J. Organomet. Chem. 1989, 364, C33–C36. doi:10.1016/0022-328X(89)87156-6
Return to citation in text: [1] [2] [3] -
Dérien, S.; Duñach, E.; Périchon, J. J. Am. Chem. Soc. 1991, 113, 8447–8454. doi:10.1021/ja00022a037
Return to citation in text: [1] [2] [3] -
Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. J. Org. Chem. 1993, 58, 2578–2588. doi:10.1021/jo00061a038
Return to citation in text: [1] [2] -
Saito, S.; Nakagawa, S.; Koizumi, T.; Hirayama, K.; Yamamoto, Y. J. Org. Chem. 1999, 64, 3975–3978. doi:10.1021/jo982443f
Return to citation in text: [1] [2] -
Yuan, G.-Q.; Jiang, H.-F.; Lin, C. Tetrahedron 2008, 64, 5866–5872. doi:10.1016/j.tet.2008.04.053
Return to citation in text: [1] [2] [3] -
Li, C.; Yuan, G.; Jiang, H. Chin. J. Chem. 2010, 28, 1685–1689. doi:10.1002/cjoc.201090285
Return to citation in text: [1] [2] -
Köster, F.; Dinjus, E.; Duñach, E. Eur. J. Org. Chem. 2001, 2507–2511. doi:10.1002/1099-0690(200107)2001:13<2507::AID-EJOC2507>3.0.CO;2-P
Return to citation in text: [1] -
Senboku, H.; Komatsu, H.; Fujimura, Y.; Tokuda, M. Synlett 2001, 418–420. doi:10.1055/s-2001-11417
Return to citation in text: [1] -
Wang, H.; Lin, M.-Y.; Fang, H.-J.; Chen, T.-T.; Lu, J.-X. Chin. J. Chem. 2007, 25, 913–916. doi:10.1002/cjoc.200790177
Return to citation in text: [1] -
Yuan, G.-Q.; Jiang, H.-F.; Lin, C.; Liao, S.-J. Electrochim. Acta 2008, 53, 2170–2176. doi:10.1016/j.electacta.2007.09.023
Return to citation in text: [1] [2] -
Gambino, S.; Gennaro, A.; Filardo, G.; Silvestri, G.; Vianello, E. J. Electrochem. Soc. 1987, 134, 2172–2175. doi:10.1149/1.2100846
Return to citation in text: [1] -
Orsini, M.; Feroci, M.; Sotgiu, G.; Inesi, A. Org. Biomol. Chem. 2005, 3, 1202–1208. doi:10.1039/b500570a
Return to citation in text: [1] [2] -
Wang, H.; Zhang, G.; Liu, Y.; Luo, Y.; Lu, J. Electrochem. Commun. 2007, 9, 2235–2239. doi:10.1016/j.elecom.2007.06.031
Return to citation in text: [1] [2] -
Wang, H.; Du, Y.-F.; Lin, M.-Y.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1745–1748. doi:10.1002/cjoc.200890316
Return to citation in text: [1] [2] [3] [4] -
Lin, M.-Y.; Wang, H.; Zhang, A.-J.; Zhang, G.-R.; Lu, J.-X. Chin. J. Org. Chem. 2008, 28, 1572–1577.
http://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract337403.shtml
Return to citation in text: [1] [2] -
Gambino, S.; Silvestri, G. Tetrahedron Lett. 1973, 14, 3025–3028. doi:10.1016/S0040-4039(01)96310-5
Return to citation in text: [1] [2] -
Tyssee, D. A.; Baizer, M. M. J. Org. Chem. 1974, 39, 2819–2823. doi:10.1021/jo00933a001
Return to citation in text: [1] [2] -
Tyssee, D. A. Electrolytic monocarboxylation of activated olefins. U.S. Patent 4,028,201, June 7, 1977.
Return to citation in text: [1] [2] -
Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Scialdone, O.; Amatore, C.; Galia, A.; Filardo, G. J. Electroanal. Chem. 2006, 592, 163–174. doi:10.1016/j.jelechem.2006.04.009
Return to citation in text: [1] [2] [3] [4] [5] -
Pletcher, D.; Slevin, L. J. Chem. Soc., Perkin Trans. 2 1996, 217–220. doi:10.1039/p29960000217
Return to citation in text: [1] [2] [3] -
Doherty, A. P. Electrochim. Acta 2002, 47, 2963–2967. doi:10.1016/S0013-4686(02)00196-2
Return to citation in text: [1] [2] -
Wawzonek, S.; Gundersen, A. J. Electrochem. Soc. 1960, 107, 537–540. doi:10.1149/1.2427738
Return to citation in text: [1] -
Rieu, J.-P.; Boucherle, A.; Cousse, H.; Mouzin, G. Tetrahedron 1986, 42, 4095–4131. doi:10.1016/S0040-4020(01)87634-1
Return to citation in text: [1] -
Zhao, S.-F.; Wu, L.-X.; Wang, H.; Lu, J.-X.; Bond, A. M.; Zhang, J. Green Chem. 2011, 13, 3461–3468. doi:10.1039/c1gc15929a
Return to citation in text: [1] [2] [3] -
Zhao, S.-F.; Horne, M.; Bond, A. M.; Zhang, J. Green Chem. 2014, 16, 2242–2251. doi:10.1039/c3gc42404a
Return to citation in text: [1] [2] [3] [4] -
Feng, Q.; Huang, K.; Liu, S.; Yu, J.; Liu, F. Electrochim. Acta 2011, 56, 5137–5141. doi:10.1016/j.electacta.2011.03.061
Return to citation in text: [1] [2] [3] [4] -
Silvestri, G.; Gambino, S.; Filardo, G. Tetrahedron Lett. 1986, 27, 3429–3430. doi:10.1016/S0040-4039(00)84814-5
Return to citation in text: [1] [2] -
Chan, A. S. C.; Huang, T. T.; Wagenknecht, J. H.; Miller, R. E. J. Org. Chem. 1995, 60, 742–744. doi:10.1021/jo00108a047
Return to citation in text: [1] [2] [3] -
Datta, A. K.; Marron, P. A.; King, C. J. H.; Wagenknecht, J. H. J. Appl. Electrochem. 1998, 28, 569–577. doi:10.1023/A:1003289800341
Return to citation in text: [1] [2] [3] [4] -
Lateef, S. K.; Raju, R. R.; Mohan, S. K.; Reddy, S. J. Synth. Commun. 2006, 36, 31–36. doi:10.1080/00397910500328811
Return to citation in text: [1] [2] -
Zhang, L.; Xiao, L. P.; Niu, D. F.; Luo, Y. W.; Lu, J. X. Chin. J. Chem. 2008, 26, 35–38. doi:10.1002/cjoc.200890034
Return to citation in text: [1] -
Zhang, K.; Wang, H.; Wu, L.; Zhang, J.; Lu, J. Chin. J. Chem. 2010, 28, 509–513. doi:10.1002/cjoc.201090104
Return to citation in text: [1] [2] -
Zhao, S.-F.; Wang, H.; Lan, Y.-C.; Liu, X.; Lu, J.-X.; Zhang, J. J. Electroanal. Chem. 2012, 664, 105–110. doi:10.1016/j.jelechem.2011.11.001
Return to citation in text: [1] [2] [3] -
Scialdone, O.; Galia, A.; Isse, A. A.; Gennaro, A.; Sabatino, M. A.; Leone, R.; Filardo, G. J. Electroanal. Chem. 2007, 609, 8–16. doi:10.1016/j.jelechem.2007.02.014
Return to citation in text: [1] [2] -
Silvestri, G.; Gambino, S.; Filardo, G. Process for the electrocarboxylation of carbonyl compounds, for producing alpha-hydoxycarboxylic acids. U.S. Patent 4,708,780, Nov 24, 1987.
Return to citation in text: [1] [2] -
Maspero, F.; Piccolo, O.; Romano, U.; Gambino, S. New process for the preparation of 2-aryl-propionic acids. U.S. Patent 5,089,661, Feb 18, 1992.
Return to citation in text: [1] [2] -
Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone. U.S. Patent 4,582,577, April 15, 1986.
Return to citation in text: [1] [2] [3] [4] [5] -
Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone and other aryl ketones. U.S. Patent 4,601,797, July 22, 1986.
Return to citation in text: [1] [2] [3] [4] [5] -
Zhang, K.; Wang, H.; Zhao, S.-F.; Niu, D.-F.; Lu, J.-X. J. Electroanal. Chem. 2009, 630, 35–41. doi:10.1016/j.jelechem.2009.02.013
Return to citation in text: [1] [2] -
Zhao, S.-F.; Zhu, M.-X.; Zhang, K.; Wang, H.; Lu, J.-X. Tetrahedron Lett. 2011, 52, 2702–2705. doi:10.1016/j.tetlet.2011.03.076
Return to citation in text: [1] [2] -
Chen, B.-L.; Tu, Z.-Y.; Zhu, H.-W.; Sun, W.-W.; Wang, H.; Lu, J.-X. Electrochim. Acta 2014, 116, 475–483. doi:10.1016/j.electacta.2013.11.001
Return to citation in text: [1] [2] -
Doherty, A. P.; Diaconu, L.; Marley, E.; Spedding, P. L.; Barhdadi, R.; Troupel, M. Asia-Pac. J. Chem. Eng. 2012, 7, 14–23. doi:10.1002/apj.529
Return to citation in text: [1] -
Barrosse-Antle, L. E.; Compton, R. G. Chem. Commun. 2009, 3744–3746. doi:10.1039/b906320j
Return to citation in text: [1] -
Snuffin, L. L.; Whaley, L. W.; Yu, L. J. Electrochem. Soc. 2011, 158, F155–F158. doi:10.1149/1.3606487
Return to citation in text: [1] -
Weinberg, N. L.; Hoffmann, A. K.; Reddy, T. B. Tetrahedron Lett. 1971, 12, 2271–2274. doi:10.1016/S0040-4039(01)96837-6
Return to citation in text: [1] -
Hess, U. Z. Chem. 1980, 20, 148–149.
