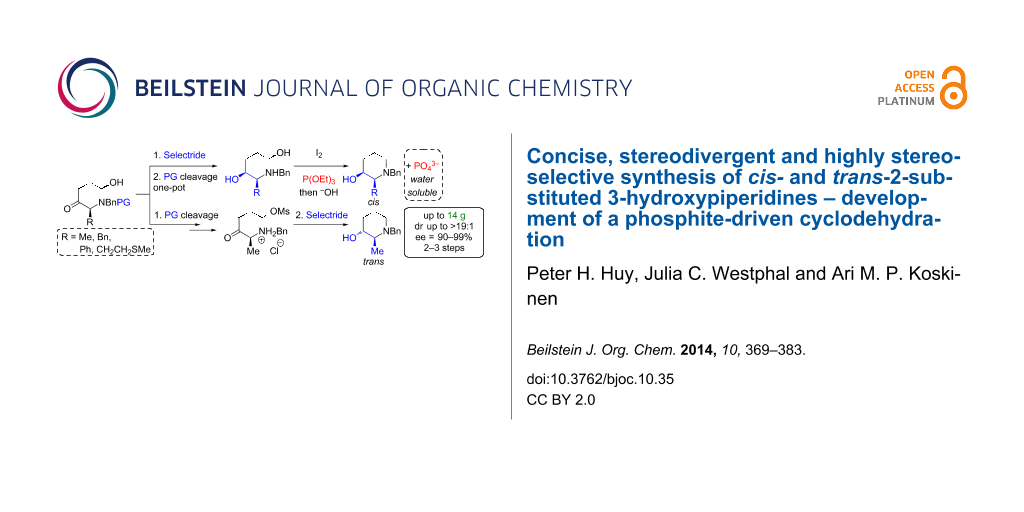
Abstract
A concise (5 to 6 steps), stereodivergent, highly diastereoselective (dr up to >19:1 for both stereoisomers) and scalable synthesis (up to 14 g) of cis- and trans-2-substituted 3-piperidinols, a core motif in numerous bioactive compounds, is presented. This sequence allowed an efficient synthesis of the NK-1 inhibitor L-733,060 in 8 steps. Additionally, a cyclodehydration-realizing simple triethylphosphite as a substitute for triphenylphosphine is developed. Here the stoichiometric oxidized P(V)-byproduct (triethylphosphate) is easily removed during the work up through saponification overcoming separation difficulties usually associated to triphenylphosphine oxide.

Graphical Abstract
Introduction
1,2-Amino alcohols of the type A (Figure 1) represent a frequent core motif of many pharmacologically active natural products [1-9], chiral auxiliaries [10,11] and catalysts for asymmetric synthesis [12-14]. Especially the 2-substituted 3-hydroxypiperidine scaffold of the general structure B (as one type of an 1,2-amino alcohol) can be found in numerous natural products and other bioactive compounds [1-7]. Selected examples are given in Figure 1: The non-peptidic human neurokinin-1 (NK1) substance P receptor antagonists L-733,060 [15,16] and CP-99,994 [17-19], the natural product febrifugine (antimalarial) [20,21] and antiprotozoal agent halofuginone (commercial trade names Halocur® (lactate salt) and Stenorol® (hydrobromide salt)) [22]. Other relevant examples are 3-hydroxypipecolic acids, which serve as (conformationally restricted) substitutes of proline and serine [23,24] and have been incorporated into diverse bioactive peptidomimetics [25,26], and the iminosugar swainsonine, a new potential chemotherapeutic agent [27,28]. Recently, analogs of halofuginone were discovered as inhibitors of tRNA synthetases [29-31].
![[1860-5397-10-35-1]](/bjoc/content/figures/1860-5397-10-35-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Natural products and other bioactive piperidine derivatives of type B.
Figure 1: Natural products and other bioactive piperidine derivatives of type B.
The majority of the reported syntheses [2-4,32-35] are elaborate (far more than 10 steps), specific on one of the targets depicted in Figure 1 and therefore on one relative configuration (either cis- or trans), and have not been proven to be scalable. In our opinion the following examples represent the most efficient synthesis of 3-piperidinols of type B in terms of step-economy (<10 steps to establish the core motif B): 1. Charette [36] prepared the less bioactive enantiomers of L-733,060 and CP-99,994 through nucleophilic additions to chiral pyridylium salts and subsequent hydrogenation. 2. Cossy and Pardo [37,38] synthesized L-733,060 (formal total synthesis) and two (epimeric) hydroxypipecolic acid through ring expansions of 2-(α-hydroxyalkyl)pyrrolidines deduced to proline. 3. Based on the ring expansion of Cossy and Pardo [37], O´Brien [39] realized the synthesis of L-733,060 (80% ee). The 2-(α-hydroxyalkyl)pyrrolidine precursor was prepared from a protected pyrrolidine through sparteine-mediated enantioselective lithiation and subsequent hydroxyalkylation. 4. Recently, Pansare [40] reported the synthesis of L-733,060, CP-99,994 and a hydroxypipecolic acid through asymmetric organocatalytic vinylogous aldol addition as the key step.
While the syntheses by Charette [38] and Pansare [40] are restricted to piperidinols B in cis-configuration (dr >19:1 and 8:1, respectively), the sequences of Cossy and Pardo [35] and O´Brien [39] are stereodivergent. Nevertheless, the observed diastereomeric ratios are low (1:1 and 2.3:1 for trans-B, respectively) at least for one of the epimers. Considering the versatile pharmacological activities of compounds based on the 3-piperidinol scaffold, a step-economic, scalable and stereodivergent synthesis of both cis- and trans-diastereomers of B in good diastereoselectivities is highly desirable.
In the syntheses of potentially new drug candidates scalability is a significant factor to provide sufficient substance amounts for clinical tests [41,42]. Additionally, alternatives in reactions driven by the formation of phosphine oxides from phosphines (e.g. the Appel and Mitsunobu reaction) are highly desired to improve atom economy (reduced waste amounts) and to circumvent difficulties in the separation of these by-products as demonstrated by a number of reviews [43-45]. Numerous protocols have been developed to improve these issues, mostly based on polymer supported or otherwise modified (more complex) phosphines [43-45]. Surprisingly, in this context simple and inexpensive phosphites (P(OR)3) have only been applied as phosphine substitutes in one single example: Beal [46] utilized tri-isopropylphosphite in a Mitsunobu condensation of a guanine-derived nucleoside analog with benzylic alcohols providing simplified byproduct separation through improved water solubility (of O=P(OiPr)3). In our case we were not able to remove stoichiometric amounts of OP(OEt)3 (which is more hydrophilic than OP(OiPr)3) through an aqueous work up (without saponification). Moreover, pentavalent P(OEt)5 prepared from P(OEt)3 with diethylperoxide and ethylbenzenesulfonate, respectively, in an additional step, was reported to effect cyclodehydration of diols to furans and pyrans [47,48] (for recent examples for cyclodehydration protocols see [49,50]). Thereby, the volatile products were separated from O=P(OEt)3 through distillation.
After our initial short communication [51] about the step-economic and stereodivergent synthesis of trans- and cis-2-substituted 3-piperidinols B, we want to report the development of this sequence in more detail with a focus on the phosphite-mediated cyclodehydration. Additionally, the synthesis of a side chain functionalized piperidinol derived from methionine and studies towards the preparation of glutamic and aspartic acid derived heterocycles are presented.
