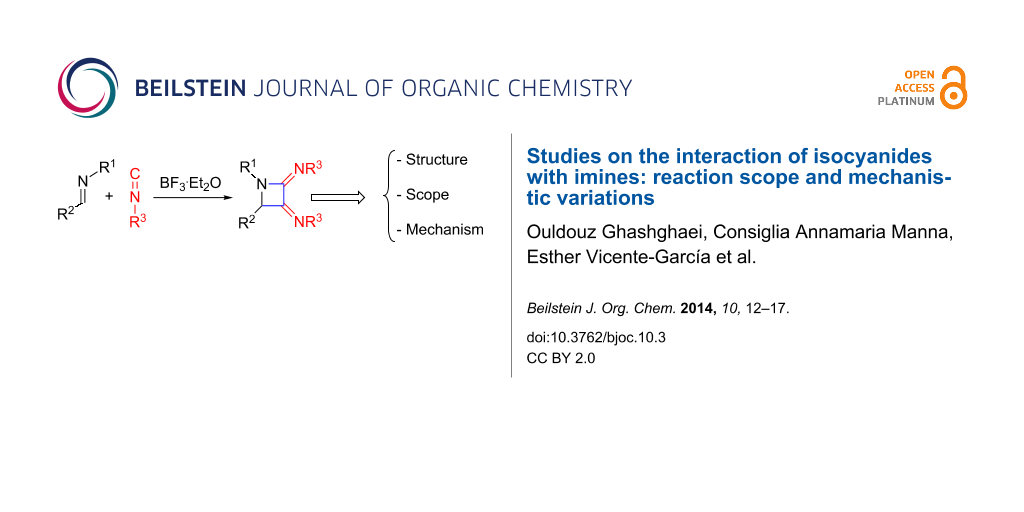
Abstract
The interaction of imines with isocyanides has been studied. The main product results from a sequential process involving the attack of two units of isocyanide, under Lewis acid catalysis, upon the carbon–nitrogen double bond of the imine to form the 4-membered ring system. The scope of the reaction regarding the imine and isocyanide ranges has been determined, and also some mechanistic variations and structural features have been described.

Graphical Abstract
Introduction
The interaction of imines with isocyanides is mainly focused on to the well-known Ugi multicomponent reaction (MCR) [1]. This fundamental process features the participation of a carboxylic acid group which attacks the intermediate nitrilium ion thus leading, after the Mumm rearrangement, to α-amidoamides. However, the direct reaction of imines and isocyanides has been considerably less studied and, in the absence of a carboxylate, the mechanistic outcome is considerably modified [2]. A relevant precedent was described by Deyrup in the late sixties, demonstrating the double incorporation of an isocyanide moiety to an imine [3,4]. Interestingly, the 3CR between a carbonyl, an amine and an isocyanide, taking place through the intermediacy of the in situ generated imine, leads to α-aminoamidines, resulting from the trapping of the nitrilium cation by the remaining amine [5-8]. Taking into account the intrinsic interest in the azetidine scaffold in medicinal chemistry [9], we decided to study in detail the formation of bis(imino)azetidines 3 from the interaction of imines 1 and isocyanides 2 (Scheme 1), including the scope of the reaction and mechanistic features of this interesting ABB’ process [10].
![[1860-5397-10-3-i1]](/bjoc/content/inline/1860-5397-10-3-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Azetidine formation from the interaction of imines with isocyanides.
Scheme 1: Azetidine formation from the interaction of imines with isocyanides.
Results and Discussion
Reaction scope
In this section we analyze the reaction conditions, the structural features of the products and the scope of the reactants.