Return to citation in text: [1] -
Hess, U.; Thiele, R. J. Prakt. Chem. 1982, 324, 385–399. doi:10.1002/prac.19823240306
Return to citation in text: [1] -
Silvestri, G.; Gambino, S.; Filardo, G.; Tedeschi, F. J. Appl. Electrochem. 1989, 19, 946–948. doi:10.1007/BF01007947
Return to citation in text: [1] [2] [3] -
Koshechko, V. G.; Titov, V. E.; Bondarenko, V. N.; Pokhodenko, V. D. J. Fluorine Chem. 2008, 129, 701–706. doi:10.1016/j.jfluchem.2008.06.010
Return to citation in text: [1] -
Titov, V. E.; Bondarenko, V. N.; Koshechko, V. G.; Pokhodenko, V. D. Theor. Exp. Chem. 2010, 46, 8–13. doi:10.1007/s11237-010-9113-6
Return to citation in text: [1] -
Wang, H.; Zhang, K.; Chen, B. L.; Li, R. N.; Zhao, J. Q.; Lu, J. X. Int. J. Electrochem. Sci. 2011, 6, 1720–1729.
Return to citation in text: [1] -
Lehmann, T.; Schneider, R.; Weckbecker, C.; Dunach, E.; Olivera, S. Process for the production of 2-hydroxy-4-methylmercaptobutyric acid. U.S. Patent 6,475,370, Nov 5, 2002.
Return to citation in text: [1] -
Reufer, C.; Hateley, M.; Lehmann, T.; Weckbecker, C.; Sanzenbacher, R.; Bilz, J. Process for the preparation of α-substituted carboxylic acids from the series comprising α-hydroxycarboxylic acids and n-substituted-α-aminocarboxylic acids. U.S. Patent 7,332,067, Feb 19, 2008.
Return to citation in text: [1] [2] -
Hoppe, C.-F.; Nordschild, A.; Jakob, H.; Weckbecker, C.; Roth, P.; Imad, M. Preparing alpha-substituted carboxylic acids, comprises cathodic carboxylation of a compound in a conducting salt and an organic solvent containing catholyte with carbon dioxide at a diamond cathode layer. Ger. Patent 102,011,078,468, Jan 3, 2013.
Return to citation in text: [1] [2] [3] -
Engels, R.; Smit, C. J.; van Tilborg, W. J. M. Angew. Chem., Int. Ed. Engl. 1983, 22, 492–493. doi:10.1002/anie.198304921
Return to citation in text: [1] -
Ikeda, Y.; Manda, E. Chem. Lett. 1984, 13, 453–454. doi:10.1246/cl.1984.453
Return to citation in text: [1] -
Baizer, M. M.; Chruma, J. L. J. Org. Chem. 1972, 37, 1951–1960. doi:10.1021/jo00977a020
Return to citation in text: [1] [2] -
Wawzonek, S.; Shradel, J. M. J. Electrochem. Soc. 1979, 126, 401–403. doi:10.1149/1.2129051
Return to citation in text: [1] [2] -
Silvestri, G.; Gambino, S.; Filardo, G.; Gulotta, A. Angew. Chem., Int. Ed. Engl. 1984, 23, 979–980. doi:10.1002/anie.198409791
Return to citation in text: [1] [2] -
Sock, O.; Troupel, M.; Périchon, J. Tetrahedron Lett. 1985, 26, 1509–1512. doi:10.1016/S0040-4039(00)98538-1
Return to citation in text: [1] [2] -
Heintz, M.; Sock, O.; Saboureau, C.; Périchon, J. Tetrahedron 1988, 44, 1631–1636. doi:10.1016/S0040-4020(01)86724-7
Return to citation in text: [1] [2] -
Isse, A. A.; Gennaro, A. Chem. Commun. 2002, 2798–2799. doi:10.1039/b206746c
Return to citation in text: [1] [2] -
Gennaro, A.; Sánchez-Sánchez, C. M.; Isse, A. A.; Montiel, V. Electrochem. Commun. 2004, 6, 627–631. doi:10.1016/j.elecom.2004.04.019
Return to citation in text: [1] [2] [3] -
Isse, A. A.; Ferlin, M. G.; Gennaro, A. J. Electroanal. Chem. 2005, 581, 38–45. doi:10.1016/j.jelechem.2005.04.007
Return to citation in text: [1] [2] -
Aishah, A. J.; Hartini, M. A.; Normala, S.; Norhuda, A. M.; Hanis, H. H. N.; Razif, H. M.; Sugeng, T. J. Nat. Gas Chem. 2007, 16, 273–277. doi:10.1016/S1003-9953(07)60059-0
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Niu, D.-F.; Xiao, L.-P.; Zhang, A.-J.; Zhang, G.-R.; Tan, Q.-Y.; Lu, J.-X. Tetrahedron 2008, 64, 10517–10520. doi:10.1016/j.tet.2008.08.093
Return to citation in text: [1] [2] -
Niu, D.; Zhang, J.; Zhang, K.; Xue, T.; Lu, J. Chin. J. Chem. 2009, 27, 1041–1044. doi:10.1002/cjoc.200990174
Return to citation in text: [1] [2] [3] [4] -
Lan, Y.-C.; Wang, H.; Wu, L.-X.; Zhao, S.-F.; Gu, Y.-Q.; Lu, J.-X. J. Electroanal. Chem. 2012, 664, 33–38. doi:10.1016/j.jelechem.2011.10.011
Return to citation in text: [1] [2] [3] [4] -
Hiejima, Y.; Hayashi, M.; Uda, A.; Oya, S.; Kondo, H.; Senboku, H.; Takahashi, K. Phys. Chem. Chem. Phys. 2010, 12, 1953–1957. doi:10.1039/b920413j
Return to citation in text: [1] [2] [3] -
Feng, Q.; Huang, K.; Liu, S.; Wang, X. Electrochim. Acta 2010, 55, 5741–5745. doi:10.1016/j.electacta.2010.05.010
Return to citation in text: [1] -
Scialdone, O.; Galia, A.; Silvestri, G.; Amatore, C.; Thouin, L.; Verpeaux, J.-N. Chem. – Eur. J. 2006, 12, 7433–7447. doi:10.1002/chem.200501499
Return to citation in text: [1] [2] [3] [4] -
Troupel, M.; Rollin, Y.; Périchon, J.; Fauvarque, J. F. Nouv. J. Chim. 1981, 2, 621–625.
Return to citation in text: [1] [2] -
Amatore, C.; Jutand, A. J. Am. Chem. Soc. 1991, 113, 2819–2825. doi:10.1021/ja00008a003
Return to citation in text: [1] [2] -
Gennaro, A.; Isse, A. A.; Maran, F. J. Electroanal. Chem. 2001, 507, 124–134. doi:10.1016/S0022-0728(01)00373-4
Return to citation in text: [1] [2] -
Amatore, C.; Jutand, A.; Khalil, F.; Nielsen, M. F. J. Am. Chem. Soc. 1992, 114, 7076–7085. doi:10.1021/ja00044a018
Return to citation in text: [1] [2] -
Folest, J.-C.; Duprilot, J.-M.; Périchon, J.; Robin, Y.; Devynck, J. Tetrahedron Lett. 1985, 26, 2633–2636. doi:10.1016/S0040-4039(00)98122-X
Return to citation in text: [1] [2] -
Isse, A. A.; Gennaro, A.; Vianello, E. J. Chem. Soc., Dalton Trans. 1996, 1613–1618. doi:10.1039/dt9960001613
Return to citation in text: [1] [2] -
Zheng, G.; Stradiotto, M.; Li, L. J. Electroanal. Chem. 1998, 453, 79–88. doi:10.1016/S0022-0728(98)00173-9
Return to citation in text: [1] [2] -
Chung, W.-H.; Guo, P.; Wong, K.-Y.; Lau, C.-P. J. Electroanal. Chem. 2000, 486, 32–39. doi:10.1016/S0022-0728(00)00125-X
Return to citation in text: [1] [2] -
Fabre, P.-L.; Reynes, O. Electrochem. Commun. 2010, 12, 1360–1362. doi:10.1016/j.elecom.2010.07.020
Return to citation in text: [1] [2] [3] [4] -
Chaussard, J.; Troupel, M.; Robin, Y.; Jacob, G.; Juhasz, J. P. J. Appl. Electrochem. 1989, 19, 345–348. doi:10.1007/BF01015234
Return to citation in text: [1] [2] -
Chanfreau, S.; Cognet, P.; Camy, S.; Condoret, J.-S. J. Supercrit. Fluids 2008, 4, 156–162. doi:10.1016/j.supflu.2008.04.003
Return to citation in text: [1] [2] -
Fauvarque, J. F.; Jutand, A.; Francois, M. J. Appl. Electrochem. 1988, 18, 109–115. doi:10.1007/BF01016213
Return to citation in text: [1] [2] -
Fauvarque, J. F.; De Zelicourt, Y.; Amatore, C.; Jutand, A. J. Appl. Electrochem. 1990, 20, 338–340. doi:10.1007/BF01033614
Return to citation in text: [1] [2] -
Olloqui-Sariego, J. L.; Molina, V. M.; González-Arjona, D.; Roldán, E.; Domínguez, M. J. Electrochem. Soc. 2010, 157, E64–E68. doi:10.1149/1.3299365
Return to citation in text: [1] [2] -
Olloqui-Sariego, J. L.; Molina, V. M.; González-Arjona, D.; Roldán, E.; Domínguezz, M. J. Electrochem. Soc. 2008, 155, E157–E161. doi:10.1149/1.2971028
Return to citation in text: [1] [2] -
Grinberg, K. A.; Koch, T. A.; Mazin, E. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1997, 46, 1560–1564. doi:10.1007/BF02502939
Return to citation in text: [1] [2] [3] -
Scialdone, O.; Galia, A.; Belfiore, C.; Filardo, G.; Silvestri, G. Ind. Eng. Chem. Res. 2004, 43, 5006–5014. doi:10.1021/ie034275+
Return to citation in text: [1] [2] -
Isse, A. A.; Gennaro, A. J. Electrochem. Soc. 2002, 149, D113–D117. doi:10.1149/1.1490358
Return to citation in text: [1] [2] [3] -
Batanero, B.; Barba, F.; Sánchez-Sánchez, C. M.; Aldaz, A. J. Org. Chem. 2004, 69, 2423–2426. doi:10.1021/jo0358473
Return to citation in text: [1] [2] [3] -
Scialdone, O.; Sabatino, M. A.; Galia, A.; Filardo, G.; Silvestri, G. J. Electroanal. Chem. 2008, 614, 175–178. doi:10.1016/j.jelechem.2007.11.012
Return to citation in text: [1] -
Tyssee, D. A. Electrolytic carboxylation of acetonitrile and alpha-substituted acetonitriles. U.S. Patent 3,945,896, March 23, 1976.
