Abstract
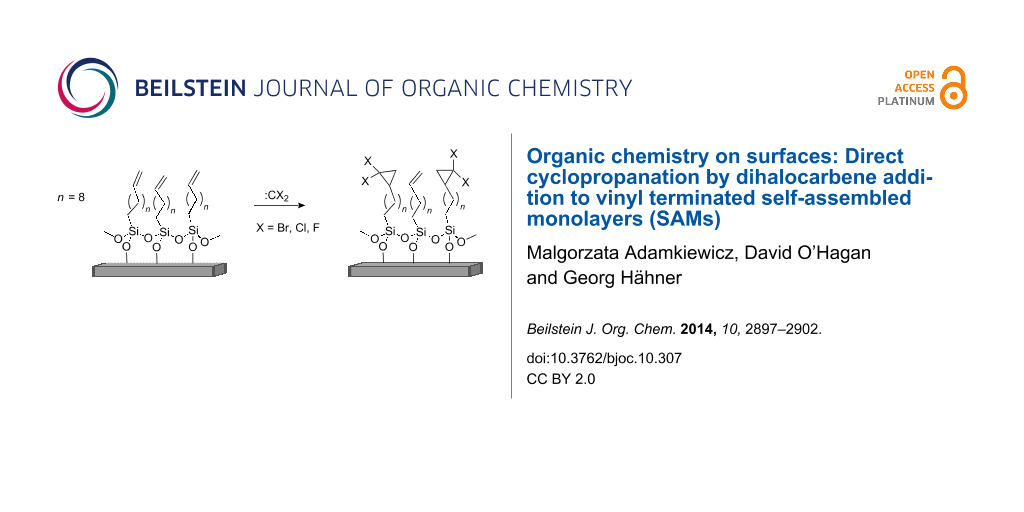
C11-Vinyl-terminated self-assembled monolayers (SAMs) on silica surfaces are successfully modified in C–C bond forming reactions with dihalocarbenes to generate SAMs, terminated with dihalo- (fluoro, chloro, bromo) cyclopropane motifs with about 30% surface coverage.

Graphical Abstract
Introduction
Self-assembled monolayers (SAMs) are increasingly being used as a means of surface modification to alter properties in a tuneable manner [1-3]. The major classes of SAMs are those with adsorbed long chain alkyl thiols on gold surfaces/nanoparticles [4,5], or long chain alkylsilanes on silica surfaces [6,7]. Two general approaches are taken to achieve surface modification as illustrated in Figure 1. The first involves incorporating pre-functionalised alkylsilanes/alkylthiols carrying functional groups (FG) to generate the SAM directly, whereas the second approach involves chemical modification of a pre-assembled monolayer carrying reactive groups (RG), as a means to introduce the SAM carrying the FGs [8]. Both approaches present challenges. In the former the desired functionality (FG) requires to be robust and orthogonal in reactivity to the chemistry involved in securing the substrate to the organic film (e.g., FG-Alkyl-SiCl3 and silicon substrate). In the latter chemical modification of the reactive groups of the pre-coated SAM has to be efficient enough such that a reasonable conversion can be obtained, with chemical specificity and lack of surface degradation. In this respect ‘click’ reactions have become attractive including azide–alkyne cycloadditions [9,10], Diels–Alder reactions [11,12], maleimide–thiol reactions [13], thiol–ene additions [14], and imine/oxime conjugations [15]. In this article we demonstrate that dihalocarbenes can be used to generate dihalocyclopropanes on olefin terminated SAMs.
![[1860-5397-10-307-1]](/bjoc/content/figures/1860-5397-10-307-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: General strategies for incorporating functional groups (FGs) on the surface of self-assembled monolayers (SAMs), illustrated for alkylsilanes onto silica.
Figure 1: General strategies for incorporating functional groups (FGs) on the surface of self-assembled monol...