Reaction conditions
We began our studies with the experimental screening of the solvents, catalysts and temperatures suitable for this transformation. In this respect, taking imine 1a (R1 = p-MeOC6H4; R2 = p-ClC6H4) and isocyanide 2a (R3 = t-Bu), we tested the standard reaction in THF, MeCN, and CH2Cl2 as solvents using a variety of Lewis and Brønsted acid activating agents (20–100 mol %) including: InCl3, Sc(OTf)3, AuCl3, AgOTf, GaCl3, NbCl5, camphorsulfonic acid, I2, Br2·SMe2 and BF3·OEt2, at temperatures ranging from rt to 80 °C. The transformations were tested under standard heating or microwave irradiation, with reaction times lasting form 30 min to 48 h. The imine 1a was generated in situ, using MS 4 Å, or previously prepared by condensation of the corresponding aldehyde and aniline. It was found that the best conditions were obtained using BF3·Et2O as the activating agent in stoichiometric amounts in THF, at rt for 24 hours or under MW irradiation for 30 min at 65 °C, allowing the formation of the expected azetidine 3a in 43% and 48%, respectively. Compounds 4 and 5 could not be detected (Scheme 2). When the process was run as a true MCR (mixing the amine, the aldehyde and two equivalents of isocyanide 2a), the adduct 3a was produced in trace amounts and the main product was the α-amino-amidine 4, in good agreement with previous reports [5-8]. In a different experiment, the addition of a 9-fold excess of isocyanide 2a to the imine 1a under the usual conditions led to detection of tris(imino)pyrrolidine 5 (9%) as the minor product, and azetidine 3a as the major component (24%, Scheme 2).
![[1860-5397-10-3-i2]](/bjoc/content/inline/1860-5397-10-3-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Structural elucidation
Although we could confirm the constitution of the azetidine 3a by spectroscopic methods (NMR, MS), the stereochemistry of the C=N bonds present in the structure remained unsolved. Furthermore, no conclusive nOe’s were observed to assign these stereogenic centers, and there were no reports in the literature regarding this point. A monocrystal of the bis(imino)azetidine 3a was subjected to X-ray diffraction analysis and the solid state structure is depicted in Figure 1 [11].
![[1860-5397-10-3-1]](/bjoc/content/figures/1860-5397-10-3-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray diffraction analysis of azetidine 3a.
Figure 1: X-ray diffraction analysis of azetidine 3a.
This result confirms the structural features associated to this scaffold, and also raises some questions on the origin of the stereochemistry associated to the C=N moieties. First of all, the process can be explained by an attack of one isocyanide equivalent to the Lewis acid (LA)-activated imine, leading to a first nitrilium intermediate (Scheme 3), then a subsequent attack would give raise to a second nitrilium ion [12-15], which is trapped by the nucleophilic nitrogen of the imine. This last step is formally a disfavoured nucleophilic 4-exo-dig process [16], although recent results and calculations show that it should be feasible and some examples have been disclosed [17]. Interestingly, the expected anti-addition mode towards the carbon–nitrogen triple bonds should generate Z configurations [18,19], which are not observed in the solid-sate, thus suggesting isomerization processes affecting the C=N moieties, likely mediated by acid-catalyzed prototropy or other tautomerization steps. Computational calculations (MMFF, AM1 and BL3YP/6-31G* performed in a Spartan suite) suggest that the differences in the heat of formation among some geometrical isomers are small. Furthermore, NMR spectra nearly always display a single set of signals, thus discarding further isomerization events once the compounds are detected or isolated.
![[1860-5397-10-3-i3]](/bjoc/content/inline/1860-5397-10-3-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Stepwise mechanism for the formation of azetidine 3a.
Scheme 3: Stepwise mechanism for the formation of azetidine 3a.
Reactant scope
Next, the scope of the reaction was investigated, and a variety of imines was subjected to the interaction with a range of isocyanides under the optimized conditions to determine the generality of the process and to detect possible restrictions. Results are depicted in Table 1.
Table 1: Scope of the imine and isocyanide starting materials.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-3-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | R1 | R2 | R3 | yield |
|---|---|---|---|---|
| 1 | 4-MeOC6H4 | 4-ClC6H4 | t-Bu | 3a (48%) |
| 2 | 4-MeOC6H4 | 4-ClC6H4 | c-C6H11 | 3b (41%)a |
| 3 | 4-MeOC6H4 | 4-ClC6H4 | Bn | 3c (63%)a |
| 4 | 4-MeC6H4 | 4-ClC6H4 | 4-MeOC6H4 | 3d (19%)b |
| 5 | 3-MeOC6H4 | 4-ClC6H4 | t-Bu | 3e (19%) + 6 (27%) |
| 6 | 4-MeOC6H4 | 2-ClC6H4 | t-Bu | 3f (12%) + 3f' (15%) |
| 7 | 2-MeC6H4 | 4-ClC6H4 | t-Bu | 3g (31%) |
| 8 | n-C3H7 | 4-ClC6H4 | t-Bu | 3h' (32%) |
| 9 | 4-MeOC6H4 | EtOCO | t-Bu | 3i (9%)a + 7 (34%) |
| 10 | 4-MeOC6H4 | EtOCO | c-C6H11 | 3j (34%)a |
| 11 | 4-MeOC6H4 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-3-i6.svg?max-width=637&scale=1.0)
|
t-Bu | 3k (14%)b + 8 (17%) |
| 12 | 4-MeC6H4 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-3-i7.svg?max-width=637&scale=1.0)
|
t-Bu | 3l (28%) |
| 13 | 4-MeOC6H4 | 4-ClC6H4 | Ts-CH2 | – |
| 14 | 4-MeOC6H4 | 4-ClC6H4 | MeO2C-CH2 | – |
aThe reaction was performed at rt. 20% of catalyst was used. bThe adduct could not be isolated in pure form.