Return to citation in text: [1] -
Otero, M. D.; Batanero, B.; Barba, F. Tetrahedron Lett. 2006, 47, 2171–2173. doi:10.1016/j.tetlet.2006.01.113
Return to citation in text: [1] -
Hallcher, R. C.; Baizer, M. M.; White, D. A. Electrolytic carboxylation of carbon acids via electrogenerated bases. U.S. Patent 4,072,583, Feb 7, 1978.
Return to citation in text: [1] [2] [3] [4] -
Riemenschneider, W.; Tanifuji, M. Oxalic acid. In Ullmann's Encyclopedia of Industrial Chemistry; Elvers, B.; Noethe, H., Eds.; Wiley-VCH: Weinheim, Germany, 2002. doi:10.1002/14356007.a18_247
Return to citation in text: [1] -
Goodridge, F.; Presland, G. J. Appl. Electrochem. 1984, 14, 791–796. doi:10.1007/BF00615269
Return to citation in text: [1] [2] [3]
| 56. | Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 352, 239–246. doi:10.1016/0022-328X(88)83038-9 |
| 56. | Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 352, 239–246. doi:10.1016/0022-328X(88)83038-9 |
| 55. | Labbé, E.; Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 353, C51–C56. doi:10.1016/0022-328X(88)80330-9 |
| 56. | Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 352, 239–246. doi:10.1016/0022-328X(88)83038-9 |
| 57. | Duñach, E.; Dérien, S.; Périchon, J. J. Organomet. Chem. 1989, 364, C33–C36. doi:10.1016/0022-328X(89)87156-6 |
| 58. | Dérien, S.; Duñach, E.; Périchon, J. J. Am. Chem. Soc. 1991, 113, 8447–8454. doi:10.1021/ja00022a037 |
| 59. | Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. J. Org. Chem. 1993, 58, 2578–2588. doi:10.1021/jo00061a038 |
| 60. | Saito, S.; Nakagawa, S.; Koizumi, T.; Hirayama, K.; Yamamoto, Y. J. Org. Chem. 1999, 64, 3975–3978. doi:10.1021/jo982443f |
| 61. | Yuan, G.-Q.; Jiang, H.-F.; Lin, C. Tetrahedron 2008, 64, 5866–5872. doi:10.1016/j.tet.2008.04.053 |
| 62. | Li, C.; Yuan, G.; Jiang, H. Chin. J. Chem. 2010, 28, 1685–1689. doi:10.1002/cjoc.201090285 |
| 63. | Köster, F.; Dinjus, E.; Duñach, E. Eur. J. Org. Chem. 2001, 2507–2511. doi:10.1002/1099-0690(200107)2001:13<2507::AID-EJOC2507>3.0.CO;2-P |
| 64. | Senboku, H.; Komatsu, H.; Fujimura, Y.; Tokuda, M. Synlett 2001, 418–420. doi:10.1055/s-2001-11417 |
| 65. | Wang, H.; Lin, M.-Y.; Fang, H.-J.; Chen, T.-T.; Lu, J.-X. Chin. J. Chem. 2007, 25, 913–916. doi:10.1002/cjoc.200790177 |
| 66. | Yuan, G.-Q.; Jiang, H.-F.; Lin, C.; Liao, S.-J. Electrochim. Acta 2008, 53, 2170–2176. doi:10.1016/j.electacta.2007.09.023 |
| 67. | Gambino, S.; Gennaro, A.; Filardo, G.; Silvestri, G.; Vianello, E. J. Electrochem. Soc. 1987, 134, 2172–2175. doi:10.1149/1.2100846 |
| 68. | Orsini, M.; Feroci, M.; Sotgiu, G.; Inesi, A. Org. Biomol. Chem. 2005, 3, 1202–1208. doi:10.1039/b500570a |
| 69. | Wang, H.; Zhang, G.; Liu, Y.; Luo, Y.; Lu, J. Electrochem. Commun. 2007, 9, 2235–2239. doi:10.1016/j.elecom.2007.06.031 |
| 70. | Wang, H.; Du, Y.-F.; Lin, M.-Y.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1745–1748. doi:10.1002/cjoc.200890316 |
| 71. |
Lin, M.-Y.; Wang, H.; Zhang, A.-J.; Zhang, G.-R.; Lu, J.-X. Chin. J. Org. Chem. 2008, 28, 1572–1577.
http://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract337403.shtml |
| 72. | Gambino, S.; Silvestri, G. Tetrahedron Lett. 1973, 14, 3025–3028. doi:10.1016/S0040-4039(01)96310-5 |
| 73. | Tyssee, D. A.; Baizer, M. M. J. Org. Chem. 1974, 39, 2819–2823. doi:10.1021/jo00933a001 |
| 74. | Tyssee, D. A. Electrolytic monocarboxylation of activated olefins. U.S. Patent 4,028,201, June 7, 1977. |
| 55. | Labbé, E.; Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 353, C51–C56. doi:10.1016/0022-328X(88)80330-9 |
| 56. | Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 352, 239–246. doi:10.1016/0022-328X(88)83038-9 |
| 57. | Duñach, E.; Dérien, S.; Périchon, J. J. Organomet. Chem. 1989, 364, C33–C36. doi:10.1016/0022-328X(89)87156-6 |
| 58. | Dérien, S.; Duñach, E.; Périchon, J. J. Am. Chem. Soc. 1991, 113, 8447–8454. doi:10.1021/ja00022a037 |
| 59. | Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. J. Org. Chem. 1993, 58, 2578–2588. doi:10.1021/jo00061a038 |
| 60. | Saito, S.; Nakagawa, S.; Koizumi, T.; Hirayama, K.; Yamamoto, Y. J. Org. Chem. 1999, 64, 3975–3978. doi:10.1021/jo982443f |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 68. | Orsini, M.; Feroci, M.; Sotgiu, G.; Inesi, A. Org. Biomol. Chem. 2005, 3, 1202–1208. doi:10.1039/b500570a |
| 69. | Wang, H.; Zhang, G.; Liu, Y.; Luo, Y.; Lu, J. Electrochem. Commun. 2007, 9, 2235–2239. doi:10.1016/j.elecom.2007.06.031 |
| 70. | Wang, H.; Du, Y.-F.; Lin, M.-Y.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1745–1748. doi:10.1002/cjoc.200890316 |
| 71. |
Lin, M.-Y.; Wang, H.; Zhang, A.-J.; Zhang, G.-R.; Lu, J.-X. Chin. J. Org. Chem. 2008, 28, 1572–1577.