We recently reported the formation of high quality vinyl-terminated SAMs generated from the vapour phase by adsorption of octadecyltrichlorosilanes onto silicon wafers [16]. With access to these SAMs it became an objective to explore functional group modification of the vinyl double bond. Carbon–carbon bond formation of vinyl-terminated SAMs has been demonstrated, e.g., through surface modification of radicals generated by C–O bond thermolysis [17] and in a more controlled sense via olefin cross metathesis/enyne metathesis [18] of mixed vinyl and acetylenyl-terminated SAMs followed by Diels–Alder modifications of the resultant dienes [19]. We are not aware however of straightforward carbene additions having been explored with olefin-terminated SAMs. The potential for product cyclopropanes offered a modification of limited steric impact, but if suitably substituted may be used to tune surface properties. Therefore dihalocyclopropanes emerged as an attractive controlled modification particularly as the precursor dihalocarbenes are relatively easily generated [20]. In this context we report dihalocyclopropanation of pre-assembled vinyl-terminated SAMs. Three dihalocarbene modifications were explored involving dibromo- (:CBr2), dichloro- (:CCl2) and difluoro- (:CF2) carbenes [21-23]. The resultant SAMs were analysed by X-ray photoelectron spectroscopy (XPS), contact angle goniometry, ellipsometry, and atomic force microscopy.
Results and Discussion
After exposure to carbenes the vinyl-terminated SAMs were characterised by XPS, contact angle measurements and ellipsometry (see Supporting Information File 1). With XPS elements such as silicon, carbon and oxygen were expected in all cases [16]. In each case control reactions were also carried out on the C18-methyl (Me)-terminated SAMs, to ensure that only the vinyl group was responsible for surface reactivity. The resultant XPS analyses are shown for the vinyl-terminated SAMs for each carbene in Figure 2, and directly underneath, the lower traces illustrate the corresponding analyses after exposure of the carbene solutions to the C18-Me-terminated SAMs.
![[1860-5397-10-307-2]](/bjoc/content/figures/1860-5397-10-307-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: XPS scans after reactions with a) :CBr2; b) :CCl2 and c) :CF2. In each case the upper traces are scans of C11-vinyl SAMs, and the lower traces are C18-Me-terminated SAMs each treated with the relevant carbene reaction solution. The expanded regions on the right hand side, associated with each C11-vinyl SAM spectrum, show the key halide specific XPS signals.
Figure 2: XPS scans after reactions with a) :CBr2; b) :CCl2 and c) :CF2. In each case the upper traces are sc...
Figure 2a represents a surface after chemical modification with :CBr2 generated from CHBr3. New peaks appeared at binding energies of 71 and 182 eV in all of the samples. These were assigned to Br 3d and Br 3p signals, respectively [24,25]. Figure 2b shows the results obtained from SAMs after modification with :CCl2 generated from CHCl3. New signals at binding energies of 201 and 270 eV were detected. These were assigned to Cl 2p and Cl 2s [26]. Finally Figure 2c represents the surface after chemical modification with :CF2 generated from TMSCF3. A new signal at binding energy of 688.7 eV was detected and assigned to F 1s, consistent with a CF2 group present on the surface [27].
It was anticipated that gem-dibromo-, gem-dichloro- and gem-difluorocyclopropane-terminated SAMs will be formed, following the usual transformations of these carbenes with double bonds. To add further support to this expectation, model reactions were carried out under each of the reaction conditions with dec-1-ene (1, Scheme 1). All of the cyclopropane products 2a-c were obtained cleanly and in moderate yields (see Supporting Information File 1). The results of the model reactions demonstrate that formation of the dihalocyclopropane rings is a relatively clean process for this long chain terminal vinyl substrate. The absence of any side products gives confidence that only dihalocyclopropanes will be formed in the surface reactions.
![[1860-5397-10-307-i1]](/bjoc/content/inline/1860-5397-10-307-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Model reactions of dec-1-ene (1) with dihalocarbenes in the liquid phase. a) and b) NaOH, BTEAC, CHX3, DCM, 8 h, 25 °C; c) TMSCF3, THF, NaI, 3 h, 65 °C.
Scheme 1: Model reactions of dec-1-ene (1) with dihalocarbenes in the liquid phase. a) and b) NaOH, BTEAC, CHX...
