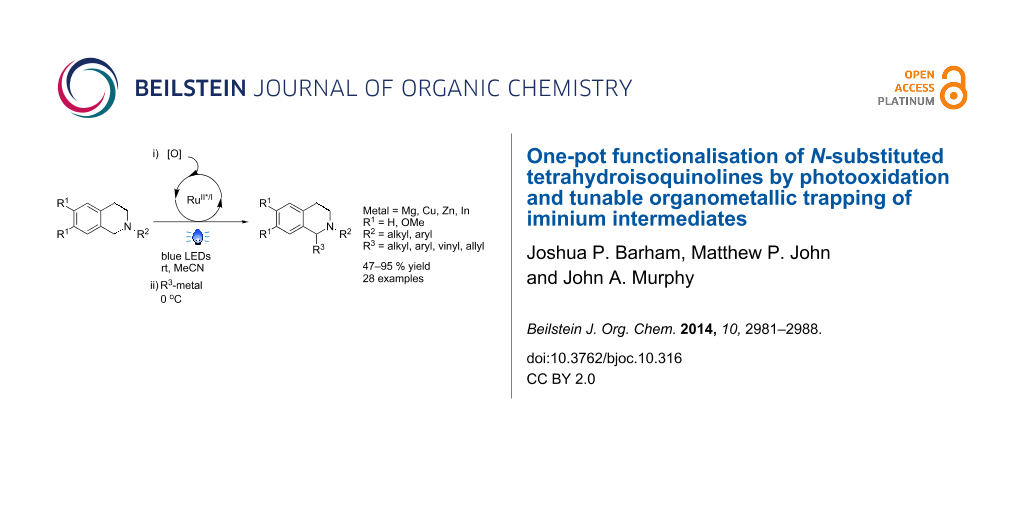
Abstract
Nucleophilic trapping of iminium salts generated via oxidative functionalisation of tertiary amines is well established with stabilised carbon nucleophiles. The few reports of organometallic additions have limited scope of substrate and organometallic nucleophile. We report a novel, one-pot methodology that functionalises N-substituted tetrahydroisoquinolines by visible light-assisted photooxidation, followed by trapping of the resultant iminium ions with organometallic nucleophiles. This affords 1,2-disubstituted tetrahydroisoquinolines in moderate to excellent yields.

Graphical Abstract
Introduction
Tetrahydroisoquinolines (THIQs) are structural motifs prominent within biologically active natural products and pharmaceutical compounds [1,2]. From (−)-carnegine (1, a monoamine oxidase A inhibitor) [3] to (+)-solifenacin (2, a bladder-selective muscarinic M3 receptor antagonist) [4] to (±)-methopholine (3, an opioid analgesic) [5,6], a 1,2-disubstituted THIQ motif occurs throughout (Figure 1).
![[1860-5397-10-316-1]](/bjoc/content/figures/1860-5397-10-316-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of biologically active 1,2-disubstituted tetrahydroisoquinolines.
Figure 1: Examples of biologically active 1,2-disubstituted tetrahydroisoquinolines.
Environmental consciousness has initiated the development of methods which construct THIQs using green technologies, in mild conditions and with high atom efficiencies. Catalytic oxidative functionalisation of the C–H bond α- to the amine function is one such methodology. Iminium salts generated in this way can be intercepted by a nucleophile in a one-pot reaction (Scheme 1). Alternatively, the α-amino radicals can be trapped by electrophiles [7-9]. Oxidative C–H functionalisation of THIQs is reported using Cu(I) [10,11], Fe(III) [12], V(IV) [13] and I2 [14,15] catalysts, but also with heterogeneous [16], metal-organic [17,18] and organic [19,20] photocatalysts. Such catalysts are used in combination with various stoichiometric oxidants including oxygen [16,21].
![[1860-5397-10-316-i1]](/bjoc/content/inline/1860-5397-10-316-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Oxidative C–H functionalisation and examples of previously reported nucleophilic trappings.
Scheme 1: Oxidative C–H functionalisation and examples of previously reported nucleophilic trappings.
However, nucleophilic trappings of resultant iminium salts are typically exemplified with highly stabilised carbon nucleophiles such as cyanides, nitronates, enolate equivalents and heterocycles (Scheme 1) [14,22-24]. Reports of organometallic additions to THIQs in this context are limited to aryl [25-27] and alkynyl [22,28-30] nucleophiles and the substrate scope is generally limited to N-aryl THIQs [31]. However, Li reported a hypervalent iodine mediated N-aryl THIQ oxidation which tolerated a wide range of organometallic nucleophiles [32] and Yu developed methodology for THIQ alkynylation which does not require N-aryl motifs [33].