http://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract337403.shtml |
| 73. | Tyssee, D. A.; Baizer, M. M. J. Org. Chem. 1974, 39, 2819–2823. doi:10.1021/jo00933a001 |
| 74. | Tyssee, D. A. Electrolytic monocarboxylation of activated olefins. U.S. Patent 4,028,201, June 7, 1977. |
| 72. | Gambino, S.; Silvestri, G. Tetrahedron Lett. 1973, 14, 3025–3028. doi:10.1016/S0040-4039(01)96310-5 |
| 61. | Yuan, G.-Q.; Jiang, H.-F.; Lin, C. Tetrahedron 2008, 64, 5866–5872. doi:10.1016/j.tet.2008.04.053 |
| 62. | Li, C.; Yuan, G.; Jiang, H. Chin. J. Chem. 2010, 28, 1685–1689. doi:10.1002/cjoc.201090285 |
| 56. | Duñach, E.; Périchon, J. J. Organomet. Chem. 1988, 352, 239–246. doi:10.1016/0022-328X(88)83038-9 |
| 57. | Duñach, E.; Dérien, S.; Périchon, J. J. Organomet. Chem. 1989, 364, C33–C36. doi:10.1016/0022-328X(89)87156-6 |
| 58. | Dérien, S.; Duñach, E.; Périchon, J. J. Am. Chem. Soc. 1991, 113, 8447–8454. doi:10.1021/ja00022a037 |
| 61. | Yuan, G.-Q.; Jiang, H.-F.; Lin, C. Tetrahedron 2008, 64, 5866–5872. doi:10.1016/j.tet.2008.04.053 |
| 66. | Yuan, G.-Q.; Jiang, H.-F.; Lin, C.; Liao, S.-J. Electrochim. Acta 2008, 53, 2170–2176. doi:10.1016/j.electacta.2007.09.023 |
| 75. | Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246 |
| 76. | Scialdone, O.; Amatore, C.; Galia, A.; Filardo, G. J. Electroanal. Chem. 2006, 592, 163–174. doi:10.1016/j.jelechem.2006.04.009 |
| 77. | Pletcher, D.; Slevin, L. J. Chem. Soc., Perkin Trans. 2 1996, 217–220. doi:10.1039/p29960000217 |
| 102. | Weinberg, N. L.; Hoffmann, A. K.; Reddy, T. B. Tetrahedron Lett. 1971, 12, 2271–2274. doi:10.1016/S0040-4039(01)96837-6 |
| 103. | Hess, U. Z. Chem. 1980, 20, 148–149. |
| 104. | Hess, U.; Thiele, R. J. Prakt. Chem. 1982, 324, 385–399. doi:10.1002/prac.19823240306 |
| 105. | Silvestri, G.; Gambino, S.; Filardo, G.; Tedeschi, F. J. Appl. Electrochem. 1989, 19, 946–948. doi:10.1007/BF01007947 |
| 106. | Koshechko, V. G.; Titov, V. E.; Bondarenko, V. N.; Pokhodenko, V. D. J. Fluorine Chem. 2008, 129, 701–706. doi:10.1016/j.jfluchem.2008.06.010 |
| 107. | Titov, V. E.; Bondarenko, V. N.; Koshechko, V. G.; Pokhodenko, V. D. Theor. Exp. Chem. 2010, 46, 8–13. doi:10.1007/s11237-010-9113-6 |
| 108. | Wang, H.; Zhang, K.; Chen, B. L.; Li, R. N.; Zhao, J. Q.; Lu, J. X. Int. J. Electrochem. Sci. 2011, 6, 1720–1729. |
| 75. | Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246 |
| 76. | Scialdone, O.; Amatore, C.; Galia, A.; Filardo, G. J. Electroanal. Chem. 2006, 592, 163–174. doi:10.1016/j.jelechem.2006.04.009 |
| 77. | Pletcher, D.; Slevin, L. J. Chem. Soc., Perkin Trans. 2 1996, 217–220. doi:10.1039/p29960000217 |
| 86. | Datta, A. K.; Marron, P. A.; King, C. J. H.; Wagenknecht, J. H. J. Appl. Electrochem. 1998, 28, 569–577. doi:10.1023/A:1003289800341 |
| 70. | Wang, H.; Du, Y.-F.; Lin, M.-Y.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1745–1748. doi:10.1002/cjoc.200890316 |
| 105. | Silvestri, G.; Gambino, S.; Filardo, G.; Tedeschi, F. J. Appl. Electrochem. 1989, 19, 946–948. doi:10.1007/BF01007947 |
| 75. | Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246 |
| 105. | Silvestri, G.; Gambino, S.; Filardo, G.; Tedeschi, F. J. Appl. Electrochem. 1989, 19, 946–948. doi:10.1007/BF01007947 |
| 70. | Wang, H.; Du, Y.-F.; Lin, M.-Y.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1745–1748. doi:10.1002/cjoc.200890316 |
| 96. | Zhang, K.; Wang, H.; Zhao, S.-F.; Niu, D.-F.; Lu, J.-X. J. Electroanal. Chem. 2009, 630, 35–41. doi:10.1016/j.jelechem.2009.02.013 |
| 97. | Zhao, S.-F.; Zhu, M.-X.; Zhang, K.; Wang, H.; Lu, J.-X. Tetrahedron Lett. 2011, 52, 2702–2705. doi:10.1016/j.tetlet.2011.03.076 |
| 98. | Chen, B.-L.; Tu, Z.-Y.; Zhu, H.-W.; Sun, W.-W.; Wang, H.; Lu, J.-X. Electrochim. Acta 2014, 116, 475–483. doi:10.1016/j.electacta.2013.11.001 |
| 52. | van Tilborg, W. J. M.; Smit, C. J.; Engels, R. A process for the electroreductive preparation of organic compounds. Eur. Patent 0,028,430, May 13, 1981. |
| 92. | Silvestri, G.; Gambino, S.; Filardo, G. Process for the electrocarboxylation of carbonyl compounds, for producing alpha-hydoxycarboxylic acids. U.S. Patent 4,708,780, Nov 24, 1987. |
| 93. | Maspero, F.; Piccolo, O.; Romano, U.; Gambino, S. New process for the preparation of 2-aryl-propionic acids. U.S. Patent 5,089,661, Feb 18, 1992. |
| 94. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone. U.S. Patent 4,582,577, April 15, 1986. |
| 95. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone and other aryl ketones. U.S. Patent 4,601,797, July 22, 1986. |
| 86. | Datta, A. K.; Marron, P. A.; King, C. J. H.; Wagenknecht, J. H. J. Appl. Electrochem. 1998, 28, 569–577. doi:10.1023/A:1003289800341 |
| 85. | Chan, A. S. C.; Huang, T. T.; Wagenknecht, J. H.; Miller, R. E. J. Org. Chem. 1995, 60, 742–744. doi:10.1021/jo00108a047 |
| 86. | Datta, A. K.; Marron, P. A.; King, C. J. H.; Wagenknecht, J. H. J. Appl. Electrochem. 1998, 28, 569–577. doi:10.1023/A:1003289800341 |
| 90. | Zhao, S.-F.; Wang, H.; Lan, Y.-C.; Liu, X.; Lu, J.-X.; Zhang, J. J. Electroanal. Chem. 2012, 664, 105–110. doi:10.1016/j.jelechem.2011.11.001 |
| 79. | Wawzonek, S.; Gundersen, A. J. Electrochem. Soc. 1960, 107, 537–540. doi:10.1149/1.2427738 |
| 80. | Rieu, J.-P.; Boucherle, A.; Cousse, H.; Mouzin, G. Tetrahedron 1986, 42, 4095–4131. doi:10.1016/S0040-4020(01)87634-1 |
| 78. | Doherty, A. P. Electrochim. Acta 2002, 47, 2963–2967. doi:10.1016/S0013-4686(02)00196-2 |
| 78. | Doherty, A. P. Electrochim. Acta 2002, 47, 2963–2967. doi:10.1016/S0013-4686(02)00196-2 |
| 94. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone. U.S. Patent 4,582,577, April 15, 1986. |
| 95. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone and other aryl ketones. U.S. Patent 4,601,797, July 22, 1986. |
| 41. | van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 114. | Baizer, M. M.; Chruma, J. L. J. Org. Chem. 1972, 37, 1951–1960. doi:10.1021/jo00977a020 |
| 115. | Wawzonek, S.; Shradel, J. M. J. Electrochem. Soc. 1979, 126, 401–403. doi:10.1149/1.2129051 |
| 116. | Silvestri, G.; Gambino, S.; Filardo, G.; Gulotta, A. Angew. Chem., Int. Ed. Engl. 1984, 23, 979–980. doi:10.1002/anie.198409791 |
| 117. | Sock, O.; Troupel, M.; Périchon, J. Tetrahedron Lett. 1985, 26, 1509–1512. doi:10.1016/S0040-4039(00)98538-1 |
| 118. | Heintz, M.; Sock, O.; Saboureau, C.; Périchon, J. Tetrahedron 1988, 44, 1631–1636. doi:10.1016/S0040-4020(01)86724-7 |
| 119. | Isse, A. A.; Gennaro, A. Chem. Commun. 2002, 2798–2799. doi:10.1039/b206746c |
| 120. | Gennaro, A.; Sánchez-Sánchez, C. M.; Isse, A. A.; Montiel, V. Electrochem. Commun. 2004, 6, 627–631. doi:10.1016/j.elecom.2004.04.019 |
| 121. | Isse, A. A.; Ferlin, M. G.; Gennaro, A. J. Electroanal. Chem. 2005, 581, 38–45. doi:10.1016/j.jelechem.2005.04.007 |
| 122. | Aishah, A. J.; Hartini, M. A.; Normala, S.; Norhuda, A. M.; Hanis, H. H. N.; Razif, H. M.; Sugeng, T. J. Nat. Gas Chem. 2007, 16, 273–277. doi:10.1016/S1003-9953(07)60059-0 |
| 123. | Niu, D.-F.; Xiao, L.-P.; Zhang, A.-J.; Zhang, G.-R.; Tan, Q.-Y.; Lu, J.-X. Tetrahedron 2008, 64, 10517–10520. doi:10.1016/j.tet.2008.08.093 |
| 124. | Niu, D.; Zhang, J.; Zhang, K.; Xue, T.; Lu, J. Chin. J. Chem. 2009, 27, 1041–1044. doi:10.1002/cjoc.200990174 |
| 125. | Lan, Y.-C.; Wang, H.; Wu, L.-X.; Zhao, S.-F.; Gu, Y.-Q.; Lu, J.-X. J. Electroanal. Chem. 2012, 664, 33–38. doi:10.1016/j.jelechem.2011.10.011 |