Turning to the C11-vinyl-terminated SAMs products. In each case the presence of gem-dihalocyclopropane groups on the surface is supported by the ratios of the C 1s signals to the Br 3d, Cl 2p or F 1s signal, respectively. The theoretical and experimental ratios between the carbon and halogen XPS signals are summarised in Table 1. In all cases the ratios are consistent with a modification coverage of ~30%, with a slightly lower conversion rate in case of F, which might be due to its higher electronegativity and an associated higher repulsion between the terminal groups after cyclopropanation. Conversion rates were determined by correcting the experimental C3/(C1 + C2) ratios from Table 1 with a factor of d/(λ(1 − exp(−d/λ)), where d is the film thickness (determined with ellipsometry) and λ the mean free path of the electrons. This accounts for the partial attenuation of the C3 XPS carbon signal. The water contact angles (CAs) of Br, Cl and F carbene treated surfaces were recorded and the CA values obtained of 80°, 85° and 104°, respectively, are in good agreement with the literature [28-30]. Notably the fluorinated SAM has the largest contact angle as expected, however, the increase and the final contact angle values are clearly lower than that for a pure CF3 terminated film (~118–120°) [31], but this is not surprising given that the halogen functional group density is lower.
Table 1: Assignment of the C 1s XPS signals after treatment of C11-vinyl SAMs with the respective dihalocarbene. Theoretical and experimental ratios of the Br 3d to C 1s, Cl 2p to C 1s and F 1s to C 1s XPS signals of modified C11-vinyl SAMs.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-307-i2.svg?max-width=637&scale=1.0)
|
|||||
| Ratios | |||||
|---|---|---|---|---|---|
| X:C1 | X:C2 | C1:C2 | C3:(C1 + C2) | ||
| Theor. | :CX2 | 2:1 | 1:1 | 1:2 | 3:1 |
| Exp. | :CBr2 | 1.9:1 | 0.9:1 | 1:2.1 | 7.6:1 |
| Exp. | :CCl2 | 2.2:1 | 1.1:1 | 1:2 | 7.4:1 |
| Exp. | :CF2 | 2.1:1 | 1:1 | 1:2 | 8.5:1 |
Finally AFM images were recorded for the three dihalocyclopropane modified surfaces and they are shown in Figure 3a–c. In all cases the images are smooth and defect free. There was no excess of material observed from reagents after washing, and in each case the RMS surface roughness values did not exceeded 150 pm. It is clear that there is no detectable change observed in the film after each modification, and that the films are of good integrity.
![[1860-5397-10-307-3]](/bjoc/content/figures/1860-5397-10-307-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: AFM images of 5 μm × 5 μm area of C11-vinyl SAMs modified with a) :CBr2 carbene, RMS 93 pm; b) :CCl2 carbene, RMS 101 pm; c) TMSCF3, RMS 79 pm.
Figure 3: AFM images of 5 μm × 5 μm area of C11-vinyl SAMs modified with a) :CBr2 carbene, RMS 93 pm; b) :CCl2...
An alternative approach, also illustrated in Figure 1 is to prepare SAMs using pure samples of pre-halogenated cyclopropyl chains, with deposition directly onto the surface. This presents the obvious challenge of obtaining highly ordered films after direct deposition. The current approach establishes films of good integrity, which then become chemically modified. There is good evidence that this is less straightforward with functionalised surfactants [32].
Conclusion
In summary we have been able to demonstrate that vinyl-terminated SAMs can be chemically modified by a range of dihalocarbenes to generate surfaces carrying the corresponding dihalocyclopropane motifs. The reactions demonstrate that these organic chemical transformations, which have been relatively widely used in solution reactions of olefins, can be extended to surface reactions of SAMs. This opens up prospects too of modifying surfaces in this manner with carbenes carrying more elaborate functional groups, and thus a more dramatic change to the surface properties.
Experimental
Bromoform (CHBr3), chloroform (CHCl3) and the Ruppert–Prakash reagent (CF3Si(CH3)2) [33,34] were used as the carbene precursors for surface modification, with the resultant carbenes generated in solution. For dibromo- and dichlorocarbene generation a solution of NaOH with CHBr3 or CHCl3 was stirred with a solution of benzyltriethylammonium chloride (BTEAC, 0.1 mmol) in dichloromethane for 10 min at 0 °C. Pre-coated silicon wafers (1 cm × 1.5 cm) with C11-vinyl-terminated SAMs, were immersed in the reaction mixture and the liquids were stirred at room temperature for fixed periods of time (see Supporting Information File 1). SAMs on silicon substrates form stable films [6,7], however, they can be vulnerable to chemical degradation particularly in aqueous base [28,35,36]. For this reason the NaOH concentration and reaction time required to be optimised. The reaction temperature was kept at 25 °C and the phase-transfer catalyst, benzyltriethylammonium chloride (BTEAC) was chosen to generate the :CX2 carbenes, and minimise exposure of the wafers to the base.