We sought a general procedure for organometallic trapping of iminium salts generated by oxidative functionalisation; a methodology amenable to a range of tertiary amine substrates and unstabilised carbon nucleophiles. Recently, visible-light photoredox catalysis has gained interest as a technique for oxidative functionalisation [34,35]. An important feature of photoredox catalysis is that different photocatalysts have different redox potentials upon accessing the excited state [18,34,36]. The ability to adjust oxidising power through photocatalyst choice renders the transformation substrate-tunable. Thus, we selected photoredox catalysis as an oxidative functionalisation whose substrate scope might be extended (by catalyst selection) in future investigations. Herein, we report a one-pot, tandem visible-light powered oxidative functionalisation of N-substituted THIQs and organometallic trapping of iminium intermediates.
Results and Discussion
Initially, a blue LED strip (λmax = 458 nm), Ru(bpy)3(PF6)2 (1 mol %) and BrCCl3 (3.0 equiv) facilitated oxidation of N-phenyl THIQ (4a) (1 mmol) to its corresponding iminium salt (5a, Table 1) in anhydrous MeCN. As observed by Stephenson [37], photoredox activation of 4a under these conditions required long reaction times (14–16 h) to reach full conversion. As reactions progressed, we observed precipitation and were able to collect half (by mass) of the crude iminium salt 5a by filtration. Precipitation acts to stall reactions by shielding the photocatalyst from the light. Addition of vinylmagnesium bromide directly to the reaction mixture led to a complex mixture of products by HPLC.
Table 1: Organometallic additions to iminium salts generated via visible-light photoredox catalysis.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-316-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | R-Metal | Y | R | Product | Yielda |
|---|---|---|---|---|---|
| 1 | RMgBrb | – | vinyl | 6aa | 80 |
| 2 | RMgBrb | – | Me | 6ab | 78 |
| 3 | RMgClb | – | Et | 6ac | 75 |
| 4 | RMgClb | – | iPr | 6ad | 78 |
| 5 | RMgBrb | – | cyclopropyl | 6ae | 66 |
| 6 | RMgBrb,c | – | Bn | 6af | 69 |
| 7 | RMgBrb | – | Ph | 6ag | 90 |
| 8 | RMgBrb | CuBrd | Ph | 6ag | 77 |
| 9 | RMgBrb | – | 4-FC6H4 | 6ah | 72 |
| 10 | RMgBrb | – | 4-MeOC6H4 | 6ai | 62 |
| 11e | RMgBrb | – | allyl | 6aj | – |
| 12f | RTMS | – | allyl | 6aj | – |
| 13 | RMgBrb | ZnCl2g | allyl | 6aj | 37, 88h |
| 14 | RI | Ini | allyl | 6aj | 92, 68j |
| 15e | RMgBrb | – | 2-methylallyl | 6ak | – |
| 16 | RMgBrb | ZnCl2g | 2-methylallyl | 6ak | 90 |
| 17 | RMgClb | ZnCl2g | 2-butenyl | 6alk | 92 |
aIsolated (%) yields after chromatography. bCommercially available solutions in THF or Et2O. c6 equiv used. dGrignard (2.0 equiv) premixed with CuBr (2.6 equiv). eComplex mixture. fNo reaction. gGrignard (2.0 equiv) premixed with a solution of ZnCl2 (2.6 equiv). hSolvent switched to THF. iAllyl iodide (3.0 equiv) premixed with In powder (2.0 equiv). jDirect addition of R-metal without solvent switch. k6al is a 1:1 mixture of diastereomers where R = 1-methyl-2-propenyl, see Supporting Information File 1.
We found that BrCCl3 and its byproduct CHCl3 [22] were responsible for poor organometallic reaction profiles. Generation of radical intermediates or carbenes upon reacting Grignard reagents or magnesium salts with BrCCl3 or CHCl3 are evidenced in the literature [38,39]. Initially, we took advantage of the volatilities of BrCCl3 and CHCl3 by removing them under vacuum and replacing the solvent. A solvent switch was also reported when photoredox activation of 4a was combined with thiourea-catalysed enantioselective alkylation [37]. The enantioselectivity of the thiourea-catalysed alkylation was optimal in non-polar solvents, yet low photocatalyst solubility in these solvents precluded photoactivation of 4a. Thus, a solvent switch was used to capitalise on the beneficial properties of both solvents.
At this stage, we employed an MeCN/H2O (4:1) solvent system and Ru(bpy)3Cl2 in photoactivations which facilitated full conversion of 4a to 5a in 2 h (Table 1). Here, MeCN/H2O (4:1) was chosen because MeCN forms an azeotrope with water [40] such that it could be easily dried by concentration. After dissolving resultant crude 5a in anhydrous MeCN and shielding from ambient light, alkyl, aryl and vinyl Grignard reagents added virtually instantaneously to 5a, affording 6aa–6ai in good to excellent (62–90%) yields (Table 1). In general, the enhanced electrophilicity of 5a compared to MeCN results in faster reaction of the Grignard with 5a despite the solvent (MeCN) being present in vast excess.