Following the retrosynthetic analysis in Figure 2 the relative configuration of B (cis/trans) should be controlled through targeted protecting group (PG) manipulation: Reduction of the common precursor ketone D (derived from commercial available amino acids) should deliver the syn-amino alcohol C proceeding though a Felkin–Anh transition state [52,53] (due to the sterically demanding –NBnPG function). Further PG cleavage and cyclodehydration would give rise to cis-B. On the other hand, initial deprotection of D (to liberate the Lewis-basic –NHBn moiety) and subsequent reduction passing through a Cram-chelate transition state [54] should deliver the anti-amino alcohol C.
![[1860-5397-10-35-2]](/bjoc/content/figures/1860-5397-10-35-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis of piperidines B (X = OH or leaving group, PG = protecting group).
Figure 2: Retrosynthetic analysis of piperidines B (X = OH or leaving group, PG = protecting group).
After subsequent cyclisation trans-B would result. Noteworthy, this strategy would completely circumvent configurationally labile amino aldehyde intermediates [55,56]. Basically any carbamate or amide protecting group (= PG) orthogonal to the benzyl moiety would be suitable for this strategy. Furthermore, we decided to surrender protection of the OH functions in the synthesis of intermediates D and C in order to minimize the number of steps of the sequence. Thus far only four examples following a related strategy to establish the syn- and anti-configuration of 1,2-aminoalcohol motifs have been reported [57-60].
Results and Discussion
Synthesis of hydroxyketone intermediates D
In the first step L-alanine, phenylalanine, phenylglycine and methionine 1a–d were converted to their N-benzyl-N-carbamate-protected derivatives 2a–d (PG = Cbz, Boc) in a practical one pot procedure through combination of Quitt´s reductive benzylation protocol [61] and a Schotten–Baumann acylation [62,63] in 70–95% yield (Scheme 1). While we choose a Cbz protecting group for the amino acids 1a–c due to the mild cleavage conditions (hydrogenolysis), we decided to introduce a Boc group at the N-terminus of methionine 1d to avoid desulfuration (–EtSMe → –Et) in the later deprotection.
![[1860-5397-10-35-i1]](/bjoc/content/inline/1860-5397-10-35-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the protected amino acids 2. (a) KOH for 1b. b) PG–X = Cbz–Cl (1a–c), Boc2O (1d).
Scheme 1: Synthesis of the protected amino acids 2. (a) KOH for 1b. b) PG–X = Cbz–Cl (1a–c), Boc2O (1d).
In order to suppress the formation of the only carbamate protected amino acid derivatives 3 (remaining as impurity in the isolated products 2), quantitative benzylation (1→I) was ensured by successive addition of three portions of benzaldehyde/NaBH4 (Quitt´s procedure [61] → two portions) and by maintaining the pH at a value of 10–11. The extractive separation (washing of a basic aqueous solution of the acids 2) of the two equivalents of BnOH formed during the reductive amination (1→I) proved to be challenging: Due to the high amphiphilicity of carboxylate salts of 2, mixtures in water and an organic solvent tend to form three distinct phases separating poorly. Nevertheless, washing of an aqueous solution of the polar lithium salts of 2 (crude 2 in aq. LiOH) with organic solvents of increasing polarity (Et2O→EtOAc) allowed to remove BnOH almost completely (<10% of BnOH referred to 2). Residual benzyl alcohol diminished the yield in the following amidation (2→5) due to competitive formation of the corresponding benzyl esters. Depending on the NaBH4 batch the dibenzyl-protected derivatives 4 formed as side products (<10% referred to 2). The residual impurities (BnOH and 4) were separated either through work up (of amide 5a) or chromatographic purification (of amides 5b–d) after the following amidation (2→5).
Importantly, through this procedure not only one isolation step was avoided, but also the overall yield was improved significantly: For the transformation from 1a to 2a (I isolated as free acid) under (optimized) literature conditions we achieved a yield of 44% over two steps (59% and 74%, respectively) compared to 70% according to our direct conversion from 1a to 2a.
Next the carboxylic acids 2 were transformed to the Weinreb amides 5 with DCC and MeONHMe in 77–91% yield (Scheme 2). While no racemisation occurred with substrates 2a, 2b and 2d (Et3N/DMAP or NMM as bases; Table 1, entries 1 and 2), the protected phenylglycine derivative 2c showed high configurational lability: Under standard conditions (DCC/Et3N/0.3 equiv DMAP) the amide 5c was obtained in a diminished ee of 49% (Table 1, entry 3). HOBt as nucleophilic catalyst proved to be even worse than DMAP, because under otherwise identical conditions the product 5c was isolated almost as a racemate (5% ee, entry 4). By replacing Et3N with the less basic NMM (in the presence of DMAP) the ee increased clearly (49→75%, entry 5). However, without any nucleophilic catalyst the desired amide 5c was isolated with a very good ee of 95% (entry 6) [64].
![[1860-5397-10-35-i2]](/bjoc/content/inline/1860-5397-10-35-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of hydroxy ketones 7 (R = Me (a), Bn (b), Ph (c) and EtSMe (d); PG = Cbz (a–c), Boc (d)).
Scheme 2: Synthesis of hydroxy ketones 7 (R = Me (a), Bn (b), Ph (c) and EtSMe (d); PG = Cbz (a–c), Boc (d)).
Table 1: Ees and isolated yields of Weinreb amides 5.
| entry | substrate | R | reagents | eea | yield |
|---|---|---|---|---|---|
| 1 | 2a | –Me | DCC, NMM | 99% | 91%b |
| 2 | 2b | –Bn | DCC, Et3N, DMAP (cat.)d | 99% | 77%c |
| 3 | 2c | DCC, Et3N, DMAP (cat.)d | 49% | n.d. | |
| 4 | DCC, Et3N, HOBt (cat.)d | 5% | n.d. | ||
| 5 | –Ph | DCC, NMM, DMAP (cat.)d | 75% | n.d. | |
| 6 | DCC, NMM | 95% | 81%b | ||
| 7 | T3P, pyridinee | 74% | n.d. | ||
| 8 | 2d | –EtSMe | DCC, NMM | 99%f | 91%b |
aThe ee was determined via HPLC on a chiral stationary phase. bIsolated crude yield, purity >90% according 1H NMR. cIsolated yield after chromatography. dcat. = 0.3 equiv. eSolvent EtOAc instead of CH2Cl2. fThe ee was determined to be ≥99% at the later stage the piperidinol cis-11d. DCC = N,N´-dicyclohexylcarbodiimide; NMM = N-methylmorpholine; DMAP = N,N-dimethylaminopyridine, HOBt = 1-hydroxybenzotriazole; T3P = n-propylphosphonic acid anhydride; n.d. = not determined.
The high racemisation sensitivity of 2c is further underlined by a T3P/pyridine (n-propylphosphonic acid anhydride) [65,66] mediated amidation, which has been reported to suppress racemisation [67]. With substrate 2c the condensation product 5c was formed in a moderate ee of 74% (Table 1, entry 7). The configurational lability of the phenylglycine derivative was not surprising, as phenylglycine itself is 60 times more prone to racemisation than alanine [68]. The optical purity of amides 5a–c, hydroxyketone 7c and piperidins cis-11a–d and trans-11a (Table 2, Scheme 6 and 7) was determined with HPLC on a chiral stationary phase and comparison with racemic samples (alanine 1a and phenylglycine 1c derived substrates) or in analogy to the aforementioned amino acid derivatives (phenylalanine 1b and methionine 1d deduced substrates).