The scan of isocyanides shows that their nucleophilicity plays a determining role in the reactivity, since tert-butyl, cyclohexyl and benzyl isocyanides (Table 1, entries 1–3, 5–12) show a relatively high conversion, whereas the aromatic isocyanide was less reactive (entry 4) and the weaker nucleophiles TOSMIC and methyl isocyanoacetate were not productive (Table 1, entries 13 and 14) [20]. Additionally, with respect to imines, the reaction works well for substrates generated from aromatic aldehydes and anilines displaying o-, m- and p-substituents (Table 1, entries 1–7). Moreover, in one case the 3-aminoindole 6 was detected (27%), in agreement with a recent report (Table 1, entry 5) [21].
N-Alkylimines seem to react appropriately (Table 1, entry 8). Furthermore, we have observed some tert-butyl eliminations, probably due to competing reactions under the acidic conditions (Table 1, entries 5, 6 and 8). In general, imines containing electron-rich N-aryl moieties showed higher reactivities, and we were not able to isolate azetidine adducts 3 from the reaction of arylimines containing strong electron-withdrawing groups at the aniline moiety (p-CF3, p-F and p-COOEt), likely because of their low conversions. However, the presence of such groups linked to the carbon of the imine did not seem to disturb their reactivity (Table 1, entries 1–8). Interestingly, the reaction of glyoxylate imine (Table 1, entry 9) with tert-butyl isocyanide led to the formation of minor amounts of the azetidine adduct 3i (9%) whereas the α-aminoamide 7 (34%) was the major component. The formation of amidoamides has been reported in the p-toluenesulfonic acid-catalyzed interaction of anilines, amines and isocyanides [8]. On the other hand, the reaction of the same imine with cyclohexyl isocyanide gave azetidine 3j (34%) in a selective manner (Table 1, entry 10), without traces of the corresponding α-aminoamide. Different types of activated substrates displaying C=N bonds (N-sulfinylimines, oximes and hydrazones) were studied, but none of them reacted productively with isocyanides under the described conditions. Finally, the reaction with isatinimines led to the formation of the spiro-azetidines 3k (14%) and 3l (28%, Table 1, entries 11 and 12). Remarkably, in the former case an intramolecular cyclization of the nitrilium ion upon the electron rich p-methoxyphenyl group took place and led to the formation of the bis(imino)tetrahydroquinoline 8 (17%, Table 1, entry 11).
Mechanistic analysis
Taking into account the structural variety observed in this family of reactions, a rational explanation is needed to understand the formation of such products. Here we describe a simplified hypothesis based on the well-known nucleophilic addition of isocyanides to Lewis acid-activated imines (Scheme 3). The first nitrilium intermediate can evolve through a second addition and ring-closure to yield the azetidine adduct 3 (Scheme 4, route i), or can also be trapped by water or amine/imine nucleophiles leading to α-aminoamides 7 and α-aminoamidines 4, respectively (Scheme 4, route ii). The formation of aminoamide 7 was restricted to the use of glyoxylate imines, and happens only with tert-butyl isocyanide, but not with cyclohexyl isocyanide. On the other hand, the aminoamidines 4, the standard adducts from the amine–aldehyde–isocyanide 3CR were observed in some occasions [5-7] under our conditions presumably by attack of the unreacted imine upon the nitrilium cation. These facts suggest that either a good nucleophile (amines, imines) may act intermolecularly to undergo fast addition to this intermediate or, when an alkyl carboxylate group is present it may stabilize the nitrilium intermediate precluding further addition events and leading to the aminoamides 7 after the final aqueous treatment.