Notably, allyl and 2-methylallyl Grignard additions (Table 1, entries 11 and 15) were exceptions and resulted in complex mixtures of products. We reasoned that use of a less reactive organometallic reagent would supress undesirable pathways. However, allyltrimethylsilane does not react with 5a (Table 1, entry 12) [16,22]. As organometallics of intermediate reactivity, allylzinc halides were explored. Indeed, 2-methylallylzinc and 2-butenylzinc reagents added to 5a to afford 6ak and 6al in 90% and 92% isolated yields, respectively. Conversely, addition of the allylzinc reagent to 5a afforded side-products 7a and 8a in addition to 6aj in a 1:4:4 ratio by LC–MS, respectively (Figure 2).
![[1860-5397-10-316-2]](/bjoc/content/figures/1860-5397-10-316-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Products from allylzinc reagent addition to 5a and 5b.
Figure 2: Products from allylzinc reagent addition to 5a and 5b.
First, we sought to rule out the possibility of Ru(bpy)3Cl2 promoting these undesired pathways. Although Ru(bpy)3Cl2 could not be separated from 5a after photoactivation due to their similar polarities, we successfully separated the less polar iminium salt 5b. In absence of Ru(bpy)3Cl2, addition of the allylzinc reagent to 5b gave 6ba and side-products 7b and 9 (Figure 2), and we now explored the origin of these byproducts.
The possibility of 6aj or 6ba being intermediates in these side-reactions was ruled out when 6aj was exposed to the allylzinc reagent and no reaction was observed. When the allylzinc reagent was premixed (1:1) with MeCN before adding to 5a, 6aj was not observed. Instead, enone 8a was observed as the sole product. Conversely, when the allylzinc reagent was added to 5a, suspended in anhydrous THF, 8a was not observed. The ratio of 6aj:7a was 8:1 by LC–MS and an 88% yield of 6aj resulted.
We propose that allylzinc reagents are reactive enough to trap MeCN in competition with 5a. The allylzinc reagent adds to MeCN to form an imine salt that is transformed in situ into a conjugated dienamine intermediate (Figure 3). Vinylogous nucleophilic addition of the enamine to 5a generates 8a. This hypothesis is supported by the reaction of 5a with crotonaldehyde in the presence of a MacMillan-type imidazolidinone catalyst [41] and TFA which delivers 8b. Formation of cyclic products 8b and 9 is rationalised by intramolecular electrophilic aromatic substitution at the 2-position of the N-aryl moiety. (The isolation of enone 8a and the fact that 5a does not react with oct-1-ene under the same conditions rules out a Diels–Alder-type pathway to 8b.)
![[1860-5397-10-316-3]](/bjoc/content/figures/1860-5397-10-316-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Proposed mechanism for formation of side-product 8a. Analogous reactivity in the formation of cyclic product 8b under enamine catalysis. LC–MS (%) yields in parenthesis. Isolated yields, %, after chromatography not in parenthesis.
Figure 3: Proposed mechanism for formation of side-product 8a. Analogous reactivity in the formation of cycli...
Formation of side-products 7a and 7b can be rationalised by dimerisation of allylzinc halides as has been previously reported [42]. The authors describe generation of a bis-organozinc species which, upon addition to an electrophile, generates an intermediate which can undergo β-hydride delivery to a second electrophile. In this case addition to 5a generates an intermediate organozinc species which, following β-hydride delivery to a second iminium (5a), generates 7a and 4a (Figure 4). Heating the allylzinc halide to promote dimerisation [42], prior to addition to 5a in THF, altered the ratio of 6aj:7a from 8:1 to 3:2 by LC–MS. In further support of this mechanism, reduction of 5a to 4a was also observed (the ratio of 6aj:7a:4a = 5:3:1). However, our observations cannot rule out β-hydride addition as the first step. Consistent with our observations, the authors report that the more sterically hindered 2-methylallylzinc and 2-butenylzinc halides do not dimerise even after 48 h under reflux [42].
![[1860-5397-10-316-4]](/bjoc/content/figures/1860-5397-10-316-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Mechanism for dimerisation of the allylzinc halide and β-hydride addition to 5a [36].
Figure 4: Mechanism for dimerisation of the allylzinc halide and β-hydride addition to 5a [36].
To avoid the pathways outlined in Figure 3 and Figure 4, we sought a less reactive allyl organometallic than the allylzinc reagent. Notably, allylindium reagents have attracted attention for their tolerance to water [43]. Such reagents have mediated reactions where allyl Grignards and allylzinc reagents have failed [44]. Gratifyingly, an allylindium reagent [45] appeared inert to pathways available to the allyl Grignard and allylzinc reagent, affording a 92% yield of 6aj. Strikingly, the same reagent was added without a solvent switch and tolerated BrCCl3, CHCl3 and water, affording 6aj albeit in lower yield (68%).
Murthy and Blechert and their respective co-workers reported allylation of THIQs under aerobic conditions using allyltrialkylstannanes [12,16] (Blechert’s studies also included success with allylboron reagents). Whilst our reactions are carried out under N2, the indium metal used, allylindium reagents generated and indium trihalide salt byproducts are non-toxic [43]. Our conditions benefit from the absence of amide side-products typically effected by peroxide intermediates in aerobic photoactivation of THIQs [16,22] and so our methodology serves to complement existing strategies in the literature.