In the next step, addition of a slight excess of the 3-chloropropanol-derived Grignard reagent 6 (final concentration 0.25–0.3 mol/L), which was found to be superior in the final concentration to the reported ClMgnPrOMgCl derivative (0.1–0.2 mol/L in our hands) [69], to amides 5a–d resulted in ketones 7a–d in good to excellent yields (86–97%, Scheme 2). In contrast to the reported procedure (2–3 h of reflux) [69], short reflux times to form 6 (20–30 min) were crucial to avoid decomposition of this dianionic reagent. Furthermore, a slight excess of MeMgBr in the deprotonation step of 3-chloropropanol was found to assist the Grignard formation. Notably, with this strategy we saved additional protection and deprotection steps of the free hydroxy group of 7. Thereby, only the phenylglycine derivative 7c displayed a slight decrease of ee (95→92%), the other hydroxy ketones 7a, 7b and 7d did not show any racemisation at all, not even after a longer time of storage. For the latter three the enantiomeric excess was not determined on this step: Further conversion as depicted in Scheme 4 and Table 2 delivered piperidinols 11a, 11b and 11d in ≥99% ee. As no intermediate in the conversion of 7 to 11 was crystallized, the ketones 7a, 7b and 7d must have been enantiopure.
Residual 3-chloropropanol (<15 mol % referred to 7), which was difficult to separate chromatographically, was removed after the next step either during the work up (diols 9) or chromatographically (on the sequence leading to 15a). Interestingly, the ketones 7a–d existed in an equilibrium with their cyclic hemiacetal tautomers as shown in Scheme 2. The predominant keto form possessed a clear singlet around 205–210 ppm for the quaternary carbonyl carbon and the furan form was indicated by two weak signals at 105–110 ppm for the hemiacetal carbon (two diastereomers) in the 13C NMR.
Additionally, a short 3 step route to the functionalized glutamic and aspartic acid derived Weinreb amides 5e and 5f was coined as outlined in Scheme 3: At the outset both amino acids (1e and 1f) were subjected to esterification of the sterically less hindered side chain carboxyl function with acetyl chloride in MeOH [70-72]. Neutralization of the reaction mixture with K2CO3 and reductive benzylation [73] in one-pot then delivered the benzyl amines 8e and 8f. While glutamic acid showed selective mono esterification after 3 h at room temperature, the aspartic acid mono ester was obtained in a 5:1 ratio with the diester (not shown) after approximately 18 h of reaction time. Shorter reaction times in the esterification step of 1f decreased the yield of 8f.
![[1860-5397-10-35-i3]](/bjoc/content/inline/1860-5397-10-35-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of amides 5e and 5f and ketone 7e.
Scheme 3: Synthesis of amides 5e and 5f and ketone 7e.
The amines 8e and 8f were converted in a straightforward manner to the carbamates 2e and 2f under Schotten–Baumann conditions [62,63]. Thus, K2CO3 (→ pH = 10) as the base prevented saponification of the side chain methyl ester functions (as observed with hydroxide salts). Nevertheless, in the acylation of the glutamic acid derivative 8e the corresponding pyroglutamic acid derivative 2g resulting from lactamisation of the ester function was observed as a side product in traces (<5%). Next, amidation of the acids 2e and 2f gave the amides 5e and 5f in good yields under standard coupling conditions. Unfortunately, the ketone 7e was obtained in only 24% yield through addition of the Grignard reagent 6 to amide 5e (along with 30% of reisolated starting material), illustrating the tendency of reagent 6 to attack the side chain ester moiety.
Synthesis of syn-amino alcohols C
Already the NaBH4 reduction (in MeOH at −40/0 °C) of ketones 7a and 7b and subsequent hydrogenolysis of the Cbz-group in one-pot delivered the syn-amino alcohols 9a and 9b in good diastereomeric ratios (around 11:1 syn/anti, compare Scheme 4). In contrast the NaBH4 reduction of ketone 7c under identical conditions gave diol 9c without any selectivity (dr = 1.3:1). To our delight, reduction of the ketones 7a and 7b with L-Selectride [74] (or N-Selectride) and subsequent hydrogenolysis of the Cbz-group in one-pot delivered the benzyl amines syn-9a and 9b in accordance with the Felkin–Anh model in excellent yields and as pure diastereomers as indicated by 400 MHz 1H NMR (Scheme 4). Unfortunately, L-Selectride proved to be too unreactive at low temperatures towards the phenylglycine derived ketone 7c, at higher temperatures mainly decomposition of the starting material was observed. Nevertheless, Superhydride [75,76] reduction and successive Cbz-cleavage gave the desired amino alcohol syn-9c in useful diastereomeric ratios of 3.7–4:1 syn/anti. The slightly diminished yield of 9c (74–77%) compared to 9a and 9b (85–93%) is rationalized by formation of the oxazolidinone 10c as sideproduct. This cyclic carbamate results from the condensation of the alcoholate moiety of II with the Cbz-function passing the syn-periplanar conformation indicated in Scheme 4. With a bulky phenyl substituent (= R) this conformation is higher populated than with smaller R groups (e.g. Me and Bn) due to increased steric repulsion between the n-PrOH and R moiety. This explains the more facile formation of oxazolidinones of type 10 with the phenylglycine derived carbamate 7c. Indeed, if the excess of Selectride was quenched with acetaldehyde after reduction of 7a, the oxazolidine 10a was obtained (under the basic reaction conditions) as the major product.
![[1860-5397-10-35-i4]](/bjoc/content/inline/1860-5397-10-35-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of amino alcohols syn-9a–d and oxazolidinone 10a. (for 7a–c conditions A: H2 (1 atm), Pd/C, HCl, MeOH (THF/MeOH 1:1), rt, 3 h; for 7d conditions B: HCl (aq.; 4 N HCl in THF/H2O), rt, 6 h) aR´= sBu for 7a, 7b and 7d (L-Selectride); R´= Et for 7c (Superhydride). bdr ≥ 19:1. cdr = 3.7–4:1.)
Scheme 4: Synthesis of amino alcohols syn-9a–d and oxazolidinone 10a. (for 7a–c conditions A: H2 (1 atm), Pd/...
The outcome of the Cbz-cleavage strongly depended on the activity of the commercial Pd/C batch: While an active catalyst led to a quantitative cleavage after 3 h (all substrates), less reactive batches led to considerably increased reaction times (2 d for 7a). For 7b and 7c the isolated yields of the amines 9 even dropped to 10–20% after 2 d reaction time due to incomplete Cbz-cleavage. However, Pd/C freshly prepared from Pd(OAc)2 and activated charcoal according to Felpin [77] delivered consistent results (reaction time < 3 h). Although the Cbz-cleavage beside a benzyl moiety in alcohols as solvent is known (for selected examples of the chemoselective Cbz-cleavage of Bn, Cbz-protected amines see [78-80]), we only achieved high chemoselectivities in the presence of an excess of HCl.
In a similar manner the methionine derived Boc-protected ketone 7d was converted through L-Selectride reduction and Boc-cleavage induced by aqueous HCl solution in one pot to the syn-aminoalcohol 9d as a single diastereomer according to 1H NMR. Importantly for up-scaling, only a slight excess of L-Selectride (1.3 equiv) or Superhydride (1.5 equiv), respectively, was required for the quantitative conversion of 7a–d to the intermediate II. Therefore the deprotonation of the free OH-group of 7 must be significantly slower than the desired reduction of the carbonyl function. Practically, the residual 3-chloropropanol remaining from the prior Grignard addition 5→7 step (vide supra) and sec-Bu3B/BEt3 from the reduction were easily separated through washing with Et2O of an (HCl) acidic, aqueous phase of the products 9 resulting in the crude diols 9a–d in a circa 90% purity according 1H NMR. Hence, the crude amino alcohols 9 were not further purified before the following cyclodehydration.