| 126. | Hiejima, Y.; Hayashi, M.; Uda, A.; Oya, S.; Kondo, H.; Senboku, H.; Takahashi, K. Phys. Chem. Chem. Phys. 2010, 12, 1953–1957. doi:10.1039/b920413j |
| 127. | Feng, Q.; Huang, K.; Liu, S.; Wang, X. Electrochim. Acta 2010, 55, 5741–5745. doi:10.1016/j.electacta.2010.05.010 |
| 128. | Scialdone, O.; Galia, A.; Silvestri, G.; Amatore, C.; Thouin, L.; Verpeaux, J.-N. Chem. – Eur. J. 2006, 12, 7433–7447. doi:10.1002/chem.200501499 |
| 129. | Troupel, M.; Rollin, Y.; Périchon, J.; Fauvarque, J. F. Nouv. J. Chim. 1981, 2, 621–625. |
| 130. | Amatore, C.; Jutand, A. J. Am. Chem. Soc. 1991, 113, 2819–2825. doi:10.1021/ja00008a003 |
| 131. | Gennaro, A.; Isse, A. A.; Maran, F. J. Electroanal. Chem. 2001, 507, 124–134. doi:10.1016/S0022-0728(01)00373-4 |
| 132. | Amatore, C.; Jutand, A.; Khalil, F.; Nielsen, M. F. J. Am. Chem. Soc. 1992, 114, 7076–7085. doi:10.1021/ja00044a018 |
| 133. | Folest, J.-C.; Duprilot, J.-M.; Périchon, J.; Robin, Y.; Devynck, J. Tetrahedron Lett. 1985, 26, 2633–2636. doi:10.1016/S0040-4039(00)98122-X |
| 134. | Isse, A. A.; Gennaro, A.; Vianello, E. J. Chem. Soc., Dalton Trans. 1996, 1613–1618. doi:10.1039/dt9960001613 |
| 135. | Zheng, G.; Stradiotto, M.; Li, L. J. Electroanal. Chem. 1998, 453, 79–88. doi:10.1016/S0022-0728(98)00173-9 |
| 136. | Chung, W.-H.; Guo, P.; Wong, K.-Y.; Lau, C.-P. J. Electroanal. Chem. 2000, 486, 32–39. doi:10.1016/S0022-0728(00)00125-X |
| 137. | Fabre, P.-L.; Reynes, O. Electrochem. Commun. 2010, 12, 1360–1362. doi:10.1016/j.elecom.2010.07.020 |
| 138. | Chaussard, J.; Troupel, M.; Robin, Y.; Jacob, G.; Juhasz, J. P. J. Appl. Electrochem. 1989, 19, 345–348. doi:10.1007/BF01015234 |
| 139. | Chanfreau, S.; Cognet, P.; Camy, S.; Condoret, J.-S. J. Supercrit. Fluids 2008, 4, 156–162. doi:10.1016/j.supflu.2008.04.003 |
| 140. | Fauvarque, J. F.; Jutand, A.; Francois, M. J. Appl. Electrochem. 1988, 18, 109–115. doi:10.1007/BF01016213 |
| 141. | Fauvarque, J. F.; De Zelicourt, Y.; Amatore, C.; Jutand, A. J. Appl. Electrochem. 1990, 20, 338–340. doi:10.1007/BF01033614 |
| 142. | Olloqui-Sariego, J. L.; Molina, V. M.; González-Arjona, D.; Roldán, E.; Domínguez, M. J. Electrochem. Soc. 2010, 157, E64–E68. doi:10.1149/1.3299365 |
| 143. | Olloqui-Sariego, J. L.; Molina, V. M.; González-Arjona, D.; Roldán, E.; Domínguezz, M. J. Electrochem. Soc. 2008, 155, E157–E161. doi:10.1149/1.2971028 |
| 144. | Grinberg, K. A.; Koch, T. A.; Mazin, E. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1997, 46, 1560–1564. doi:10.1007/BF02502939 |
| 145. | Scialdone, O.; Galia, A.; Belfiore, C.; Filardo, G.; Silvestri, G. Ind. Eng. Chem. Res. 2004, 43, 5006–5014. doi:10.1021/ie034275+ |
| 146. | Isse, A. A.; Gennaro, A. J. Electrochem. Soc. 2002, 149, D113–D117. doi:10.1149/1.1490358 |
| 41. | van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113 |
| 52. | van Tilborg, W. J. M.; Smit, C. J.; Engels, R. A process for the electroreductive preparation of organic compounds. Eur. Patent 0,028,430, May 13, 1981. |
| 122. | Aishah, A. J.; Hartini, M. A.; Normala, S.; Norhuda, A. M.; Hanis, H. H. N.; Razif, H. M.; Sugeng, T. J. Nat. Gas Chem. 2007, 16, 273–277. doi:10.1016/S1003-9953(07)60059-0 |
| 124. | Niu, D.; Zhang, J.; Zhang, K.; Xue, T.; Lu, J. Chin. J. Chem. 2009, 27, 1041–1044. doi:10.1002/cjoc.200990174 |
| 125. | Lan, Y.-C.; Wang, H.; Wu, L.-X.; Zhao, S.-F.; Gu, Y.-Q.; Lu, J.-X. J. Electroanal. Chem. 2012, 664, 33–38. doi:10.1016/j.jelechem.2011.10.011 |
| 126. | Hiejima, Y.; Hayashi, M.; Uda, A.; Oya, S.; Kondo, H.; Senboku, H.; Takahashi, K. Phys. Chem. Chem. Phys. 2010, 12, 1953–1957. doi:10.1039/b920413j |
| 128. | Scialdone, O.; Galia, A.; Silvestri, G.; Amatore, C.; Thouin, L.; Verpeaux, J.-N. Chem. – Eur. J. 2006, 12, 7433–7447. doi:10.1002/chem.200501499 |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 122. | Aishah, A. J.; Hartini, M. A.; Normala, S.; Norhuda, A. M.; Hanis, H. H. N.; Razif, H. M.; Sugeng, T. J. Nat. Gas Chem. 2007, 16, 273–277. doi:10.1016/S1003-9953(07)60059-0 |
| 124. | Niu, D.; Zhang, J.; Zhang, K.; Xue, T.; Lu, J. Chin. J. Chem. 2009, 27, 1041–1044. doi:10.1002/cjoc.200990174 |
| 125. | Lan, Y.-C.; Wang, H.; Wu, L.-X.; Zhao, S.-F.; Gu, Y.-Q.; Lu, J.-X. J. Electroanal. Chem. 2012, 664, 33–38. doi:10.1016/j.jelechem.2011.10.011 |
| 126. | Hiejima, Y.; Hayashi, M.; Uda, A.; Oya, S.; Kondo, H.; Senboku, H.; Takahashi, K. Phys. Chem. Chem. Phys. 2010, 12, 1953–1957. doi:10.1039/b920413j |
| 128. | Scialdone, O.; Galia, A.; Silvestri, G.; Amatore, C.; Thouin, L.; Verpeaux, J.-N. Chem. – Eur. J. 2006, 12, 7433–7447. doi:10.1002/chem.200501499 |
| 1. | Proceedings of “CO2 Emission from Fuels Combustion: Highlights 2012”, October, 2012, Paris, France. http://www.iea.org/publications/freepublications/publication/CO2emissionfromfuelcombustionhighlightsMarch2013.pdf (accessed October 27, 2014). |
| 110. | Reufer, C.; Hateley, M.; Lehmann, T.; Weckbecker, C.; Sanzenbacher, R.; Bilz, J. Process for the preparation of α-substituted carboxylic acids from the series comprising α-hydroxycarboxylic acids and n-substituted-α-aminocarboxylic acids. U.S. Patent 7,332,067, Feb 19, 2008. |
| 111. | Hoppe, C.-F.; Nordschild, A.; Jakob, H.; Weckbecker, C.; Roth, P.; Imad, M. Preparing alpha-substituted carboxylic acids, comprises cathodic carboxylation of a compound in a conducting salt and an organic solvent containing catholyte with carbon dioxide at a diamond cathode layer. Ger. Patent 102,011,078,468, Jan 3, 2013. |
| 110. | Reufer, C.; Hateley, M.; Lehmann, T.; Weckbecker, C.; Sanzenbacher, R.; Bilz, J. Process for the preparation of α-substituted carboxylic acids from the series comprising α-hydroxycarboxylic acids and n-substituted-α-aminocarboxylic acids. U.S. Patent 7,332,067, Feb 19, 2008. |
| 111. | Hoppe, C.-F.; Nordschild, A.; Jakob, H.; Weckbecker, C.; Roth, P.; Imad, M. Preparing alpha-substituted carboxylic acids, comprises cathodic carboxylation of a compound in a conducting salt and an organic solvent containing catholyte with carbon dioxide at a diamond cathode layer. Ger. Patent 102,011,078,468, Jan 3, 2013. |
| 52. | van Tilborg, W. J. M.; Smit, C. J.; Engels, R. A process for the electroreductive preparation of organic compounds. Eur. Patent 0,028,430, May 13, 1981. |
| 94. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone. U.S. Patent 4,582,577, April 15, 1986. |
| 95. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone and other aryl ketones. U.S. Patent 4,601,797, July 22, 1986. |
| 112. | Engels, R.; Smit, C. J.; van Tilborg, W. J. M. Angew. Chem., Int. Ed. Engl. 1983, 22, 492–493. doi:10.1002/anie.198304921 |
| 113. | Ikeda, Y.; Manda, E. Chem. Lett. 1984, 13, 453–454. doi:10.1246/cl.1984.453 |
| 111. | Hoppe, C.-F.; Nordschild, A.; Jakob, H.; Weckbecker, C.; Roth, P.; Imad, M. Preparing alpha-substituted carboxylic acids, comprises cathodic carboxylation of a compound in a conducting salt and an organic solvent containing catholyte with carbon dioxide at a diamond cathode layer. Ger. Patent 102,011,078,468, Jan 3, 2013. |
| 10. | Guyer, A. Process for the manufacture of urea. U.S. Patent 2,854,482, Sept 30, 1958. |
| 11. | Krase, N. W.; Gaddy, V. L. Ind. Eng. Chem. 1922, 14, 611–615. doi:10.1021/ie50151a009 |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 7. | Tomishige, K.; Yasuda, H.; Yoshida, Y.; Nurunnabi, M.; Li, B.; Kunimori, K. Green Chem. 2004, 6, 206–214. doi:10.1039/b401215a |
| 8. | Ion, A.; Parvulescu, V.; Jacobs, P.; De Vos, D. Green Chem. 2007, 9, 158–161. doi:10.1039/b612403h |