The substrate scope of our methodology is outlined in Table 2. Iminium salts derived from a range of electronically diverse N-aryl THIQs (4b and 11a–14b) were trapped with PhMgBr to afford products (6bb and 11b–14b in fair to excellent (47–95%) yields. Substrates with both electron-rich (12a) and electron-poor (13a,14a) N-aryl substituents were tolerated. Although Ru(bpy)3Cl2 was ineffective at catalysing oxidation of N-Boc protected THIQ 17a, we are pleased to report the first examples of Ru(bpy)3Cl2 catalysed oxidative functionalisation of N-alkyl THIQs. Subjecting 15a and 16a to photoactivation for 16 h furnished in both cases their corresponding benzylic endo-iminium salts, which were trapped by PhMgBr to afford 15b and 16b in 58% and 81% yield, respectively.
Table 2: Substrate scope of organometallic additions to iminium salts generated via visible-light photoredox catalysis.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-316-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | R1/R2 | Substrate | Product | Yielda |
|---|---|---|---|---|
| 1b,c | H/Ph | 4a | 6ag | 90 |
| 2d | H/2-Naphthyl | 4b | 6bb | 47 |
| 3c | OMe/Ph | 11a | 11b | 95 |
| 4c | H/4-MeOC6H4 | 12a | 12b | 52 |
| 5c | H/4-BrC6H4 | 13a | 13b | 53 |
| 6c | H/4-NO2C6H4 | 14a | 14b | 77 |
| 7e | H/Me | 15a | 15b | 58 |
| 8e | OMe/Me | 16a | 16b | 81 |
| 9f | H/CO2t-Bu | 17a | 17b | – |
aIsolated (%) yields after chromatography. bEntry 7, Table 1 given for comparison. cPhotoactivation time of 2 h. dPhotoactivation time of 4 h. ePhotoactivation time of 16 h. fNo reaction/photoactivation observed.
Encouraged by these results, we decided to apply our methodology to THIQ 16a using Grignard 18 in a synthesis of methopholine (3, Scheme 2). Previous syntheses of 3 involving oxidative functionalisation of 16a have used (4-chlorophenyl)acetylene as a pronucleophile [13,46]. However, isolation and hydrogenation of the resulting THIQ intermediate are required to access 3. Photoactivation of 16a and trapping of the resultant benzylic endo-iminium salt with 18 resulted in a 56% yield of 3. To our knowledge, this concise synthesis of 3 is higher yielding (based on THIQ 16a) than previously reported syntheses [13,46,47] with no intermediate isolations required.
![[1860-5397-10-316-i2]](/bjoc/content/inline/1860-5397-10-316-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: A concise synthesis of methopholine (3).
Scheme 2: A concise synthesis of methopholine (3).
Revisiting the concept of direct organometallic addition after photoactivation (thus far precluded by the use of BrCCl3), compatibility might be accomplished in two ways. Firstly, moderate the reactivity of the organometallic to tolerate BrCCl3 or secondly, find alternative oxidants which tolerate the organometallic. Whilst the former looked promising with the allylindium example, the latter approach was thought to be more general in terms of increasing nucleophile scope.
Stephenson reported diethyl bromomalonate as an effective oxidant to regenerate Ru(II) [22]. General application of this alkyl halide oxidant was ruled out due to potential side-reactions of the malonyl radical and diethyl malonate. We explored alternative alkyl halide oxidants that would form inert byproducts. No reaction was observed when substituting BrCCl3 with ClCH2CN (−0.72 V vs SCE [48]) but to our delight, BrCH2CN (−0.60 V vs SCE [48]) resulted in near-quantitative (90%) conversion of 4a to 5a in 3 h (anhydrous conditions). According to the mechanism proposed by Stephenson for BrCCl3 [22], BrCH2CN forms MeCN as an inert product. Grignard additions were unaffected by traces of residual BrCH2CN and a selection of substrates (4a, 11a–13a) and Grignard reagents were employed, affording the products (6aa, 6ab, 6ag, 11b–13b) in encouraging (50–77%) yields (Table 3).
Table 3: Direct one-pot organometallic additions to iminium salts generated via visible-light photoredox catalysis.
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-316-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | R1/R2/R3 | Substrate | Product | Yielda |
|---|---|---|---|---|
| 1b | H/Ph/vinyl | 4a | 6aa | 77 |
| 2b | H/Ph/Me | 4a | 6ab | 73 |
| 3b | H/Ph/Ph | 4a | 6ag | 61 |
| 4b | OMe/Ph/Ph | 11a | 11b | 72 |
| 5b | H/4-MeOC6H4/Ph | 12a | 12b | 73 |
| 6c,d | H/4-BrC6H4/Ph | 13a | 13b | 50 |
aIsolated (%) yields after chromatography. bPhotoactivation time of 3 h. cPhotoactivation time of 5 h. dHeating required to solubilise substrate.