Synthesis of cis-piperidinols B
Cyclodehydration of amino alcohol syn-9a to piperidinol cis-11a under Appel conditions (I2, PPh3) [81,82] surpassed by far Mitsunobu conditions and sulfonation (with MsCl, TsCl) induced cyclisations (see Supporting Information File 1 for more details): Under optimized conditions (1.1 equiv I2, Et3N in MeCN at −40 °C) the cyclic products 11a–c were isolated in 68–77% yield and 90–99% ee (Table 2 entry 1, 10, 12). For work up the reaction mixture was simply absorbed on silica gel (ca. 9 fold amount of starting material 9) through evaporation of the solvent and directly subjected to chromatographic purification.
Table 2: Cyclodehydration of amino alcohols 9a–d to piperidinols 11a–d.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-35-i9.svg?max-width=637&scale=1.0)
|
|||||
| entry | substrate | R | PR3 | deviation from standard conditions | yield |
|---|---|---|---|---|---|
| 1 | syn-9a | –Me | PPh3 | – | 77%a,b,c |
| 2 | P(OEt)3 | – | 82%a,b,c (14 g) | ||
| 3 | P(OEt)3 | reflux instead of rt in saponification | 52%a,c | ||
| 4 | P(OEt)3 | DIPEA instead of Et3Ne | 57%c,d | ||
| 5 | P(OEt)3 | lutidine instead of Et3Ne | 50%c,d | ||
| 6 | P(OEt)3 | imidazole instead of Et3Ne | 30%c,d | ||
| 7 | P(OEt)3 | Et3N/CH2Cl2 1:10 | 79%c,d | ||
| 8 | P(OEt)3 | THF instead of CH2Cl2e | 58%c,d | ||
| 9 | P(OEt)3 | −20 °C then warming to 0 °C | 41%a,c | ||
| 10 | syn-9b | –Bn | PPh3 | – | 68%a,b,c |
| 11 | P(OEt)3 | – | 74%a,b,c | ||
| 12 | syn-9c | –Ph | PPh3 | – | 77%a,e,f (2.3 g) |
| 13 | P(OEt)3 | Et3N/CH2Cl2 1:1.3 | 68%a,e,f | ||
| 14 | P(OEt)3 | – | 49%a,f | ||
| 15 | P(OEt)3 | Et3N/CH2Cl2 1:3 | 22%a,f | ||
| 16 | syn-9d | –EtSMe | P(OEt)3 | – | 74%a,b,c |
aIsolated yield after chromatographic purification. bee ≥ 99%, determined by HPLC on a chiral stationary phase. cdr >19:1 cis/trans. dYield determined with naphthalene as NMR-standard. eRatio base/solvent 1:10. fee = 90%; dr = 4.0:1 (entry 12), 5.3:1 (entry 13), 3.7.1 (entry 14), 3.0:1 (entry 15) cis/trans. DIPEA = di-isopropylethylamine; lutidine = 2,6-dimethylpyridine. All reactions were run until full conversion of the starting material 9.
Unfortunately, the stoichiometric byproduct triphenyl phosphine oxide was only separable by chromatography requiring significantly increased amounts of silica gel (the crude product weight usually obtained 300–400% of the theoretical yield after aqueous work up). Attempts to crystallize OPPh3 beside the piperidine 11a or of the fumaric acid salt of 11a for instance failed, only an oily mixture of 11a and OPPh3 precipitated. Separation of OPPh3 through washing of an aqueous solution of hydrochlorides of 11 led to a significant loss of piperidines 11 (especially with 11b and 11c bearing lipophilic side chains R).
In general, the reaction of alcohols with alkyl phosphites (P(OR)3), activated through oxidants such as iodine, have been reported to give the corresponding phosphates in a Michaelis–Arbuzow type reaction [83-85] (Scheme 5). In order to improve atom economy and side product separation, we rationalized that in the phosphonium intermediate III (originating from activation of the diol/amino alcohol E) the intramolecular substitution by the amino function (delivering the desired heterocycles of type G) should be significantly faster than the bimolecular reaction of the iodide ion with intermediate III as indicated (resulting in the formation of the corresponding undesired phosphates F). The formation of heterocycles G passing phosphates F through substitution of the phosphate leaving group by the nucleophilic YH moiety would be in principal also a plausible explanation for the formation of F, but can most likely be excluded (see Supporting Information File 1).
![[1860-5397-10-35-i5]](/bjoc/content/inline/1860-5397-10-35-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Competition between the Michaelis–Arbuzow process and the desired cyclodehydration of amino alcohols and diols E (Y = e.g. O, NBn).
Scheme 5: Competition between the Michaelis–Arbuzow process and the desired cyclodehydration of amino alcohol...
Indeed, under optimized conditions (1.2 equiv I2, P(OEt)3, Et3N (5–6 equiv)/CH2Cl2 1:2; −78 °C then warming to rt) the piperidinols 11a–d were isolated after saponification (during work up) of triethylphosphate and chromatographic purification in 68–82% yield and high enantiomeric excess (90–99%) (Table 2, entries 2, 11, 13 and 16).
For both cyclodehydration methods (with PPh3 and P(OEt)3) neat iodine was simply added to a solution of the amino alcohols 9a–d, the phosphorus reagent and Et3N in the reaction solvent at the indicated temperature (−40 °C and −78 °C, respectively) and stirred at this temperature, until I2 had dissolved completely (1–2 h with PPh3 and 2–5 h with P(OEt)3). Then the usually heterogeneous mixture (through precipitated Et3NH+Cl−) was allowed to warm to room temperature to reach full conversion of the starting material 9. Due to the ammonium salt precipitation and the solid iodine for the large scale preparation of piperidinol 11a (14 g, Table 2, entry 2) mechanical stirring was preferred. For saponification, saturated KOH solution in MeOH (ca. 4 N) was chosen, because aqueous KOH or NaOH solution would result in biphasic mixtures. A quantitative hydrolysis of the triethylphosphate side product required evaporation of CH2Cl2 and Et3N (and MeOH) after the addition of the methanolic KOH-solution in vacuo, dilution with MeOH, stirring at room temperature for ca. 1 h, and a second concentration under reduced pressure. Most likely, the solubility of KOH in the CH2Cl2/Et3N/MeOH mixture is too low resulting in incomplete hydrolysis of the phosphate without evaporation of the solvent. Saponification at reflux on the other hand (in order to circumvent the solvent evaporations) led to partial decomposition of the product 11a (isolated yield 52% after several hours of reflux, see Table 2, entry 3). Chromatographic purification of the crude piperidinols 11a–d, which were already isolated in 85–90% purity (from the phosphite cyclodehydration), was best achieved with halogen free iPrOH/Et3N/hexane eluent mixtures (ratio 0.8–3:2–4:100 depending on the polarity of the product). Although the diastereomers of piperidinol 11c (dr ca. 4:1 cis/trans) were chromatographically separable, we decided to separate them after protecting group exchange at the stage of the Boc carbamate 16c (see Scheme 8), because the epimers of the latter one were much easier to isolate.
Interestingly, we observed a high specificity in the base: With pyridine, NMM, DMAP and DBU (1,8-diaza[5.4.0]bicyclo-undec-7-ene), respectively, (instead of Et3N) the desired heterocycle 11a was only formed in traces. DIPEA, lutidine and imidazole delivered 11a in clearly diminished yields of 30–57% (Table 2, entries 4–6) compared to Et3N (79%, entry 7). For comparison we ran the condensation of amino alcohol 9a to piperidinol 11a in the same concentration as in Table 2, entries 4–6 and 8 (as opposed to the standard conditions) and determined the yield also through NMR-standard: The yield assigned in that way (79%, entry 7) shows a decent match to the 82% isolated yield obtained under standard conditions (entry 2). Hence, the comparability of Table 2 entry 2 to entries 4–8 is established. In conclusion NMM, pyridine and imidazole might be too weak bases, DBU might be too strong and a base favoring other reaction pathways then the desired cyclodehydration. DIPEA and lutidine could be sterically too hindered and DMAP too nucleophilic inducing side reactions.