The Ruppert–Prakash reagent (TMSCF3) was used to prepare the gem-difluorocyclopropane-terminated SAMs by generating difluorocarbene, following the procedure of Wang et al. [22] for small molecule transformations. This involved stirring a solution of NaI (0.2 equiv) and TMSCF3 in THF (2 mL), and then immersing the silicon wafers (1 cm × 1.5 cm) into the reaction mixture at 65 °C for a fixed period of time (see Supporting Information File 1). The experimental set-up for the surface modification with the three different carbenes is shown in Figure 4. Details of the surface analytical techniques used are given in the Supporting Information File 1.
![[1860-5397-10-307-4]](/bjoc/content/figures/1860-5397-10-307-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The experimental set-up for the dibromo-, dichloro- and difluorocarbene reactions performed on C11-vinyl SAMs.
Figure 4: The experimental set-up for the dibromo-, dichloro- and difluorocarbene reactions performed on C11-...
Supporting Information
| Supporting Information File 1: Synthesis protocols and additional surface analysis data. | ||
| Format: PDF | Size: 339.3 KB | Download |
References
-
Herzer, N.; Haensch, C.; Hoeppener, S.; Schubert, U. S. Langmuir 2010, 26, 8358–8365. doi:10.1021/la9047837
Return to citation in text: [1] -
Love, J. C.; Estroff, L. A.; Kriebel, J. K.; Nuzzo, R. G.; Whitesides, G. M. Chem. Rev. 2005, 105, 1103–1170. doi:10.1021/cr0300789
Return to citation in text: [1] -
Ulman, A. Chem. Rev. 1996, 96, 1533–1554. doi:10.1021/cr9502357
Return to citation in text: [1] -
Häkkinen, H. Nat. Chem. 2012, 4, 443–455. doi:10.1038/nchem.1352
Return to citation in text: [1] -
Vericat, C.; Vela, M. E.; Benitez, G.; Carro, P.; Salvarezza, R. C. Chem. Soc. Rev. 2010, 39, 1805–1834. doi:10.1039/b907301a
Return to citation in text: [1] -
Haensch, C.; Hoeppener, S.; Schubert, U. S. Chem. Soc. Rev. 2010, 39, 2323–2334. doi:10.1039/b920491a
Return to citation in text: [1] [2] -
Onclin, S.; Ravoo, B. J.; Reinhoudt, D. N. Angew. Chem., Int. Ed. 2005, 44, 6282–6304. doi:10.1002/anie.200500633
Return to citation in text: [1] [2] -
Nicosia, C.; Huskens, J. Mater. Horiz. 2014, 1, 32–45. doi:10.1039/c3mh00046j
See for a recent excellent overview see.
Return to citation in text: [1] -
Orski, S. V.; Poloukhtine, A. A.; Arumugam, S.; Mao, L.; Popik, V. V.; Locklin, J. J. Am. Chem. Soc. 2010, 132, 11024–11026. doi:10.1021/ja105066t
Return to citation in text: [1] -
Collman, J. P.; Devaraj, N. K.; Chidsey, C. E. D. Langmuir 2004, 20, 1051–1053. doi:10.1021/la0362977
Return to citation in text: [1] -
Arumugam, S.; Popik, V. V. J. Am. Chem. Soc. 2011, 133, 15730–15736. doi:10.1021/ja205652m
Return to citation in text: [1] -
Yousaf, M. N.; Mrksich, M. J. Am. Chem. Soc. 1999, 121, 4286–4287. doi:10.1021/ja983529t
Return to citation in text: [1] -
Houseman, B. T.; Gawalt, E. S.; Mrksich, M. Langmuir 2003, 19, 1522–1531. doi:10.1021/la0262304