Conclusion
We have developed a user-friendly, one-pot methodology which combines visible-light photoredox catalysis and organometallic addition to deliver 1,2-disubstituted THIQs. It is rapid, performed under practical conditions and custom-made or commercially available organometallic solutions can be used directly. Highly reactive carbon nucleophiles (for example, allyl) have been harnessed by varying the organometallic species. Overall, this methodology is synthetically valuable for two reasons. Firstly, a virtually limitless host of carbon nucleophiles may be employed via organometallic chemistry (compounds 6aa–6af are novel compounds derived from unstabilised carbon nucleophiles). Secondly, photoredox catalysis can be substrate tailored through photocatalyst selection. Having demonstrated the former reason herein, investigation of the latter is underway to extend the substrate scope beyond benzylic tertiary amines.
Supporting Information
| Supporting Information File 1: Experimental procedures, 1H and 13C spectra of all novel compounds and HPLC/LC–MS data from which conclusions were drawn. | ||
| Format: PDF | Size: 2.9 MB | Download |
References
-
Antkiewicz-Michaluk, L.; Wąsik, A.; Michaluk, J. Neurotoxic. Res. 2014, 25, 1–12. doi:10.1007/s12640-013-9402-7
Return to citation in text: [1] -
Scott, J. D.; Williams, R. M. Chem. Rev. 2002, 102, 1669–1730. doi:10.1021/cr010212u
Return to citation in text: [1] -
Bembenek, M. E.; Abell, C. W.; Chrisey, L. A.; Rozwadowska, M. D.; Gessner, W.; Brossi, A. J. Med. Chem. 1990, 33, 147–152. doi:10.1021/jm00163a025
Return to citation in text: [1] -
Naito, R.; Yonetoku, Y.; Okamoto, Y.; Toyoshima, A.; Ikeda, K.; Takeuchi, M. J. Med. Chem. 2005, 48, 6597–6606. doi:10.1021/jm050099q
Return to citation in text: [1] -
Wanner, K. T.; Praschak, I.; Höfner, G.; Beer, H. Arch. Pharm. 1996, 329, 11–22. doi:10.1002/ardp.19963290104
Return to citation in text: [1] -
Cass, L. J.; Frederik, W. S. Am. J. Med. Sci. 1963, 246, 550–557. doi:10.1097/00000441-196311000-00005
Return to citation in text: [1] -
Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t
Return to citation in text: [1] -
Ruiz Espelt, L.; Wiensch, E. M.; Yoon, T. P. J. Org. Chem. 2013, 78, 4107–4114. doi:10.1021/jo400428m
Return to citation in text: [1] -
Prier, C. K.; MacMillan, D. W. C. Chem. Sci. 2014, 5, 4173–4178. doi:10.1039/C4SC02155J
Return to citation in text: [1] -
Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 6968–6969. doi:10.1021/ja0516054
Return to citation in text: [1] -
Boess, E.; Schmitz, C.; Klussmann, M. J. Am. Chem. Soc. 2012, 134, 5317–5325. doi:10.1021/ja211697s
Return to citation in text: [1] -
Kumaraswamy, G.; Murthy, A. N.; Pitchaiah, A. J. Org. Chem. 2010, 75, 3916–3919. doi:10.1021/jo1005813
Return to citation in text: [1] [2] -
Jones, K. M.; Karier, P.; Klussmann, M. ChemCatChem 2012, 4, 51–54. doi:10.1002/cctc.201100324
Return to citation in text: [1] [2] [3] -
Dhineshkumar, J.; Lamani, M.; Alagiri, K.; Prabhu, K. R. Org. Lett. 2013, 15, 1092–1095. doi:10.1021/ol4001153
Return to citation in text: [1] [2] -
Nobuta, T.; Tada, N.; Fujiya, A.; Kariya, A.; Miura, T.; Itoh, A. Org. Lett. 2013, 15, 574–577. doi:10.1021/ol303389t
Return to citation in text: [1] -
Möhlmann, L.; Blechert, S. Adv. Synth. Catal. 2014, 356, 2825–2829. doi:10.1002/adsc.201400551
Return to citation in text: [1] [2] [3] [4] [5] -
Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y
Return to citation in text: [1] -
Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x
Return to citation in text: [1] [2] -
Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852–3855. doi:10.1021/ol201376v
Return to citation in text: [1] -
Rueping, M.; Vila, C.; Bootwicha, T. ACS Catal. 2013, 3, 1676–1680. doi:10.1021/cs400350j
Return to citation in text: [1] -
Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234
Return to citation in text: [1] -
Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Chen, C.; Xia, W. Chem. Commun. 2012, 48, 2337–2339. doi:10.1039/c2cc17130a
Return to citation in text: [1] -
Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f
Return to citation in text: [1] -
Baslé, O.; Li, C.-J. Org. Lett. 2008, 10, 3661–3663. doi:10.1021/ol8012588
Return to citation in text: [1] -
Muramatsu, W.; Nakano, K.; Li, C.-J. Org. Lett. 2013, 15, 3650–3653. doi:10.1021/ol401534g
Return to citation in text: [1] -
Singh, K. N.; Kessar, S. V.; Singh, P.; Singh, P.; Kaur, M.; Batra, A. Synthesis 2014, 46, 2644–2650. doi:10.1055/s-0034-1378337
Return to citation in text: [1] -
Li, Z.; Li, C.-J. Org. Lett. 2004, 6, 4997–4999. doi:10.1021/ol047814v
Return to citation in text: [1] -
Fu, W.; Guo, W.; Zou, G.; Xu, C. J. Fluorine Chem. 2012, 140, 88–94. doi:10.1016/j.jfluchem.2012.05.009
Return to citation in text: [1] -
Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050
Return to citation in text: [1] -
There are two reports containing examples of N-alkyl THIQ oxidative functionalisation [27,30]; aryl [27] and alkynyl [30] organometallic nucleophiles are reported.