Moreover, we observed a strong influence of the ratio of Et3N/CH2Cl2 with substrate 9c: The phenylglycine-derived hydroxypiperidine 11c was obtained in 68% yield in Et3N/CH2Cl2 in a 1:1.3 ratio, while a 1:3 solvent mixture gave 11c in only 22% yield (Table 2, entries 13–15). Indeed, the furan 13h (see Table 2) resulting from nucleophilic substitution of the primary (activated) OH-function through the secondary hydroxy group of 9c formed in significant amounts in the cyclisation of amino diol 9c. This is explained by steric shielding of the amino function (→ decreased nucleophilicity) through the bulky phenyl group in α-position (compared to the less demanding Me and Bn side chains R of substrates 9a and 9c). As the R-substituent is in the β-position of the secondary OH-function of diols 9, this hydroxy moiety is less shielded. For substrate 9c the yield could not be improved further, because in higher ratios Et3N/CH2Cl2 iodine did not completely dissolve (react) at −78 °C in a reasonable time (<12 h).
This strong influence of the ratio of Et3N/CH2Cl2 can be attributed to the lower solubility of iodine in Et3N (which leads to a slower and thus more selective reaction) and general base catalysis: Simultaneous deprotonation (through Et3N) in the cyclisation step strongly favours the desired reaction pathway to piperidines 11. Due to this effect we chose a ratio of Et3N/CH2Cl2 1:2 as standard conditions for the phosphite mediated cyclodehydration. Typically, 5 to 6 equivalents of Et3N resulting in a reasonable concentration of the substrates 11 and 12 (see Table 3) of 0.4–0.5 mol/L (in Et3N/CH2Cl2) were sufficient enough to guarantee magnetically stirring, as the viscosity of the reaction mixture increases through ammonium salt precipitation during the preceding reaction. Noteworthy, Et3N is as cheap as common solvents such as THF and CH2Cl2.
Furthermore, in THF the yield of 11a decreased to 58% (Table 2, entry 8), whereas in MeCN low conversions were observed most likely through reaction of the solvent with the I–P(OEt)3+ intermediate and in Et3N as the sole solvent the solubility of iodine is too low. Even at room temperature iodine did not dissolve (= react) completely. Also a larger excess of iodine (>1.2 equiv) has a negative effect on the yield, because the secondary hydroxy function probably is activated as well. With the more atom economic P(OMe)3 complex product mixtures were obtained (in the case of substrate 9a). Most likely the differentiation between the Michaelis–Arbuzow and cyclodehydration pathway (see Scheme 5) is declined. Moreover, the reaction temperature has a strong influence: When the condensation of 9a had been performed at −20 °C for instance, piperidine 11a was isolated in only 41% yield (Table 2, entry 9). Interestingly, the activation of the primary OH function of substrates 9 in the presence of the secondary OH group proceeded (with PPh3 and P(OEt)3) in very high chemoselectivity most likely due to steric effects. The corresponding aziridine of 9a clearly resulting from activation of the secondary hydroxy group and subsequent fast 3-exo-trig cyclisation was only obtained as a sideproduct at higher temperatures (I2, PPh3, imidazole, CH2Cl2 at 0 °C; see Supporting Information File 1 for more details).
Significantly, 14 g of alanine derivative cis-11a were obtained (cyclodehydration with P(OEt)3) in one batch with no purification of the intermediates (2a, 5a, 7a and syn-9a) at all and an overall yield of 44%, demonstrating the scalability and high practicability of our sequence. The relative configurations of compounds 9 and 11 were proven by NOE spectroscopy of piperidinols cis-11a–d, trans-11a, cis- and trans-16c, L-733,060.HCl and of oxazolidinones trans-10a and trans-10c (see Supporting Information File 1 for more details).
Synthesis of other heterocycles through phosphite-mediated cyclodehydration
We next wanted to demonstrate the generality of the phosphite-mediated cyclodehydration (Table 3). Towards this end, a range of amino alcohols and diols 12a,b and 12d–h were converted in usually very good yields (>80%) to pyrrolidines, piperidines and furans 13a,b and 13d–h. The established cyclodehydration procedure only had to be slightly modified regarding the work up: As most of the products 13a,b and 13d–h are volatile, the crude reaction mixture was concentrated under reduced pressure (50 mbar) after quenching with the methanolic KOH solution and kept at this pressure at the rotatory evaporator for ca. 0.5 h. Then the residue was portioned between water and n-pentane (high volatility and low solubility of OP(OEt)3!) and the organic phase was washed with five portions of water to remove the remaining phosphate (after saponification 13 contained 20–30 mol % of the phosphate). Finally, the heterocycles 13 were isolated without a trace of the phosphoric acid ester (“traceless cyclodehydration”) in >90% purity according to 1H NMR.
Table 3: Cyclodehydration of amino alcohols and diols 12a–h to heterocycles 13a–h.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-35-i10.svg?max-width=637&scale=1.0)
|
|||||||
| entry | substrate | YH | R1 | R2 | n | deviation from standard conditions | yield |
|---|---|---|---|---|---|---|---|
| 1 | 12a | NBn | H | H | 1 | – | 80%a |
| 2 | washing of 13-HCl (aq.) with EtOAc instead of saponification | 81%b | |||||
| 3 | 12b | NBn | H | H | 2 | washing 13b-HCl (aq.) with EtOAc instead of saponification | 63%b |
| 4 | 12c | NBoc | H | H | 2 | 2 d at rt | n. r. |
| 5 | rac-12d | OH | Ph | H | 1 | – | 92%b |
| 6 | rac-12e | OH | o-ClPh | H | 1 | – | 85%b |
| 7 | rac-12f | OH | p-BrPh | H | 1 | – | 83%b |
| 8 | P(OiPr)3 instead of P(OEt)3 | 67%a | |||||
| 9 | P(OPh)3 instead of P(OEt)3 | 83%a | |||||
| 10 | rac-12g | OH | Mes | H | 1 | – | 84%b |
| 11 | 12h | OH | Ph | Ph | 1 | – | 82%b |
| 12 | PPh3/MeCN instead of P(OEt)3/CH2Cl2 | 75%a | |||||
aYield determined with naphthalene as NMR-standard. bIsolated yields, purity >90% according to crude 1H NMR.
We first investigated the amino alcohols 12a–c: Pyrrolidine 13a was formed in 80% yield after hydrolysis of the phosphate and in 81% yield after extraction of triethylphosphate with EtOAc (×5) while keeping the product 13a as the hydrochloride salt in the aqueous phase (Table 3, entries 1 and 2). Surprisingly, with the higher homologue 12b the piperidine 13b was obtained in only 63% yield (entry 3). The lower yield compared to the hydroxypiperidines 11a,b and 11d (≥74%, see Table 2) might be explained by the preference of a conformation of the phosphonium intermediate III of 11a,b and 11d (Scheme 5), which is favourable for the cyclisation. This arrangement might be stabilized through a hydrogen bridge between the NH proton and the O atom of the secondary OH group and a (weak) Thorpe–Ingold effect by the substituent R. The much less nucleophilic Boc-carbamate 12c was not converted to the desired piperidine 13c (entry 4). Here only starting material was re-isolated (probably originating from hydrolysis of the corresponding phosphate of alcohol 12c).