Return to citation in text: [1] -
Wendeln, C.; Rinnen, S.; Schulz, C.; Arlinghaus, H. F.; Ravoo, B. J. Langmuir 2010, 26, 15966–15971. doi:10.1021/la102966j
Return to citation in text: [1] -
Rozkiewicz, D. I.; Kraan, Y.; Werten, M. W. T.; de Wolf, F. A.; Subramaniam, V.; Ravoo, B. J.; Reinhoudt, D. N. Chem. – Eur. J. 2006, 12, 6290–6297. doi:10.1002/chem.200501554
Return to citation in text: [1] -
Adamkiewicz, M.; O'Hara, T.; O'Hagan, D.; Hähner, G. Thin Solid Films 2012, 520, 6719–6723. doi:10.1016/j.tsf.2012.07.054
Return to citation in text: [1] [2] -
Siegenthaler, K. O.; Schäfer, A.; Studer, A. J. Am. Chem. Soc. 2007, 129, 5826–5827. doi:10.1021/ja0686716
Return to citation in text: [1] -
Dutta, S.; Perring, M.; Barrett, S.; Mitchell, M.; Kenis, P. J. A.; Bowden, N. B. Langmuir 2006, 22, 2146–2155. doi:10.1021/la0532196
Return to citation in text: [1] -
Lee, J. K.; Shi, Y. S.; Lee, J. S.; Kim, Y.-G.; Jung, Y. H.; Oh, E.; Ko, S.-B.; Jung, H.-j.; Kang, P.-S.; Choi, I. S. Langmuir 2005, 21, 10311–10315. doi:10.1021/la051680s
Return to citation in text: [1] -
Makosza, M. Pure Appl. Chem. 2000, 72, 1399–1403. doi:10.1351/pac200072071399
Return to citation in text: [1] -
Ziyat, H.; Itto, M. Y. A.; Ali, M. A.; Riahi, A.; Karim, A.; Daran, J.-C. ARKIVOC 2006, xii, 152–160. doi:10.3998/ark.5550190.0007.c18
Return to citation in text: [1] -
Wang, F.; Luo, T.; Hu, J.; Wang, Y.; Krishnan, H. S.; Jog, P. V.; Ganesh, S. K.; Prakash, G. K. S.; Olah, G. A. Angew. Chem., Int. Ed. 2011, 50, 7153–7157. doi:10.1002/anie.201101691
Return to citation in text: [1] [2] -
Juliá, S.; Ginebreda, A. Synthesis 1977, 682–683. doi:10.1055/s-1977-24531
Return to citation in text: [1] -
Balachander, N.; Sukenik, C. N. Langmuir 1990, 6, 1621–1627. doi:10.1021/la00101a001
Return to citation in text: [1] -
Cohen, Y. S.; Vilan, A.; Ron, I.; Cahen, D. J. Phys. Chem. C 2009, 113, 6174–6181. doi:10.1021/jp9006125
Return to citation in text: [1] -
Chen, J.-K.; Hsieh, C.-Y.; Huang, C.-F.; Li, P.-M.; Kuo, S.-W.; Chang, F.-C. Macromolecules 2008, 41, 8729–8736. doi:10.1021/ma801127m
Return to citation in text: [1] -
Ohnishi, S.; Ishida, T.; Yaminsky, V. V.; Christenson, H. K. Langmuir 2000, 16, 2722–2730. doi:10.1021/la991167c
Return to citation in text: [1] -
Wasserman, S. R.; Tao, Y. T.; Whitesides, G. M. Langmuir 1989, 5, 1074–1087. doi:10.1021/la00088a035
Return to citation in text: [1] [2] -
Lo, M. K. F.; Gard, M. N.; Goldsmith, B. R.; Garcia-Garibay, M. A.; Monbouquette, H. G. Langmuir 2012, 28, 16156–16166. doi:10.1021/la302880v
Return to citation in text: [1] -
Motomatsu, M.; Mizutani, W.; Nie, H.-Y.; Tokumoto, H. Thin Solid Films 1996, 281–282, 548–551. doi:10.1016/0040-6090(96)08721-4
Return to citation in text: [1] -
Colorado, R., Jr.; Lee, T. R. Langmuir 2003, 19, 3288–3296. doi:10.1021/la0263763
Return to citation in text: [1] -
Fritz, M. C.; Hähner, G.; Spencer, N. D.; Bürli, R.; Vasella, A. Langmuir 1996, 12, 6074–6082. doi:10.1021/la960623j