Furthermore, we have detected indole 6 arising from the cyclization of electron-rich aromatic rings linked to the imine nitrogen upon the electrophilic nitrilium intermediate, in agreement with a Sorensen report (Scheme 4, route iii) [21]. Finally, the imino-nitrilium cation can be trapped by an aromatic ring when using isatin imines, leading to bis(imino)tetrahydroquinoline 8, (Scheme 4, route iv). In a reaction using a large isocyanide excess, a triple insertion of the isocyanide moiety has been observed, the adduct being the tris(imino)pyrrolidine 5 (Scheme 4, route v).
![[1860-5397-10-3-i4]](/bjoc/content/inline/1860-5397-10-3-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
As a summary, we have described structural and mechanistic features for the bis(imino)azetidines arising from the imine–isocyanide interaction, finding that the process involves a sequential double isocyanide incorporation into the C=N bond. The final step is a nucleophilic 4-exo-dig cyclization, and the anti addition modes likely lead to less stable stereoisomers which spontaneously isomerize to the observed compounds. Furthermore, we have determined the scope of the reaction, according to the imine and isocyanide starting materials, and a small collection of multicomponent adducts has been prepared. These structures bear a novel azetidine scaffold of potential interest in medicinal chemistry [22,23]. Although the yields are modest, the compounds can be conveniently prepared in a straightforward manner. A part of the azetidine structure distinct scaffolds have been obtained from the interaction of different reactant combinations: α-aminoamides, α-aminoamidines, indoles, bis(imino)tetrahydroquinolines and tris(imino)pyrrolidines. Finally, a unified reaction mechanism that can account for the production of this rich structural outcome has been proposed.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of the NMR spectra for all new compounds and X-ray views of azetidine 3a. | ||
| Format: PDF | Size: 3.4 MB | Download |
References
-
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
And references cited therein.
Return to citation in text: [1] -
El Kaïm, L.; Grimaud, L. In Isocyanide Chemistry; Nenajdenko, V. G., Ed.; Wiley-VCH: Weinheim, 2005; pp 159–194.
See for an overview of acid surrogates in isocyanide MCRs.
Return to citation in text: [1] -
Deyrup, J. A.; Vestling, M. M.; Hagan, W. V.; Yun, H. Y. Tetrahedron 1969, 25, 1467–1478. doi:10.1016/S0040-4020(01)82718-6
Return to citation in text: [1] -
Morel, G.; Marchand, E.; Malvaut, Y. Heteroat. Chem. 2000, 11, 370–376. doi:10.1002/1098-1071(2000)11:5<370::AID-HC8>3.0.CO;2-J
Return to citation in text: [1] -
McFarland, J. W. J. Org. Chem. 1963, 28, 2179–2181. doi:10.1021/jo01044a006
Return to citation in text: [1] [2] [3] -
Keung, W.; Bakir, F.; Patron, A. P.; Rogers, D.; Priest, C. D.; Darmohusodo, V. Tetrahedron Lett. 2004, 45, 733–737. doi:10.1016/j.tetlet.2003.11.051
Return to citation in text: [1] [2] [3] -
Khan, A. T.; Basha, S.; Lal, M.; Mir, M. H. RSC Adv. 2012, 2, 5506–5509. doi:10.1039/c2ra20539d
Return to citation in text: [1] [2] [3] -
Saha, B.; Frett, B.; Wang, Y.; Li, H.-Y. Tetrahedron Lett. 2013, 54, 2340–2343. doi:10.1016/j.tetlet.2013.02.055
Return to citation in text: [1] [2] [3] -
Brandi, A.; Cicchi, S.; Cordero, F. M. Chem. Rev. 2008, 108, 3988–4035. doi:10.1021/cr800325e
Return to citation in text: [1] -
Tejedor, D.; García-Tellado, F. Chem. Soc. Rev. 2007, 36, 484–491. doi:10.1039/b608164a
Return to citation in text: [1] -
CCDC 963354 contains the supplementary crystallographic data of product 3a. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Bez, G.; Zhao, C.-G. Org. Lett. 2003, 5, 4991–4993. doi:10.1021/ol0359618
Return to citation in text: [1] -
Oshita, M.; Yamashita, K.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2005, 127, 761–766. doi:10.1021/ja0450206
Return to citation in text: [1] -
Korotkov, V. S.; Larionov, O. V.; de Meijere, A. Synthesis 2006, 3542–3546. doi:10.1055/s-2006-942514
Return to citation in text: [1] -
Masdeu, C.; Gómez, E.; Williams, N. A. O.; Lavilla, R. Angew. Chem., Int. Ed. 2007, 46, 3043–3046. doi:10.1002/anie.200605070
Return to citation in text: [1] -
Baldwin, J. E. J. Chem. Soc., Chem. Commun. 1976, 734–736. doi:10.1039/C39760000734
Return to citation in text: [1] -
Alabugin, I. V.; Gilmore, K.; Manoharan, M. J. Am. Chem. Soc. 2011, 133, 12608–12623. doi:10.1021/ja203191f
And references cited therein.