The α-aryl furans 13d–f were formed in excellent yields (83–92%, Table 3, entries 5–7). Interestingly, the cyclodehydration of rac-12f was also mediated by P(OiPr)3 and P(OPh)3 in 67% and 83% yield, respectively (entries 8–9). Whereas OP(OiPr)3 was hydrolyzed in the work up, OP(OPh)3 was not saponified and therefore still remained in the isolated product 13f. However, considering the atom economy these phosphites do not represent an alternative to P(OEt)3. Even the diols rac-12g and 12h with a sterically demanding mesityl and two phenyl substituents, respectively, gave cleanly the furans rac-13g and 13h in good yields (Table 3, entries 10 and 11). Also in terms of isolated yield the phosphite-mediated cyclodehydration of substrate 12h was superior (82%, Table 3, entry 11) to the phosphine driven conditions (75%, entry 12).
Synthesis of a trans-piperidinol B
We initially investigated the diastereoselectivity of the reduction of the secondary benzylamino ketone 14a, which was synthesized from hydroxyketone 7a through Cbz-cleavage and basic work up (Scheme 6 and Table 4). According to 1H NMR the hemiacetal of 14a (ca. 1:1 ratio of its epimers) forms an equilibrium with its ketone tautomer in a 1.8:1 ratio. In contrast to hydroxyketones 7a–d the furan tautomer of 14a is thermodynamically more stable than the keto form. This might be rationalized by a lower steric strain in the heterocyclic form due to the smaller NHBn side chain (compared to NBnCbz in 7a–d).
![[1860-5397-10-35-i6]](/bjoc/content/inline/1860-5397-10-35-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Initial synthesis of the trans-piperidinol 11a in diminished enantiopurity. aThe amino alcohol 9a obtained through L-Selectride reduction according to entry 6 in Table 4 (dr = 25:1) was subjected to cyclodehydration.
Scheme 6: Initial synthesis of the trans-piperidinol 11a in diminished enantiopurity. aThe amino alcohol 9a o...
Table 4: Diastereoselectivity in the reduction of ketone 14a.
| entry | [H–] | solvent | T [°C] | dr (anti/syn) |
|---|---|---|---|---|
| 1 | NaBH4 | MeOH | 0 | 1:1a |
| 2 | NaBH4, HCl | MeOH | 0 | 2.6:1a |
| 3 | NaBH4, CeCl3 | MeOH | −78 | 2.9:1a |
| 4 | DIBALH | diversb | −78 | 1.4–2.6:1c |
| 5 | L-Selectride | THF | −78 | 6:1d |
| 6 | L-Selectride | CH2Cl2/THFd | −78 | 25:1e |
| 7 | N-Selectride | THF | −78 | >50:1e |
aThe amino alcohol 9a was isolated in 87% (entry 1), 85% (entry 2), 93% (entry 3) yield and >90% purity according to 1H NMR. bCH2Cl2, n-Hex/THF or n-Hex/CH2Cl2. c40–60% conversion were achieved, starting material was not separated. dA solution of the starting material 14a in CH2Cl2 was treated with a commercial solution of L-Selectride in THF. e60–80% conversion were achieved, starting material was not separated.
Disappointingly, in the reduction with NaBH4 no selectivity was observed at all (dr = 1:1 anti/syn, Table 4, entry 1). However, in the presence of one equivalent of HCl the diol 9a was isolated in a moderate dr of 2.6:1 (entry 2), which demonstrated the formation of a Cram chelate transition state stabilized through a hydrogen bond of the hydrochloride of 14a. Under Luche conditions (NaBH4, CeCl3) [86] a similar result was attained (dr = 2.9:1, Table 4, entry 3).
DIBALH delivered amino alcohol 9a (various solvents tested) only in poor selectivities (dr up 2.6:1, Table 4, entry 4), which is in harsh contrast to previously reported reductions of related para-methoxybenzylamino and benzylamino ketones with DIBALH giving rise of the best diastereoselectivities [59,60]). Albeit up to four equivalents of DIBALH were utilized, only 40–60% conversion of 14a was reached. Here the complexation by –AliBu2 after the initial deprotonation of the OH and NH function of 14a might shield the carbonyl group and encumber reduction.
However, L-Selectride reduction resulted in better stereoselectivities with dr = 6:1 anti/syn (Table 4, entry 5). If the starting material 14a was dissolved in the less polar CH2Cl2 (rather than THF), the dr further increased to >19:1 (entry 6). Moreover, N-Selectride provided the product 9a almost as a pure diastereomer (entry 7, solvent THF), only a very small trace of the syn-diastereomer was visible in the 1H NMR (400 MHz). Nevertheless, even with 4 equivalents of Selectride conversions of only up to 80% were observed. Unfortunately, subsequent Appel reaction of the diol anti-9a (dr = 25:1), synthesized through L-Selectride reduction in THF/CH2Cl2 (Table 4, entry 6), gave piperidinol 11a in a significantly diminished ee of 32% (determined via HPLC on a chiral stationary phase and comparison with a racemic sample). This racemisation might be rationalized by an intermolecular enamine formation of the secondary amino ketone 14b as depicted in the intermediate IV, Scheme 6.
At this point we realized that a hydrochloride of the secondary amino ketone 14a or a derivative should be unable to racemise through autocatalytic enamine formation (due to the protonation of the amino function). However, the hydrochloride of amine 14a was poorly soluble in organic solvents, so that its isolation proved to be difficult. Straightforward, mesylation of hydroxyketone 7a and subsequent Cbz-cleavage (H2, Pd/C) in the presence of HCl delivered the hydrochloride salt 15a, which was easily isolated through filtration and solvent evaporation (Scheme 7). To our delight, subsequent liberation of the free amine through DBU at low temperature, immediate L-Selectride reduction (giving intermediate V), HCl quenching and Et3N-induced cyclisation afforded the piperidine trans-11a in an excellent ee (≥99%) and as a single diastereomer according to crude 1H NMR. Although the reduction is performed in the presence of a secondary amino function bearing an N–H-proton and one equivalent of DBU-H+, only 1.5 equivalents of L-Selectride were required for a quantitative conversion. Thus we assume the Cram chelate transition state is formed through an amine N–H proton rather than an amide N–Li lithium cation as shown in Scheme 7 (which would result from deprotonation of the amino group by L-Selectride and would thus consume at least 2 equivalents of the reducing agent).
![[1860-5397-10-35-i7]](/bjoc/content/inline/1860-5397-10-35-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of trans-piperidinol 11a in excellent ee.
Scheme 7: Synthesis of trans-piperidinol 11a in excellent ee.
Synthesis of L-733,060
In order to probe the practicability of our sequence we synthesized L-733,060 as shown in Scheme 8. After cleavage of the Bn-group under 1 atm of hydrogen and subsequent Boc-protection in one pot, the diastereomers cis- and trans-16c were easily separated by flash chromatography. Thereby, we found it advantageous to perform the hydrogenolysis in the presence of HCl to protonate the released amine and then induce Boc protection after neutralisation of the acid by Et3N rather than to run the hydrogenolysis in the presence of Boc2O. As already observed in the reduction/Cbz-cleavage 7→9 (Scheme 4) the quality of the Pd/C batch had a high influence on the hydrogenolysis: No Bn cleavage was observed with Pd/C charges of a low activity, more catalytically active batches and freshly prepared Pd/C [77] led to quantitative conversion within 1–2 d (1 atm H2). The resulting alcohol cis-16c was subjected to Williamson etherification and subsequently the Boc-group was cleaved under acidic conditions (HCl in dioxane). We decided to isolate L-733,060 as its hydrochloride salt, because it is a non-hygroscopic solid (rather than an oil) and can be easily extracted with organic solvents (e.g. EtOAc) from an aqueous phase. With 8 steps, our sequence represents one of the shortest syntheses reported to date [38-40]. Additionally, with the carbamate cis-16c (synthesized in 6 rather than 8 steps) we also achieved a formal total synthesis of CP-99,994 [87].