Return to citation in text: [1] -
Prakash, G. K. S.; Yudin, A. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991
Return to citation in text: [1] -
Prakash, G. K. S.; Krishnamurti, R.; Olah, G. A. J. Am. Chem. Soc. 1989, 111, 393–395. doi:10.1021/ja00183a073
Return to citation in text: [1] -
Ciampi, S.; Harper, J. B.; Gooding, J. J. Chem. Soc. Rev. 2010, 39, 2158–2183. doi:10.1039/b923890p
Return to citation in text: [1] -
Calistri-Yeh, M.; Kramer, E. J.; Sharma, R.; Zhao, W.; Rafailovich, M. H.; Sokolov, J.; Brock, J. D. Langmuir 1996, 12, 2747–2755. doi:10.1021/la950518u
Return to citation in text: [1]
| 1. | Herzer, N.; Haensch, C.; Hoeppener, S.; Schubert, U. S. Langmuir 2010, 26, 8358–8365. doi:10.1021/la9047837 |
| 2. | Love, J. C.; Estroff, L. A.; Kriebel, J. K.; Nuzzo, R. G.; Whitesides, G. M. Chem. Rev. 2005, 105, 1103–1170. doi:10.1021/cr0300789 |
| 3. | Ulman, A. Chem. Rev. 1996, 96, 1533–1554. doi:10.1021/cr9502357 |
| 9. | Orski, S. V.; Poloukhtine, A. A.; Arumugam, S.; Mao, L.; Popik, V. V.; Locklin, J. J. Am. Chem. Soc. 2010, 132, 11024–11026. doi:10.1021/ja105066t |
| 10. | Collman, J. P.; Devaraj, N. K.; Chidsey, C. E. D. Langmuir 2004, 20, 1051–1053. doi:10.1021/la0362977 |
| 21. | Ziyat, H.; Itto, M. Y. A.; Ali, M. A.; Riahi, A.; Karim, A.; Daran, J.-C. ARKIVOC 2006, xii, 152–160. doi:10.3998/ark.5550190.0007.c18 |
| 22. | Wang, F.; Luo, T.; Hu, J.; Wang, Y.; Krishnan, H. S.; Jog, P. V.; Ganesh, S. K.; Prakash, G. K. S.; Olah, G. A. Angew. Chem., Int. Ed. 2011, 50, 7153–7157. doi:10.1002/anie.201101691 |
| 23. | Juliá, S.; Ginebreda, A. Synthesis 1977, 682–683. doi:10.1055/s-1977-24531 |
| 8. |
Nicosia, C.; Huskens, J. Mater. Horiz. 2014, 1, 32–45. doi:10.1039/c3mh00046j
See for a recent excellent overview see. |
| 16. | Adamkiewicz, M.; O'Hara, T.; O'Hagan, D.; Hähner, G. Thin Solid Films 2012, 520, 6719–6723. doi:10.1016/j.tsf.2012.07.054 |
| 6. | Haensch, C.; Hoeppener, S.; Schubert, U. S. Chem. Soc. Rev. 2010, 39, 2323–2334. doi:10.1039/b920491a |
| 7. | Onclin, S.; Ravoo, B. J.; Reinhoudt, D. N. Angew. Chem., Int. Ed. 2005, 44, 6282–6304. doi:10.1002/anie.200500633 |
| 19. | Lee, J. K.; Shi, Y. S.; Lee, J. S.; Kim, Y.-G.; Jung, Y. H.; Oh, E.; Ko, S.-B.; Jung, H.-j.; Kang, P.-S.; Choi, I. S. Langmuir 2005, 21, 10311–10315. doi:10.1021/la051680s |
| 4. | Häkkinen, H. Nat. Chem. 2012, 4, 443–455. doi:10.1038/nchem.1352 |
| 5. | Vericat, C.; Vela, M. E.; Benitez, G.; Carro, P.; Salvarezza, R. C. Chem. Soc. Rev. 2010, 39, 1805–1834. doi:10.1039/b907301a |
| 20. | Makosza, M. Pure Appl. Chem. 2000, 72, 1399–1403. doi:10.1351/pac200072071399 |
| 15. | Rozkiewicz, D. I.; Kraan, Y.; Werten, M. W. T.; de Wolf, F. A.; Subramaniam, V.; Ravoo, B. J.; Reinhoudt, D. N. Chem. – Eur. J. 2006, 12, 6290–6297. doi:10.1002/chem.200501554 |
| 17. | Siegenthaler, K. O.; Schäfer, A.; Studer, A. J. Am. Chem. Soc. 2007, 129, 5826–5827. doi:10.1021/ja0686716 |