Return to citation in text: [1] -
Nguyen, M. T.; Hegarty, A. F.; Sana, M.; Leroy, G. J. Am. Chem. Soc. 1985, 107, 4141–4145. doi:10.1021/ja00300a008
Return to citation in text: [1] -
Johnson, J. E.; Cornell, S. C. J. Org. Chem. 1980, 45, 4144–4148. doi:10.1021/jo01309a015
Return to citation in text: [1] -
Tumanov, V. V.; Tishkov, A. A.; Mayr, H. Angew. Chem., Int. Ed. 2007, 46, 3563–3566. doi:10.1002/anie.200605205
Return to citation in text: [1] -
Schneekloth, J. S., Jr.; Kim, J.; Sorensen, E. J. Tetrahedron 2009, 65, 3096–3101. doi:10.1016/j.tet.2008.08.055
Return to citation in text: [1] [2] -
Burkhard, J. A.; Wagner, B.; Fischer, H.; Schuler, F.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 3524–3527. doi:10.1002/anie.200907108
Return to citation in text: [1] -
Lowe, J. T.; Lee, M. D., IV; Akella, L. B.; Davoine, E.; Donckele, E. J.; Durak, L.; Duvall, J. R.; Gerard, B.; Holson, E. B.; Joliton, A.; Kesavan, S.; Lemercier, B. C.; Liu, H.; Marié, J.-C.; Mulrooney, C. A.; Muncipinto, G.; Welzel-O’Shea, M.; Panko, L. M.; Rowley, A.; Suh, B.-C.; Thomas, M.; Wagner, F. F.; Wei, J.; Foley, M. A.; Marcaurelle, L. A. J. Org. Chem. 2012, 77, 7187–7211. doi:10.1021/jo300974j
Return to citation in text: [1]
| 21. | Schneekloth, J. S., Jr.; Kim, J.; Sorensen, E. J. Tetrahedron 2009, 65, 3096–3101. doi:10.1016/j.tet.2008.08.055 |
| 22. | Burkhard, J. A.; Wagner, B.; Fischer, H.; Schuler, F.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 3524–3527. doi:10.1002/anie.200907108 |
| 23. | Lowe, J. T.; Lee, M. D., IV; Akella, L. B.; Davoine, E.; Donckele, E. J.; Durak, L.; Duvall, J. R.; Gerard, B.; Holson, E. B.; Joliton, A.; Kesavan, S.; Lemercier, B. C.; Liu, H.; Marié, J.-C.; Mulrooney, C. A.; Muncipinto, G.; Welzel-O’Shea, M.; Panko, L. M.; Rowley, A.; Suh, B.-C.; Thomas, M.; Wagner, F. F.; Wei, J.; Foley, M. A.; Marcaurelle, L. A. J. Org. Chem. 2012, 77, 7187–7211. doi:10.1021/jo300974j |
| 1. |
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
And references cited therein. |
| 9. | Brandi, A.; Cicchi, S.; Cordero, F. M. Chem. Rev. 2008, 108, 3988–4035. doi:10.1021/cr800325e |
| 8. | Saha, B.; Frett, B.; Wang, Y.; Li, H.-Y. Tetrahedron Lett. 2013, 54, 2340–2343. doi:10.1016/j.tetlet.2013.02.055 |
| 5. | McFarland, J. W. J. Org. Chem. 1963, 28, 2179–2181. doi:10.1021/jo01044a006 |
| 6. | Keung, W.; Bakir, F.; Patron, A. P.; Rogers, D.; Priest, C. D.; Darmohusodo, V. Tetrahedron Lett. 2004, 45, 733–737. doi:10.1016/j.tetlet.2003.11.051 |
| 7. | Khan, A. T.; Basha, S.; Lal, M.; Mir, M. H. RSC Adv. 2012, 2, 5506–5509. doi:10.1039/c2ra20539d |
| 8. | Saha, B.; Frett, B.; Wang, Y.; Li, H.-Y. Tetrahedron Lett. 2013, 54, 2340–2343. doi:10.1016/j.tetlet.2013.02.055 |