Return to citation in text: [1] -
Muramatsu, W.; Nakano, K.; Li, C.-J. Org. Biomol. Chem. 2014, 12, 2189–2192. doi:10.1039/c3ob42354a
Return to citation in text: [1] -
Zheng, Q.-H.; Meng, W.; Jiang, G.-J.; Yu, Z.-X. Org. Lett. 2013, 15, 5928–5931. doi:10.1021/ol402517e
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] [2] -
Douglas, J. J.; Nguyen, J. D.; Cole, K. P.; Stephenson, C. R. J. Aldrichimica Acta 2014, 47, 1–25.
Return to citation in text: [1] -
Ravelli, D.; Fagnoni, M.; Albini, A. Chem. Soc. Rev. 2013, 42, 97–113. doi:10.1039/c2cs35250h
Return to citation in text: [1] [2] -
Bergonzini, G.; Schindler, C. S.; Wallentin, C.-J.; Jacobsen, E. N.; Stephenson, C. R. J. Chem. Sci. 2014, 5, 112–116. doi:10.1039/c3sc52265b
Return to citation in text: [1] [2] -
Kharasch, M. S.; Reinmuth, O.; Urry, W. H. J. Am. Chem. Soc. 1947, 69, 1105–1110. doi:10.1021/ja01197a036
Return to citation in text: [1] -
Davis, M.; Deady, L. W.; Finch, A. J.; Smith, J. F. Tetrahedron 1973, 29, 349–352. doi:10.1016/S0040-4020(01)93300-9
Return to citation in text: [1] -
Horsley, L. H. Anal. Chem. 1947, 19, 508–600. doi:10.1021/ac60008a002
Return to citation in text: [1] -
Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 7894–7895. doi:10.1021/ja025981p
Return to citation in text: [1] -
Courtois, G.; Miginiac, L. J. Organomet. Chem. 1973, 52, 241–259. doi:10.1016/S0022-328X(00)95144-1
Return to citation in text: [1] [2] [3] -
Shen, Z.-L.; Wang, S.-Y.; Chok, Y.-K.; Xu, Y.-H.; Loh, T.-P. Chem. Rev. 2013, 113, 271–401. doi:10.1021/cr300051y
Return to citation in text: [1] [2] -
Lee, K.; Lee, P. H. Org. Lett. 2008, 10, 2441–2444. doi:10.1021/ol800719g
Return to citation in text: [1] -
Araki, S.; Shimizu, T.; Johar, P. S.; Jin, S. J.; Butsugan, Y. J. Org. Chem. 1991, 56, 2538–2542. doi:10.1021/jo00007a050
Return to citation in text: [1] -
Singh, K. N.; Singh, P.; Kaur, A.; Singh, P. Synlett 2012, 23, 760–764. doi:10.1055/s-0031-1290532
Return to citation in text: [1] [2] -
Richter, H.; Fröhlich, R.; Daniliuc, C.-G.; García Mancheño, O. Angew. Chem., Int. Ed. 2012, 51, 8656–8660. doi:10.1002/anie.201202379
Return to citation in text: [1] -
Isse, A. A.; Lin, C. Y.; Coote, M. L.; Gennaro, A. J. Phys. Chem. B 2011, 115, 678–684. doi:10.1021/jp109613t
Return to citation in text: [1] [2]
| 41. | Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 7894–7895. doi:10.1021/ja025981p |
| 42. | Courtois, G.; Miginiac, L. J. Organomet. Chem. 1973, 52, 241–259. doi:10.1016/S0022-328X(00)95144-1 |
| 42. | Courtois, G.; Miginiac, L. J. Organomet. Chem. 1973, 52, 241–259. doi:10.1016/S0022-328X(00)95144-1 |
| 1. | Antkiewicz-Michaluk, L.; Wąsik, A.; Michaluk, J. Neurotoxic. Res. 2014, 25, 1–12. doi:10.1007/s12640-013-9402-7 |
| 2. | Scott, J. D.; Williams, R. M. Chem. Rev. 2002, 102, 1669–1730. doi:10.1021/cr010212u |
| 7. | Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t |
| 8. | Ruiz Espelt, L.; Wiensch, E. M.; Yoon, T. P. J. Org. Chem. 2013, 78, 4107–4114. doi:10.1021/jo400428m |
| 9. | Prier, C. K.; MacMillan, D. W. C. Chem. Sci. 2014, 5, 4173–4178. doi:10.1039/C4SC02155J |
| 25. | Baslé, O.; Li, C.-J. Org. Lett. 2008, 10, 3661–3663. doi:10.1021/ol8012588 |
| 26. | Muramatsu, W.; Nakano, K.; Li, C.-J. Org. Lett. 2013, 15, 3650–3653. doi:10.1021/ol401534g |