| 14. | Wendeln, C.; Rinnen, S.; Schulz, C.; Arlinghaus, H. F.; Ravoo, B. J. Langmuir 2010, 26, 15966–15971. doi:10.1021/la102966j |
| 18. | Dutta, S.; Perring, M.; Barrett, S.; Mitchell, M.; Kenis, P. J. A.; Bowden, N. B. Langmuir 2006, 22, 2146–2155. doi:10.1021/la0532196 |
| 13. | Houseman, B. T.; Gawalt, E. S.; Mrksich, M. Langmuir 2003, 19, 1522–1531. doi:10.1021/la0262304 |
| 11. | Arumugam, S.; Popik, V. V. J. Am. Chem. Soc. 2011, 133, 15730–15736. doi:10.1021/ja205652m |
| 12. | Yousaf, M. N.; Mrksich, M. J. Am. Chem. Soc. 1999, 121, 4286–4287. doi:10.1021/ja983529t |
| 16. | Adamkiewicz, M.; O'Hara, T.; O'Hagan, D.; Hähner, G. Thin Solid Films 2012, 520, 6719–6723. doi:10.1016/j.tsf.2012.07.054 |
| 27. | Ohnishi, S.; Ishida, T.; Yaminsky, V. V.; Christenson, H. K. Langmuir 2000, 16, 2722–2730. doi:10.1021/la991167c |
| 24. | Balachander, N.; Sukenik, C. N. Langmuir 1990, 6, 1621–1627. doi:10.1021/la00101a001 |
| 25. | Cohen, Y. S.; Vilan, A.; Ron, I.; Cahen, D. J. Phys. Chem. C 2009, 113, 6174–6181. doi:10.1021/jp9006125 |
| 26. | Chen, J.-K.; Hsieh, C.-Y.; Huang, C.-F.; Li, P.-M.; Kuo, S.-W.; Chang, F.-C. Macromolecules 2008, 41, 8729–8736. doi:10.1021/ma801127m |
| 22. | Wang, F.; Luo, T.; Hu, J.; Wang, Y.; Krishnan, H. S.; Jog, P. V.; Ganesh, S. K.; Prakash, G. K. S.; Olah, G. A. Angew. Chem., Int. Ed. 2011, 50, 7153–7157. doi:10.1002/anie.201101691 |
| 6. | Haensch, C.; Hoeppener, S.; Schubert, U. S. Chem. Soc. Rev. 2010, 39, 2323–2334. doi:10.1039/b920491a |
| 7. | Onclin, S.; Ravoo, B. J.; Reinhoudt, D. N. Angew. Chem., Int. Ed. 2005, 44, 6282–6304. doi:10.1002/anie.200500633 |
| 28. | Wasserman, S. R.; Tao, Y. T.; Whitesides, G. M. Langmuir 1989, 5, 1074–1087. doi:10.1021/la00088a035 |
| 35. | Ciampi, S.; Harper, J. B.; Gooding, J. J. Chem. Soc. Rev. 2010, 39, 2158–2183. doi:10.1039/b923890p |
| 36. | Calistri-Yeh, M.; Kramer, E. J.; Sharma, R.; Zhao, W.; Rafailovich, M. H.; Sokolov, J.; Brock, J. D. Langmuir 1996, 12, 2747–2755. doi:10.1021/la950518u |
| 32. | Fritz, M. C.; Hähner, G.; Spencer, N. D.; Bürli, R.; Vasella, A. Langmuir 1996, 12, 6074–6082. doi:10.1021/la960623j |
| 33. | Prakash, G. K. S.; Yudin, A. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 34. | Prakash, G. K. S.; Krishnamurti, R.; Olah, G. A. J. Am. Chem. Soc. 1989, 111, 393–395. doi:10.1021/ja00183a073 |
| 28. | Wasserman, S. R.; Tao, Y. T.; Whitesides, G. M. Langmuir 1989, 5, 1074–1087. doi:10.1021/la00088a035 |
| 29. | Lo, M. K. F.; Gard, M. N.; Goldsmith, B. R.; Garcia-Garibay, M. A.; Monbouquette, H. G. Langmuir 2012, 28, 16156–16166. doi:10.1021/la302880v |
| 30. | Motomatsu, M.; Mizutani, W.; Nie, H.-Y.; Tokumoto, H. Thin Solid Films 1996, 281–282, 548–551. doi:10.1016/0040-6090(96)08721-4 |
| 31. | Colorado, R., Jr.; Lee, T. R. Langmuir 2003, 19, 3288–3296. doi:10.1021/la0263763 |
© 2014 Adamkiewicz et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)