![[1860-5397-10-35-i8]](/bjoc/content/inline/1860-5397-10-35-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Although the phenylalanine and phenylglycine-derived piperidinols 11b and 11c bear “unfunctionalized” side chains, phenyl groups represent masked carboxylic acid functions. For instance, the enantiomers of piperidine cis-11c and its N-deprotected derivative were converted to (2S,3R)-3-hydroxypipecolic acid through protecting group manipulation and oxidative cleavage of the phenyl group with RuCl3 and NaIO4 [38,40].
Conclusion
Herein we presented a highly stereodivergent (dr up to 19:1), scalable and practical (up to 14 g of cis-11a without any purification of intermediates) synthesis of cis- and trans-configured 3-piperidinols 11, which represent a key structural motive in various natural products and other bioactive target compounds. Moreover, a high step-economy (5–6 steps) was guaranteed by several novel one-pot procedures (1→2, 7→syn-9, 15a→trans-11a) and surrendering any protection of OH functions. To probe the efficiency of this sequence piperidinol 11c was converted to the NK-1 inhibitor L-733,060 in three further steps. Additionally, a unique cyclodehydration procedure replacing PPh3 through P(OEt)3 to improve atom economy (166 compared to 262 g/mol) and to allow separation of the oxidized side product (OP(OEt)3) by saponification (no similar literature precedents known) was implemented. Ongoing research is focusing on the transformation of the methionine-derived piperidinol 11d to other pharmacologically relevant targets on a gram scale.
Supporting Information
| Supporting Information File 1: Experimental and characterisation data. | ||
| Format: PDF | Size: 4.8 MB | Download |
Acknowledgements
We want to thank the German Academic Exchange Service (DAAD) for a generous scholarship for P. H., and Prof. Dr. H.-G. Schmalz for the very kind opportunity to perform a part of the project at the University of Cologne. Additionally, we want to thank Christopher Lood (Aalto University) for recording analytical data.
References
-
Karjalainen, O. K.; Koskinen, A. M. P. Org. Biomol. Chem. 2012, 10, 4311–4326. doi:10.1039/c2ob25357g
Return to citation in text: [1] [2] -
Wijdeven, M. A.; Willemsen, J.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2010, 2831–2844. doi:10.1002/ejoc.200901494
Return to citation in text: [1] [2] [3] -
Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565
Return to citation in text: [1] [2] [3] -
Nemr, A. L. Tetrahedron 2000, 56, 8579–8629. doi:10.1016/S0040-4020(00)00790-0
Return to citation in text: [1] [2] [3] -
Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6
Return to citation in text: [1] [2] -
Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. H.; Storer, R. Drug Discovery Today 2011, 16, 107–118. doi:10.1016/j.drudis.2010.08.017
Return to citation in text: [1] [2] -
Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645–651. doi:10.1016/j.tetasy.2009.02.048
Return to citation in text: [1] [2] -
Pruett, S. T.; Bushnev, A.; Hagedorn, K.; Adiga, M.; Haynes, C. A.; Sullards, M. C.; Liotta, D. C.; Merrill, A. H., Jr. J. Lipid Res. 2008, 49, 1621–1639. doi:10.1194/jlr.R800012-JLR200
Return to citation in text: [1] -
Brunner, M.; Koskinen, A. M. P. Curr. Org. Chem. 2004, 8, 1629–1645. doi:10.2174/1385272043369638
Return to citation in text: [1] -
Zappia, G.; Cancelliere, G.; Gacs-Baitz, E.; Delle Monache, G.; Misiti, D.; Nevolam, L.; Botta, B. Curr. Org. Synth. 2007, 4, 238–307. doi:10.2174/157017907781369306
Return to citation in text: [1] -
Ager, D. J.; Prakash, I.; Schaad, D. R. Chem. Rev. 1996, 96, 835–875. doi:10.1021/cr9500038
Return to citation in text: [1] -
Singh, P. K.; Singh, V. K. Pure Appl. Chem. 2012, 84, 1651–1657. doi:10.1351/PAC-CON-11-10-16
Return to citation in text: [1] -
Nishiyama, H.; Ito, J.-i. Chem. Commun. 2010, 46, 203–212. doi:10.1039/b918923h
Return to citation in text: [1] -
Desimoni, G.; Faita, G.; Jørgensen, K. A. Chem. Rev. 2006, 106, 3561–3651. doi:10.1021/cr0505324
Return to citation in text: [1] -
Baker, R.; Harrison, T.; Swain, C. J.; Williams, B. J. J. Eur. Patent 0528495A1, 1993.
Return to citation in text: [1] -
Harrison, T.; Williams, B. J.; Swain, C. J.; Ball, R. G. Bioorg. Med. Chem. Lett. 1994, 4, 2545–2550. doi:10.1016/S0960-894X(01)80280-8
Return to citation in text: [1] -
Desai, M. C.; Lefkowitz, S. L.; Thadeio, P. F.; Longo, K. P.; Snider, R. M. J. Med. Chem. 1992, 35, 4911–4913. doi:10.1021/jm00104a018
Return to citation in text: [1] -
Rosen, T.; Seeger, T. F.; McLean, S.; Desai, M. C.; Guarino, K. J.; Bryce, D.; Pratt, K.; Heym, J. J. Med. Chem. 1993, 36, 3197–3201. doi:10.1021/jm00073a022
Return to citation in text: [1] -
Data, P.; Srivastava, S.; Coutinho, E.; Govil, G. Curr. Top. Med. Chem. 2004, 4, 75–103. doi:10.2174/1568026043451636
Return to citation in text: [1] -
Koepfli, J. B.; Mead, J. F.; Brockman, J. A., Jr. J. Am. Chem. Soc. 1947, 69, 1837. doi:10.1021/ja01199a513
Return to citation in text: [1] -
Koepfli, J. B.; Mead, J. F.; Brockman, J. A., Jr. J. Am. Chem. Soc. 1949, 71, 1048–1054. doi:10.1021/ja01171a080
Return to citation in text: [1] -
Linder, M. R.; Heckeroth, A. R.; Najdrowski, M.; Daugschies, A.; Schollmeyer, D.; Miculka, C. Bioorg. Med. Chem. Lett. 2007, 17, 4140–4143. doi:10.1016/j.bmcl.2007.05.053
Return to citation in text: [1] -
Grauer, A.; König, B. Eur. J. Org. Chem. 2009, 5099–5111. doi:10.1002/ejoc.200900599
Return to citation in text: [1] -
Hanessian, S.; McNaughton-Smith, G.; Lombart, H.-G.; Lubell, W. D. Tetrahedron 1997, 53, 12789–12854. doi:10.1016/S0040-4020(97)00476-6
Return to citation in text: [1] -
Maillard, M. C.; Brookfield, F. A.; Courtney, S. M.; Eustache, F. M.; Gemkow, M. J.; Handel, R. K.; Johnson, L. C.; Johnson, P. D.; Kerry, M. A.; Krieger, F.; Meniconi, M.; Muñoz-Sanjuán, I.; Palfrey, J. J.; Park, H.; Schaertl, S.; Taylor, M. G.; Weddell, D.; Dominguez, C. Bioorg. Med. Chem. 2011, 19, 5833–5851. doi:10.1016/j.bmc.2011.08.020
Return to citation in text: [1] -
Quibell, M.; Benn, A.; Flinn, N.; Monk, T.; Ramjee, M.; Wang, Y.; Watts, J. Bioorg. Med. Chem. 2004, 12, 5689–5710. doi:10.1016/j.bmc.2004.07.054
Return to citation in text: [1] -
Guengerich, F. P.; DiMari, S. J.; Broquist, H. P. J. Am. Chem. Soc. 1973, 95, 2055–2056. doi:10.1021/ja00787a080
Return to citation in text: [1] -
Elbein, A. D. FASEB J. 1991, 5, 3055–3063.