| 27. | Singh, K. N.; Kessar, S. V.; Singh, P.; Singh, P.; Kaur, M.; Batra, A. Synthesis 2014, 46, 2644–2650. doi:10.1055/s-0034-1378337 |
| 43. | Shen, Z.-L.; Wang, S.-Y.; Chok, Y.-K.; Xu, Y.-H.; Loh, T.-P. Chem. Rev. 2013, 113, 271–401. doi:10.1021/cr300051y |
| 5. | Wanner, K. T.; Praschak, I.; Höfner, G.; Beer, H. Arch. Pharm. 1996, 329, 11–22. doi:10.1002/ardp.19963290104 |
| 6. | Cass, L. J.; Frederik, W. S. Am. J. Med. Sci. 1963, 246, 550–557. doi:10.1097/00000441-196311000-00005 |
| 22. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 28. | Li, Z.; Li, C.-J. Org. Lett. 2004, 6, 4997–4999. doi:10.1021/ol047814v |
| 29. | Fu, W.; Guo, W.; Zou, G.; Xu, C. J. Fluorine Chem. 2012, 140, 88–94. doi:10.1016/j.jfluchem.2012.05.009 |
| 30. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 16. | Möhlmann, L.; Blechert, S. Adv. Synth. Catal. 2014, 356, 2825–2829. doi:10.1002/adsc.201400551 |
| 22. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 4. | Naito, R.; Yonetoku, Y.; Okamoto, Y.; Toyoshima, A.; Ikeda, K.; Takeuchi, M. J. Med. Chem. 2005, 48, 6597–6606. doi:10.1021/jm050099q |
| 16. | Möhlmann, L.; Blechert, S. Adv. Synth. Catal. 2014, 356, 2825–2829. doi:10.1002/adsc.201400551 |
| 21. | Hu, J.; Wang, J.; Nguyen, T. H.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. doi:10.3762/bjoc.9.234 |
| 45. | Araki, S.; Shimizu, T.; Johar, P. S.; Jin, S. J.; Butsugan, Y. J. Org. Chem. 1991, 56, 2538–2542. doi:10.1021/jo00007a050 |
| 3. | Bembenek, M. E.; Abell, C. W.; Chrisey, L. A.; Rozwadowska, M. D.; Gessner, W.; Brossi, A. J. Med. Chem. 1990, 33, 147–152. doi:10.1021/jm00163a025 |
| 14. | Dhineshkumar, J.; Lamani, M.; Alagiri, K.; Prabhu, K. R. Org. Lett. 2013, 15, 1092–1095. doi:10.1021/ol4001153 |
| 22. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 23. | Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Chen, C.; Xia, W. Chem. Commun. 2012, 48, 2337–2339. doi:10.1039/c2cc17130a |
| 24. | Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f |
| 12. | Kumaraswamy, G.; Murthy, A. N.; Pitchaiah, A. J. Org. Chem. 2010, 75, 3916–3919. doi:10.1021/jo1005813 |
| 16. | Möhlmann, L.; Blechert, S. Adv. Synth. Catal. 2014, 356, 2825–2829. doi:10.1002/adsc.201400551 |
| 14. | Dhineshkumar, J.; Lamani, M.; Alagiri, K.; Prabhu, K. R. Org. Lett. 2013, 15, 1092–1095. doi:10.1021/ol4001153 |
| 15. | Nobuta, T.; Tada, N.; Fujiya, A.; Kariya, A.; Miura, T.; Itoh, A. Org. Lett. 2013, 15, 574–577. doi:10.1021/ol303389t |
| 17. | Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y |
| 18. | Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x |
| 43. | Shen, Z.-L.; Wang, S.-Y.; Chok, Y.-K.; Xu, Y.-H.; Loh, T.-P. Chem. Rev. 2013, 113, 271–401. doi:10.1021/cr300051y |
| 13. | Jones, K. M.; Karier, P.; Klussmann, M. ChemCatChem 2012, 4, 51–54. doi:10.1002/cctc.201100324 |
| 19. | Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852–3855. doi:10.1021/ol201376v |
| 20. | Rueping, M.; Vila, C.; Bootwicha, T. ACS Catal. 2013, 3, 1676–1680. doi:10.1021/cs400350j |
| 12. | Kumaraswamy, G.; Murthy, A. N.; Pitchaiah, A. J. Org. Chem. 2010, 75, 3916–3919. doi:10.1021/jo1005813 |
| 42. | Courtois, G.; Miginiac, L. J. Organomet. Chem. 1973, 52, 241–259. doi:10.1016/S0022-328X(00)95144-1 |
| 10. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 6968–6969. doi:10.1021/ja0516054 |