| 9. | Ion, A.; Van Doorslaer, C.; Parvulescu, V.; Jacobs, P.; De Vos, D. Green Chem. 2008, 10, 111–116. doi:10.1039/b711197e |
| 4. | Ion, A.; Parvulescu, V.; Jacobs, P.; de Vos, D. Appl. Catal., A 2009, 363, 40–44. doi:10.1016/j.apcata.2009.04.036 |
| 5. | Darensbourg, D. J.; Horn, A., Jr.; Moncada, A. I. Green Chem. 2010, 12, 1376–1379. doi:10.1039/c0gc00136h |
| 6. | Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N.; Gao, J.; Yin, Z.-S. Green Chem. 2012, 14, 519–527. doi:10.1039/c2gc16039k |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 109. | Lehmann, T.; Schneider, R.; Weckbecker, C.; Dunach, E.; Olivera, S. Process for the production of 2-hydroxy-4-methylmercaptobutyric acid. U.S. Patent 6,475,370, Nov 5, 2002. |
| 2. | Hoekman, S. K.; Broch, A.; Robbins, C.; Purcell, R. Int. J. Greenhouse Gas Control 2010, 4, 44–50. doi:10.1016/j.ijggc.2009.09.012 |
| 3. | Wang, W.; Wang, S.; Ma, X.; Gong, J. Chem. Soc. Rev. 2011, 40, 3703–3727. doi:10.1039/c1cs15008a |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 94. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone. U.S. Patent 4,582,577, April 15, 1986. |
| 95. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone and other aryl ketones. U.S. Patent 4,601,797, July 22, 1986. |
| 16. | Hunt, S. E.; Jones, J. I.; Lindsey, A. S.; Killoh, D. C.; Turner, H. S. J. Chem. Soc. 1958, 3152–3160. doi:10.1039/jr9580003152 |
| 17. | Markovic, Z.; Engelbrecht, J. P.; Markovic, S. Z. Naturforsch. 2002, 57, 812–818. |
| 18. | Rahim, M. A.; Matsui, Y.; Matsuyama, T.; Kosugi, Y. Bull. Chem. Soc. Jpn. 2003, 76, 2191–2195. doi:10.1246/bcsj.76.2191 |
| 19. | Kosugi, Y.; Imaoka, Y.; Gotoh, F.; Rahim, M. A.; Matsui, Y.; Sakanishi, S. Org. Biomol. Chem. 2003, 1, 817–821. doi:10.1039/b210793g |
| 20. | Marković, Z.; Marković, S.; Manojlović, N.; Predojević-Simović, J. J. Chem. Inf. Model. 2007, 47, 1520–1525. doi:10.1021/ci700068b |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 16. | Hunt, S. E.; Jones, J. I.; Lindsey, A. S.; Killoh, D. C.; Turner, H. S. J. Chem. Soc. 1958, 3152–3160. doi:10.1039/jr9580003152 |
| 17. | Markovic, Z.; Engelbrecht, J. P.; Markovic, S. Z. Naturforsch. 2002, 57, 812–818. |
| 18. | Rahim, M. A.; Matsui, Y.; Matsuyama, T.; Kosugi, Y. Bull. Chem. Soc. Jpn. 2003, 76, 2191–2195. doi:10.1246/bcsj.76.2191 |
| 19. | Kosugi, Y.; Imaoka, Y.; Gotoh, F.; Rahim, M. A.; Matsui, Y.; Sakanishi, S. Org. Biomol. Chem. 2003, 1, 817–821. doi:10.1039/b210793g |
| 20. | Marković, Z.; Marković, S.; Manojlović, N.; Predojević-Simović, J. J. Chem. Inf. Model. 2007, 47, 1520–1525. doi:10.1021/ci700068b |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 13. | Moore, E. R.; McDonald, D. C.; Willner, J.; Briggs, R. L. Carbonation of alkali metal phenates. U.S. Patent 4,171,453, Oct 16, 1979. |
| 14. | Kolbe, H. Ann. Chem. Pharm. 1860, 113, 125–127. doi:10.1002/jlac.18601130120 |
| 15. | Schmitt, R. J. Prakt. Chem. 1885, 31, 397–411. doi:10.1002/prac.18850310130 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 12. | Aresta, M.; Dibenedetto, A. Dalton Trans. 2007, 2975–2992. doi:10.1039/b700658f |
| 41. | van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 120. | Gennaro, A.; Sánchez-Sánchez, C. M.; Isse, A. A.; Montiel, V. Electrochem. Commun. 2004, 6, 627–631. doi:10.1016/j.elecom.2004.04.019 |
| 122. | Aishah, A. J.; Hartini, M. A.; Normala, S.; Norhuda, A. M.; Hanis, H. H. N.; Razif, H. M.; Sugeng, T. J. Nat. Gas Chem. 2007, 16, 273–277. doi:10.1016/S1003-9953(07)60059-0 |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 133. | Folest, J.-C.; Duprilot, J.-M.; Périchon, J.; Robin, Y.; Devynck, J. Tetrahedron Lett. 1985, 26, 2633–2636. doi:10.1016/S0040-4039(00)98122-X |
| 134. | Isse, A. A.; Gennaro, A.; Vianello, E. J. Chem. Soc., Dalton Trans. 1996, 1613–1618. doi:10.1039/dt9960001613 |
| 135. | Zheng, G.; Stradiotto, M.; Li, L. J. Electroanal. Chem. 1998, 453, 79–88. doi:10.1016/S0022-0728(98)00173-9 |
| 136. | Chung, W.-H.; Guo, P.; Wong, K.-Y.; Lau, C.-P. J. Electroanal. Chem. 2000, 486, 32–39. doi:10.1016/S0022-0728(00)00125-X |
| 137. | Fabre, P.-L.; Reynes, O. Electrochem. Commun. 2010, 12, 1360–1362. doi:10.1016/j.elecom.2010.07.020 |
| 52. | van Tilborg, W. J. M.; Smit, C. J.; Engels, R. A process for the electroreductive preparation of organic compounds. Eur. Patent 0,028,430, May 13, 1981. |
| 48. | Bringmann, J.; Dinjus, E. Appl. Organomet. Chem. 2001, 15, 135–140. doi:10.1002/1099-0739(200102)15:2<135::AID-AOC108>3.0.CO;2-L |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 138. | Chaussard, J.; Troupel, M.; Robin, Y.; Jacob, G.; Juhasz, J. P. J. Appl. Electrochem. 1989, 19, 345–348. doi:10.1007/BF01015234 |
| 128. | Scialdone, O.; Galia, A.; Silvestri, G.; Amatore, C.; Thouin, L.; Verpeaux, J.-N. Chem. – Eur. J. 2006, 12, 7433–7447. doi:10.1002/chem.200501499 |
| 119. | Isse, A. A.; Gennaro, A. Chem. Commun. 2002, 2798–2799. doi:10.1039/b206746c |
| 120. | Gennaro, A.; Sánchez-Sánchez, C. M.; Isse, A. A.; Montiel, V. Electrochem. Commun. 2004, 6, 627–631. doi:10.1016/j.elecom.2004.04.019 |
| 121. | Isse, A. A.; Ferlin, M. G.; Gennaro, A. J. Electroanal. Chem. 2005, 581, 38–45. doi:10.1016/j.jelechem.2005.04.007 |
| 122. | Aishah, A. J.; Hartini, M. A.; Normala, S.; Norhuda, A. M.; Hanis, H. H. N.; Razif, H. M.; Sugeng, T. J. Nat. Gas Chem. 2007, 16, 273–277. doi:10.1016/S1003-9953(07)60059-0 |
| 123. | Niu, D.-F.; Xiao, L.-P.; Zhang, A.-J.; Zhang, G.-R.; Tan, Q.-Y.; Lu, J.-X. Tetrahedron 2008, 64, 10517–10520. doi:10.1016/j.tet.2008.08.093 |
| 124. | Niu, D.; Zhang, J.; Zhang, K.; Xue, T.; Lu, J. Chin. J. Chem. 2009, 27, 1041–1044. doi:10.1002/cjoc.200990174 |
| 125. | Lan, Y.-C.; Wang, H.; Wu, L.-X.; Zhao, S.-F.; Gu, Y.-Q.; Lu, J.-X. J. Electroanal. Chem. 2012, 664, 33–38. doi:10.1016/j.jelechem.2011.10.011 |
| 132. | Amatore, C.; Jutand, A.; Khalil, F.; Nielsen, M. F. J. Am. Chem. Soc. 1992, 114, 7076–7085. doi:10.1021/ja00044a018 |
| 129. | Troupel, M.; Rollin, Y.; Périchon, J.; Fauvarque, J. F. Nouv. J. Chim. 1981, 2, 621–625. |
| 130. | Amatore, C.; Jutand, A. J. Am. Chem. Soc. 1991, 113, 2819–2825. doi:10.1021/ja00008a003 |
| 131. | Gennaro, A.; Isse, A. A.; Maran, F. J. Electroanal. Chem. 2001, 507, 124–134. doi:10.1016/S0022-0728(01)00373-4 |
| 114. | Baizer, M. M.; Chruma, J. L. J. Org. Chem. 1972, 37, 1951–1960. doi:10.1021/jo00977a020 |
| 115. | Wawzonek, S.; Shradel, J. M. J. Electrochem. Soc. 1979, 126, 401–403. doi:10.1149/1.2129051 |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 117. | Sock, O.; Troupel, M.; Périchon, J. Tetrahedron Lett. 1985, 26, 1509–1512. doi:10.1016/S0040-4039(00)98538-1 |
| 118. | Heintz, M.; Sock, O.; Saboureau, C.; Périchon, J. Tetrahedron 1988, 44, 1631–1636. doi:10.1016/S0040-4020(01)86724-7 |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 116. | Silvestri, G.; Gambino, S.; Filardo, G.; Gulotta, A. Angew. Chem., Int. Ed. Engl. 1984, 23, 979–980. doi:10.1002/anie.198409791 |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 51. | Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. RSC Adv. 2013, 3, 4634–4642. doi:10.1039/c3ra00129f |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 51. | Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. RSC Adv. 2013, 3, 4634–4642. doi:10.1039/c3ra00129f |
| 53. | Ballivet-Tkatchenko, D.; Folest, J.-C.; Tanji, J. Appl. Organomet. Chem. 2000, 14, 847–849. doi:10.1002/1099-0739(200012)14:12<847::AID-AOC78>3.0.CO;2-7 |