| 5. | McFarland, J. W. J. Org. Chem. 1963, 28, 2179–2181. doi:10.1021/jo01044a006 |
| 6. | Keung, W.; Bakir, F.; Patron, A. P.; Rogers, D.; Priest, C. D.; Darmohusodo, V. Tetrahedron Lett. 2004, 45, 733–737. doi:10.1016/j.tetlet.2003.11.051 |
| 7. | Khan, A. T.; Basha, S.; Lal, M.; Mir, M. H. RSC Adv. 2012, 2, 5506–5509. doi:10.1039/c2ra20539d |
| 3. | Deyrup, J. A.; Vestling, M. M.; Hagan, W. V.; Yun, H. Y. Tetrahedron 1969, 25, 1467–1478. doi:10.1016/S0040-4020(01)82718-6 |
| 4. | Morel, G.; Marchand, E.; Malvaut, Y. Heteroat. Chem. 2000, 11, 370–376. doi:10.1002/1098-1071(2000)11:5<370::AID-HC8>3.0.CO;2-J |
| 20. | Tumanov, V. V.; Tishkov, A. A.; Mayr, H. Angew. Chem., Int. Ed. 2007, 46, 3563–3566. doi:10.1002/anie.200605205 |
| 2. |
El Kaïm, L.; Grimaud, L. In Isocyanide Chemistry; Nenajdenko, V. G., Ed.; Wiley-VCH: Weinheim, 2005; pp 159–194.
See for an overview of acid surrogates in isocyanide MCRs. |
| 21. | Schneekloth, J. S., Jr.; Kim, J.; Sorensen, E. J. Tetrahedron 2009, 65, 3096–3101. doi:10.1016/j.tet.2008.08.055 |
| 12. | Bez, G.; Zhao, C.-G. Org. Lett. 2003, 5, 4991–4993. doi:10.1021/ol0359618 |
| 13. | Oshita, M.; Yamashita, K.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2005, 127, 761–766. doi:10.1021/ja0450206 |
| 14. | Korotkov, V. S.; Larionov, O. V.; de Meijere, A. Synthesis 2006, 3542–3546. doi:10.1055/s-2006-942514 |
| 15. | Masdeu, C.; Gómez, E.; Williams, N. A. O.; Lavilla, R. Angew. Chem., Int. Ed. 2007, 46, 3043–3046. doi:10.1002/anie.200605070 |
| 17. |
Alabugin, I. V.; Gilmore, K.; Manoharan, M. J. Am. Chem. Soc. 2011, 133, 12608–12623. doi:10.1021/ja203191f
And references cited therein. |
| 11. | CCDC 963354 contains the supplementary crystallographic data of product 3a. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 18. | Nguyen, M. T.; Hegarty, A. F.; Sana, M.; Leroy, G. J. Am. Chem. Soc. 1985, 107, 4141–4145. doi:10.1021/ja00300a008 |
| 19. | Johnson, J. E.; Cornell, S. C. J. Org. Chem. 1980, 45, 4144–4148. doi:10.1021/jo01309a015 |
| 5. | McFarland, J. W. J. Org. Chem. 1963, 28, 2179–2181. doi:10.1021/jo01044a006 |
| 6. | Keung, W.; Bakir, F.; Patron, A. P.; Rogers, D.; Priest, C. D.; Darmohusodo, V. Tetrahedron Lett. 2004, 45, 733–737. doi:10.1016/j.tetlet.2003.11.051 |
| 7. | Khan, A. T.; Basha, S.; Lal, M.; Mir, M. H. RSC Adv. 2012, 2, 5506–5509. doi:10.1039/c2ra20539d |
| 8. | Saha, B.; Frett, B.; Wang, Y.; Li, H.-Y. Tetrahedron Lett. 2013, 54, 2340–2343. doi:10.1016/j.tetlet.2013.02.055 |
| 10. | Tejedor, D.; García-Tellado, F. Chem. Soc. Rev. 2007, 36, 484–491. doi:10.1039/b608164a |
| 16. | Baldwin, J. E. J. Chem. Soc., Chem. Commun. 1976, 734–736. doi:10.1039/C39760000734 |
© 2014 Ghashghaei et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)