| 11. | Boess, E.; Schmitz, C.; Klussmann, M. J. Am. Chem. Soc. 2012, 134, 5317–5325. doi:10.1021/ja211697s |
| 16. | Möhlmann, L.; Blechert, S. Adv. Synth. Catal. 2014, 356, 2825–2829. doi:10.1002/adsc.201400551 |
| 36. | Ravelli, D.; Fagnoni, M.; Albini, A. Chem. Soc. Rev. 2013, 42, 97–113. doi:10.1039/c2cs35250h |
| 33. | Zheng, Q.-H.; Meng, W.; Jiang, G.-J.; Yu, Z.-X. Org. Lett. 2013, 15, 5928–5931. doi:10.1021/ol402517e |
| 31. | There are two reports containing examples of N-alkyl THIQ oxidative functionalisation [27,30]; aryl [27] and alkynyl [30] organometallic nucleophiles are reported. |
| 13. | Jones, K. M.; Karier, P.; Klussmann, M. ChemCatChem 2012, 4, 51–54. doi:10.1002/cctc.201100324 |
| 46. | Singh, K. N.; Singh, P.; Kaur, A.; Singh, P. Synlett 2012, 23, 760–764. doi:10.1055/s-0031-1290532 |
| 32. | Muramatsu, W.; Nakano, K.; Li, C.-J. Org. Biomol. Chem. 2014, 12, 2189–2192. doi:10.1039/c3ob42354a |
| 13. | Jones, K. M.; Karier, P.; Klussmann, M. ChemCatChem 2012, 4, 51–54. doi:10.1002/cctc.201100324 |
| 46. | Singh, K. N.; Singh, P.; Kaur, A.; Singh, P. Synlett 2012, 23, 760–764. doi:10.1055/s-0031-1290532 |
| 47. | Richter, H.; Fröhlich, R.; Daniliuc, C.-G.; García Mancheño, O. Angew. Chem., Int. Ed. 2012, 51, 8656–8660. doi:10.1002/anie.201202379 |
| 22. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 16. | Möhlmann, L.; Blechert, S. Adv. Synth. Catal. 2014, 356, 2825–2829. doi:10.1002/adsc.201400551 |
| 22. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 38. | Kharasch, M. S.; Reinmuth, O.; Urry, W. H. J. Am. Chem. Soc. 1947, 69, 1105–1110. doi:10.1021/ja01197a036 |
| 39. | Davis, M.; Deady, L. W.; Finch, A. J.; Smith, J. F. Tetrahedron 1973, 29, 349–352. doi:10.1016/S0040-4020(01)93300-9 |
| 27. | Singh, K. N.; Kessar, S. V.; Singh, P.; Singh, P.; Kaur, M.; Batra, A. Synthesis 2014, 46, 2644–2650. doi:10.1055/s-0034-1378337 |
| 37. | Bergonzini, G.; Schindler, C. S.; Wallentin, C.-J.; Jacobsen, E. N.; Stephenson, C. R. J. Chem. Sci. 2014, 5, 112–116. doi:10.1039/c3sc52265b |
| 30. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 37. | Bergonzini, G.; Schindler, C. S.; Wallentin, C.-J.; Jacobsen, E. N.; Stephenson, C. R. J. Chem. Sci. 2014, 5, 112–116. doi:10.1039/c3sc52265b |
| 22. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 22. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 27. | Singh, K. N.; Kessar, S. V.; Singh, P.; Singh, P.; Kaur, M.; Batra, A. Synthesis 2014, 46, 2644–2650. doi:10.1055/s-0034-1378337 |
| 30. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 34. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 35. | Douglas, J. J.; Nguyen, J. D.; Cole, K. P.; Stephenson, C. R. J. Aldrichimica Acta 2014, 47, 1–25. |
| 48. | Isse, A. A.; Lin, C. Y.; Coote, M. L.; Gennaro, A. J. Phys. Chem. B 2011, 115, 678–684. doi:10.1021/jp109613t |
| 18. | Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x |
| 34. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 36. | Ravelli, D.; Fagnoni, M.; Albini, A. Chem. Soc. Rev. 2013, 42, 97–113. doi:10.1039/c2cs35250h |
| 48. | Isse, A. A.; Lin, C. Y.; Coote, M. L.; Gennaro, A. J. Phys. Chem. B 2011, 115, 678–684. doi:10.1021/jp109613t |
© 2014 Barham et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)