| 54. | Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. Tetrahedron 1992, 48, 5235–5248. doi:10.1016/S0040-4020(01)89021-9 |
| 48. | Bringmann, J.; Dinjus, E. Appl. Organomet. Chem. 2001, 15, 135–140. doi:10.1002/1099-0739(200102)15:2<135::AID-AOC108>3.0.CO;2-L |
| 53. | Ballivet-Tkatchenko, D.; Folest, J.-C.; Tanji, J. Appl. Organomet. Chem. 2000, 14, 847–849. doi:10.1002/1099-0739(200012)14:12<847::AID-AOC78>3.0.CO;2-7 |
| 54. | Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. Tetrahedron 1992, 48, 5235–5248. doi:10.1016/S0040-4020(01)89021-9 |
| 38. | Silvestri, G.; Gambino, S.; Filardo, G. Acta Chem. Scand. 1991, 45, 987–992. doi:10.3891/acta.chem.scand.45-0987 |
| 39. | Tokuda, M. J. Nat. Gas Chem. 2006, 15, 275–281. doi:10.1016/S1003-9953(07)60006-1 |
| 40. | Silvestri, G.; Scialdone, O. Recent Scientific and Technological Developments in Electrochemical Carboxylation Based on Carbon Dioxide. In Carbon dioxide as Chemical feedstock; Aresta, M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp 317–334. doi:10.1002/9783527629916.ch12 |
| 41. | van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 137. | Fabre, P.-L.; Reynes, O. Electrochem. Commun. 2010, 12, 1360–1362. doi:10.1016/j.elecom.2010.07.020 |
| 145. | Scialdone, O.; Galia, A.; Belfiore, C.; Filardo, G.; Silvestri, G. Ind. Eng. Chem. Res. 2004, 43, 5006–5014. doi:10.1021/ie034275+ |
| 146. | Isse, A. A.; Gennaro, A. J. Electrochem. Soc. 2002, 149, D113–D117. doi:10.1149/1.1490358 |
| 43. | Scott, K. Dev. Chem. Eng. Miner. Process. 1993, 1, 71–117. doi:10.1002/apj.5500010202 |
| 144. | Grinberg, K. A.; Koch, T. A.; Mazin, E. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1997, 46, 1560–1564. doi:10.1007/BF02502939 |
| 143. | Olloqui-Sariego, J. L.; Molina, V. M.; González-Arjona, D.; Roldán, E.; Domínguezz, M. J. Electrochem. Soc. 2008, 155, E157–E161. doi:10.1149/1.2971028 |
| 142. | Olloqui-Sariego, J. L.; Molina, V. M.; González-Arjona, D.; Roldán, E.; Domínguez, M. J. Electrochem. Soc. 2010, 157, E64–E68. doi:10.1149/1.3299365 |
| 144. | Grinberg, K. A.; Koch, T. A.; Mazin, E. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1997, 46, 1560–1564. doi:10.1007/BF02502939 |
| 52. | van Tilborg, W. J. M.; Smit, C. J.; Engels, R. A process for the electroreductive preparation of organic compounds. Eur. Patent 0,028,430, May 13, 1981. |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 140. | Fauvarque, J. F.; Jutand, A.; Francois, M. J. Appl. Electrochem. 1988, 18, 109–115. doi:10.1007/BF01016213 |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 51. | Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. RSC Adv. 2013, 3, 4634–4642. doi:10.1039/c3ra00129f |
| 139. | Chanfreau, S.; Cognet, P.; Camy, S.; Condoret, J.-S. J. Supercrit. Fluids 2008, 4, 156–162. doi:10.1016/j.supflu.2008.04.003 |
| 38. | Silvestri, G.; Gambino, S.; Filardo, G. Acta Chem. Scand. 1991, 45, 987–992. doi:10.3891/acta.chem.scand.45-0987 |
| 122. | Aishah, A. J.; Hartini, M. A.; Normala, S.; Norhuda, A. M.; Hanis, H. H. N.; Razif, H. M.; Sugeng, T. J. Nat. Gas Chem. 2007, 16, 273–277. doi:10.1016/S1003-9953(07)60059-0 |
| 41. | van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 48. | Bringmann, J.; Dinjus, E. Appl. Organomet. Chem. 2001, 15, 135–140. doi:10.1002/1099-0739(200102)15:2<135::AID-AOC108>3.0.CO;2-L |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 141. | Fauvarque, J. F.; De Zelicourt, Y.; Amatore, C.; Jutand, A. J. Appl. Electrochem. 1990, 20, 338–340. doi:10.1007/BF01033614 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 46. | Neikam, W. C. Electrolytic preparation of carboxylic acids. U.S. Patent 3,344,045, Sept 26, 1967. |
| 47. | Tyssee, D. A.; Wagenknecht, J. H.; Baizer, M. M.; Chruma, J. L. Tetrahedron Lett. 1972, 13, 4809–4812. doi:10.1016/S0040-4039(01)94435-1 |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 45. | Loveland, J. W. Electrolytic production of acyclic carboxylic acids from hydrocarbons. U.S. Patent 3,032,489, May 1, 1962. |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 50. | Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochem. Commun. 2010, 12, 1698–1702. doi:10.1016/j.elecom.2010.09.028 |
| 49. | Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529–1534. doi:10.1016/j.electacta.2010.06.057 |
| 151. | Hallcher, R. C.; Baizer, M. M.; White, D. A. Electrolytic carboxylation of carbon acids via electrogenerated bases. U.S. Patent 4,072,583, Feb 7, 1978. |
| 151. | Hallcher, R. C.; Baizer, M. M.; White, D. A. Electrolytic carboxylation of carbon acids via electrogenerated bases. U.S. Patent 4,072,583, Feb 7, 1978. |
| 150. | Otero, M. D.; Batanero, B.; Barba, F. Tetrahedron Lett. 2006, 47, 2171–2173. doi:10.1016/j.tetlet.2006.01.113 |
| 151. | Hallcher, R. C.; Baizer, M. M.; White, D. A. Electrolytic carboxylation of carbon acids via electrogenerated bases. U.S. Patent 4,072,583, Feb 7, 1978. |
| 151. | Hallcher, R. C.; Baizer, M. M.; White, D. A. Electrolytic carboxylation of carbon acids via electrogenerated bases. U.S. Patent 4,072,583, Feb 7, 1978. |
| 44. | Pletcher, D.; Tietje Girault, J. J. Appl. Electrochem. 1986, 16, 791–802. doi:10.1007/BF01006524 |
| 147. | Batanero, B.; Barba, F.; Sánchez-Sánchez, C. M.; Aldaz, A. J. Org. Chem. 2004, 69, 2423–2426. doi:10.1021/jo0358473 |
| 148. | Scialdone, O.; Sabatino, M. A.; Galia, A.; Filardo, G.; Silvestri, G. J. Electroanal. Chem. 2008, 614, 175–178. doi:10.1016/j.jelechem.2007.11.012 |
| 149. | Tyssee, D. A. Electrolytic carboxylation of acetonitrile and alpha-substituted acetonitriles. U.S. Patent 3,945,896, March 23, 1976. |
| 48. | Bringmann, J.; Dinjus, E. Appl. Organomet. Chem. 2001, 15, 135–140. doi:10.1002/1099-0739(200102)15:2<135::AID-AOC108>3.0.CO;2-L |
| 146. | Isse, A. A.; Gennaro, A. J. Electrochem. Soc. 2002, 149, D113–D117. doi:10.1149/1.1490358 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 147. | Batanero, B.; Barba, F.; Sánchez-Sánchez, C. M.; Aldaz, A. J. Org. Chem. 2004, 69, 2423–2426. doi:10.1021/jo0358473 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 147. | Batanero, B.; Barba, F.; Sánchez-Sánchez, C. M.; Aldaz, A. J. Org. Chem. 2004, 69, 2423–2426. doi:10.1021/jo0358473 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 42. | Grinberg, V. A.; Koch, T. A.; Mazin, V. M.; Mysov, E. I.; Sterlin, S. R. Russ. Chem. Bull. 1999, 48, 294–299. doi:10.1007/BF02494552 |
| 41. | van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113 |
| 137. | Fabre, P.-L.; Reynes, O. Electrochem. Commun. 2010, 12, 1360–1362. doi:10.1016/j.elecom.2010.07.020 |
| 41. | van Tilborg, W. J. M.; Smit, C. J. Recl. Trav. Chim. Pays-Bas 1981, 100, 437–438. doi:10.1002/recl.19811001113 |
| 75. | Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246 |
| 76. | Scialdone, O.; Amatore, C.; Galia, A.; Filardo, G. J. Electroanal. Chem. 2006, 592, 163–174. doi:10.1016/j.jelechem.2006.04.009 |
| 82. | Zhao, S.-F.; Horne, M.; Bond, A. M.; Zhang, J. Green Chem. 2014, 16, 2242–2251. doi:10.1039/c3gc42404a |
| 91. | Scialdone, O.; Galia, A.; Isse, A. A.; Gennaro, A.; Sabatino, M. A.; Leone, R.; Filardo, G. J. Electroanal. Chem. 2007, 609, 8–16. doi:10.1016/j.jelechem.2007.02.014 |
| 85. | Chan, A. S. C.; Huang, T. T.; Wagenknecht, J. H.; Miller, R. E. J. Org. Chem. 1995, 60, 742–744. doi:10.1021/jo00108a047 |
| 84. | Silvestri, G.; Gambino, S.; Filardo, G. Tetrahedron Lett. 1986, 27, 3429–3430. doi:10.1016/S0040-4039(00)84814-5 |
| 75. | Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246 |
| 76. | Scialdone, O.; Amatore, C.; Galia, A.; Filardo, G. J. Electroanal. Chem. 2006, 592, 163–174. doi:10.1016/j.jelechem.2006.04.009 |
| 83. | Feng, Q.; Huang, K.; Liu, S.; Yu, J.; Liu, F. Electrochim. Acta 2011, 56, 5137–5141. doi:10.1016/j.electacta.2011.03.061 |
| 81. | Zhao, S.-F.; Wu, L.-X.; Wang, H.; Lu, J.-X.; Bond, A. M.; Zhang, J. Green Chem. 2011, 13, 3461–3468. doi:10.1039/c1gc15929a |
| 82. | Zhao, S.-F.; Horne, M.; Bond, A. M.; Zhang, J. Green Chem. 2014, 16, 2242–2251. doi:10.1039/c3gc42404a |
| 83. | Feng, Q.; Huang, K.; Liu, S.; Yu, J.; Liu, F. Electrochim. Acta 2011, 56, 5137–5141. doi:10.1016/j.electacta.2011.03.061 |
| 81. | Zhao, S.-F.; Wu, L.-X.; Wang, H.; Lu, J.-X.; Bond, A. M.; Zhang, J. Green Chem. 2011, 13, 3461–3468. doi:10.1039/c1gc15929a |
| 82. | Zhao, S.-F.; Horne, M.; Bond, A. M.; Zhang, J. Green Chem. 2014, 16, 2242–2251. doi:10.1039/c3gc42404a |
| 83. | Feng, Q.; Huang, K.; Liu, S.; Yu, J.; Liu, F. Electrochim. Acta 2011, 56, 5137–5141. doi:10.1016/j.electacta.2011.03.061 |
| 99. | Doherty, A. P.; Diaconu, L.; Marley, E.; Spedding, P. L.; Barhdadi, R.; Troupel, M. Asia-Pac. J. Chem. Eng. 2012, 7, 14–23. doi:10.1002/apj.529 |
| 100. | Barrosse-Antle, L. E.; Compton, R. G. Chem. Commun. 2009, 3744–3746. doi:10.1039/b906320j |
| 101. | Snuffin, L. L.; Whaley, L. W.; Yu, L. J. Electrochem. Soc. 2011, 158, F155–F158. doi:10.1149/1.3606487 |
| 75. | Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246 |
| 76. | Scialdone, O.; Amatore, C.; Galia, A.; Filardo, G. J. Electroanal. Chem. 2006, 592, 163–174. doi:10.1016/j.jelechem.2006.04.009 |
| 77. | Pletcher, D.; Slevin, L. J. Chem. Soc., Perkin Trans. 2 1996, 217–220. doi:10.1039/p29960000217 |
| 81. | Zhao, S.-F.; Wu, L.-X.; Wang, H.; Lu, J.-X.; Bond, A. M.; Zhang, J. Green Chem. 2011, 13, 3461–3468. doi:10.1039/c1gc15929a |
| 82. | Zhao, S.-F.; Horne, M.; Bond, A. M.; Zhang, J. Green Chem. 2014, 16, 2242–2251. doi:10.1039/c3gc42404a |
| 83. | Feng, Q.; Huang, K.; Liu, S.; Yu, J.; Liu, F. Electrochim. Acta 2011, 56, 5137–5141. doi:10.1016/j.electacta.2011.03.061 |
| 84. | Silvestri, G.; Gambino, S.; Filardo, G. Tetrahedron Lett. 1986, 27, 3429–3430. doi:10.1016/S0040-4039(00)84814-5 |
| 85. | Chan, A. S. C.; Huang, T. T.; Wagenknecht, J. H.; Miller, R. E. J. Org. Chem. 1995, 60, 742–744. doi:10.1021/jo00108a047 |
| 86. | Datta, A. K.; Marron, P. A.; King, C. J. H.; Wagenknecht, J. H. J. Appl. Electrochem. 1998, 28, 569–577. doi:10.1023/A:1003289800341 |
| 87. | Lateef, S. K.; Raju, R. R.; Mohan, S. K.; Reddy, S. J. Synth. Commun. 2006, 36, 31–36. doi:10.1080/00397910500328811 |
| 88. | Zhang, L.; Xiao, L. P.; Niu, D. F.; Luo, Y. W.; Lu, J. X. Chin. J. Chem. 2008, 26, 35–38. doi:10.1002/cjoc.200890034 |
| 89. | Zhang, K.; Wang, H.; Wu, L.; Zhang, J.; Lu, J. Chin. J. Chem. 2010, 28, 509–513. doi:10.1002/cjoc.201090104 |
| 90. | Zhao, S.-F.; Wang, H.; Lan, Y.-C.; Liu, X.; Lu, J.-X.; Zhang, J. J. Electroanal. Chem. 2012, 664, 105–110. doi:10.1016/j.jelechem.2011.11.001 |
| 91. | Scialdone, O.; Galia, A.; Isse, A. A.; Gennaro, A.; Sabatino, M. A.; Leone, R.; Filardo, G. J. Electroanal. Chem. 2007, 609, 8–16. doi:10.1016/j.jelechem.2007.02.014 |
| 92. | Silvestri, G.; Gambino, S.; Filardo, G. Process for the electrocarboxylation of carbonyl compounds, for producing alpha-hydoxycarboxylic acids. U.S. Patent 4,708,780, Nov 24, 1987. |
| 93. | Maspero, F.; Piccolo, O.; Romano, U.; Gambino, S. New process for the preparation of 2-aryl-propionic acids. U.S. Patent 5,089,661, Feb 18, 1992. |
| 94. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone. U.S. Patent 4,582,577, April 15, 1986. |
| 95. | Wagenknecht, J. H. Electrochemical carboxylation of p-isobutylacetophenone and other aryl ketones. U.S. Patent 4,601,797, July 22, 1986. |
| 96. | Zhang, K.; Wang, H.; Zhao, S.-F.; Niu, D.-F.; Lu, J.-X. J. Electroanal. Chem. 2009, 630, 35–41. doi:10.1016/j.jelechem.2009.02.013 |
| 97. | Zhao, S.-F.; Zhu, M.-X.; Zhang, K.; Wang, H.; Lu, J.-X. Tetrahedron Lett. 2011, 52, 2702–2705. doi:10.1016/j.tetlet.2011.03.076 |
| 98. | Chen, B.-L.; Tu, Z.-Y.; Zhu, H.-W.; Sun, W.-W.; Wang, H.; Lu, J.-X. Electrochim. Acta 2014, 116, 475–483. doi:10.1016/j.electacta.2013.11.001 |
| 29. | Cook, R. L.; MacDuff, R. C.; Sammells, A. F. J. Electrochem. Soc. 1988, 135, 1320–1326. doi:10.1149/1.2095972 |
| 153. | Goodridge, F.; Presland, G. J. Appl. Electrochem. 1984, 14, 791–796. doi:10.1007/BF00615269 |
| 30. | Olah, G. A.; Prakash, G. K. S.; Goeppert, A. J. Am. Chem. Soc. 2011, 133, 12881–12898. doi:10.1021/ja202642y |
| 153. | Goodridge, F.; Presland, G. J. Appl. Electrochem. 1984, 14, 791–796. doi:10.1007/BF00615269 |
| 27. | Agarwal, A. S.; Zhai, Y.; Hill, D.; Sridhar, N. ChemSusChem 2011, 4, 1301–1310. doi:10.1002/cssc.201100220 |
| 28. | Ganesh, I. Renewable Sustainable Energy Rev. 2014, 31, 221–257. doi:10.1016/j.rser.2013.11.045 |
| 75. | Yuan, G.; Li, Z.; Jiang, H. Chin. J. Chem. 2009, 27, 1464–1470. doi:10.1002/cjoc.200990246 |
| 89. | Zhang, K.; Wang, H.; Wu, L.; Zhang, J.; Lu, J. Chin. J. Chem. 2010, 28, 509–513. doi:10.1002/cjoc.201090104 |
| 87. | Lateef, S. K.; Raju, R. R.; Mohan, S. K.; Reddy, S. J. Synth. Commun. 2006, 36, 31–36. doi:10.1080/00397910500328811 |
| 153. | Goodridge, F.; Presland, G. J. Appl. Electrochem. 1984, 14, 791–796. doi:10.1007/BF00615269 |
| 21. | Finnegan, R. A.; Altschuld, J. W. J. Organomet. Chem. 1967, 9, 193–204. doi:10.1016/S0022-328X(00)83721-3 |
| 22. | Quirk, R. P.; Yin, J.; Fetters, L. J.; Kastrup, R. V. Macromolecules 1992, 25, 2262–2267. doi:10.1021/ma00034a030 |
| 23. | Ebert, G. W.; Juda, W. L.; Kosakowski, R. H.; Ma, B.; Dong, L.; Cummings, K. E.; Phelps, M. V. B.; Mostafa, A. E.; Luo, J. J. Org. Chem. 2005, 70, 4314–4317. doi:10.1021/jo047731s |
| 24. | Correa, A.; Martín, R. Angew. Chem., Int. Ed. 2009, 48, 6201–6204. doi:10.1002/anie.200900667 |
| 90. | Zhao, S.-F.; Wang, H.; Lan, Y.-C.; Liu, X.; Lu, J.-X.; Zhang, J. J. Electroanal. Chem. 2012, 664, 105–110. doi:10.1016/j.jelechem.2011.11.001 |
| 152. | Riemenschneider, W.; Tanifuji, M. Oxalic acid. In Ullmann's Encyclopedia of Industrial Chemistry; Elvers, B.; Noethe, H., Eds.; Wiley-VCH: Weinheim, Germany, 2002. doi:10.1002/14356007.a18_247 |
| 33. | Matthews, M. A. Pure Appl. Chem. 2001, 73, 1305–1308. doi:10.1351/pac200173081305 |
| 31. | Sequeira, C. A. C.; Santos, D. M. F. J. Braz. Chem. Soc. 2009, 20, 387–406. doi:10.1590/S0103-50532009000300002 |
| 32. | Danly, D. E. J. Electrochem. Soc. 1984, 131, 435C–442C. doi:10.1149/1.2115324 |
| 37. | Li, J.; Hu, X.; Su, Y.; Li, Q. Chem. Eng. Sci. 2007, 62, 6784–6793. doi:10.1016/j.ces.2007.02.021 |
| 31. | Sequeira, C. A. C.; Santos, D. M. F. J. Braz. Chem. Soc. 2009, 20, 387–406. doi:10.1590/S0103-50532009000300002 |
| 34. | Pütter, H.; Hannebaum, H. Preparation of phthalides. U.S. Patent 6,063,256, May 16, 2000. |
| 36. | Doherty, A. P.; Christensen, P. A.; Hamnett, A.; Scott, K. J. Electroanal. Chem. 1995, 386, 39–44. doi:10.1016/0022-0728(94)03816-L |
| 35. | Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d |
| 34. | Pütter, H.; Hannebaum, H. Preparation of phthalides. U.S. Patent 6,063,256, May 16, 2000. |
| 32. | Danly, D. E. J. Electrochem. Soc. 1984, 131, 435C–442C. doi:10.1149/1.2115324 |
| 34. | Pütter, H.; Hannebaum, H. Preparation of phthalides. U.S. Patent 6,063,256, May 16, 2000. |
© 2014 Matthessen et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)