Return to citation in text: [1] -
Teitelbaum, S. L.; Deselm, C. J. U.S. Pat. Appl. Publ. US 20110311519 A1, 2011.
Return to citation in text: [1] -
Keller, T.; Mazitschek, R.; Whitman, M.; Lee, J. US Patent 2011/0263532 A1, 2011.
Return to citation in text: [1] -
Keller, T.; Mazitschek, R.; Whitman, M. PCT Int. Appl. WO 2010019210 A2, 2010.
Return to citation in text: [1] -
Cochi, A.; Pardo, D. G.; Cossy, J. Heterocycles 2012, 86, 89–116. doi:10.3987/REV-12-SR(N)2
Review on the synthesis of L-733,060 and L-733,061.
Return to citation in text: [1] -
Cochi, A.; Pardo, D. G.; Cossy, J. Eur. J. Org. Chem. 2013, 809–829. doi:10.1002/ejoc.201201415
Reviews on the synthesis of (3-hydroxy)pipecolic acid derivatives.
Return to citation in text: [1] -
Cant, A. A.; Sutherland, A. Synthesis 2012, 44, 1935–1950. doi:10.1055/s-0031-1289767
Reviews on the synthesis of (3-hydroxy)pipecolic acid derivatives.
Return to citation in text: [1] -
Kadouri-Puchot, C.; Comesse, S. Amino Acids 2005, 29, 101–130. doi:10.1007/s00726-005-0193-x
Reviews on the synthesis of (3-hydroxy)pipecolic acid derivatives.
Return to citation in text: [1] [2] -
Lemire, A.; Grenon, M.; Pourashraf, M.; Charette, A. B. Org. Lett. 2004, 6, 3517–3520. doi:10.1021/ol048624n
Return to citation in text: [1] -
Cossy, J.; Dumas, C.; Pardo, D. G. Eur. J. Org. Chem. 1999, 1693–1699. doi:10.1002/(SICI)1099-0690(199907)1999:7<1693::AID-EJOC1693>3.0.CO;2-J
Return to citation in text: [1] [2] -
Cochi, A.; Burger, B.; Navarro, C.; Pardo, D. G.; Cossy, J.; Zhao, Y.; Cohen, T. Synlett 2009, 2157–2161. doi:10.1055/s-0029-1217568
Return to citation in text: [1] [2] [3] [4] -
Bilke, J. L.; Moore, S. P.; O'Brien, P.; Gilday, J. Org. Lett. 2009, 11, 1935–1938. doi:10.1021/ol900366m
Return to citation in text: [1] [2] [3] -
Pansare, S. A.; Paul, E. K. Org. Biomol. Chem. 2012, 10, 2119–2125. doi:10.1039/c2ob06644k
Return to citation in text: [1] [2] [3] [4] -
Shioiri, T.; Izawa, K.; Konoike, T.; Karpf, M. Pharmaceutical Process Chemistry; Wiley-VCH: Weinheim, Germany, 2010; pp 1–37.
Return to citation in text: [1] -
Roberge, D. M. Org. Process Res. Dev. 2004, 8, 1049–1053. doi:10.1021/op0400160
Return to citation in text: [1] -
Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L., Jr.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c
Return to citation in text: [1] [2] -
Dandapani, S.; Curran, D. P. Chem.–Eur. J. 2004, 10, 3130–3138. doi:10.1002/chem.200400363
Return to citation in text: [1] [2] -
Dembinski, R. Eur. J. Org. Chem. 2004, 2763–2772. doi:10.1002/ejoc.200400003
Return to citation in text: [1] [2] -
Véliz, E. A.; Beal, P. A. Tetrahedron Lett. 2006, 47, 3153–3156. doi:10.1016/j.tetlet.2006.02.138
Return to citation in text: [1] -
Chang, B. C.; Conrad, W. E.; Denney, D. B.; Denney, D. Z.; Edelman, R.; Powell, R. L.; White, D. W. J. Am. Chem. Soc. 1971, 93, 4004–4009. doi:10.1021/ja00745a031
Return to citation in text: [1] -
Denney, D. B.; Denney, D. Z.; Gigantino, J. J. J. Org. Chem. 1984, 49, 2831–2832. doi:10.1021/jo00189a044
Return to citation in text: [1] -
Kelly, B. D.; Lambert, T. H. Org. Lett. 2011, 13, 740–743. doi:10.1021/ol102980t
Return to citation in text: [1] -
Flamme, E. M.; Roush, W. R. Beilstein J. Org. Chem. 2005, 1, No. 7. doi:10.1186/1860-5397-1-7
Return to citation in text: [1] -
Huy, P. H.; Koskinen, A. M. P. Org. Lett. 2013, 15, 5178–5181. doi:10.1021/ol4026588
Return to citation in text: [1] -
Cherest, M.; Felkin, H.; Prudent, N. Tetrahedron Lett. 1968, 9, 2199–2204. doi:10.1016/S0040-4039(00)89719-1
Introduction of Felkin–Anh model.
Return to citation in text: [1] -
Stevens, C. L.; TerBeek, K. J.; Pillai, P. M. J. Org. Chem. 1974, 39, 3943–3946. doi:10.1021/jo00940a035
An early example of diastereodiscriminating reductions of amino ketones.
Return to citation in text: [1] -
Cram, D. J.; Wilson, D. R. J. Am. Chem. Soc. 1963, 85, 1245–1249. doi:10.1021/ja00892a008
Introduction of Cram chelate model.
Return to citation in text: [1] -
Rittle, K. E.; Homnick, C. F.; Ponticello, G. S.; Evans, B. E. J. Org. Chem. 1982, 47, 3016–3018. doi:10.1021/jo00136a045
Return to citation in text: [1] -
Lubell, W. D.; Rapoport, H. J. Am. Chem. Soc. 1987, 109, 236–239. doi:10.1021/ja00235a035
Return to citation in text: [1] -
Reyes, E.; Ruiz, N.; Vicario, J. L.; Badia, D.; Carrillo, L. Synthesis 2011, 443–450. doi:10.1055/s-0030-1258390
Return to citation in text: [1] -
Fraser, D. S.; Park, S. B.; Chong, J. M. Can. J. Chem. 2004, 82, 87–101. doi:10.1139/V03-165
Return to citation in text: [1] -
Chung, S.-K.; Lee, J.-M. Tetrahedron: Asymmetry 1999, 1441–1444. doi:10.1016/S0957-4166(99)00147-0
Return to citation in text: [1] [2] -
Dondoni, A.; Perrone, A. Synthesis 1993, 1162–1176. doi:10.1055/s-1993-26021
Return to citation in text: [1] [2] -
Quitt, P.; Hellerbach, J.; Vogler, K. Helv. Chim. Acta 1963, 46, 327–333. doi:10.1002/hlca.19630460133
Return to citation in text: [1] [2] -
Schotten, C. Ber. Dtsch. Chem. Ges. 1884, 17, 2544–2547. doi:10.1002/cber.188401702178
Return to citation in text: [1] [2] -
Baumann, E. Ber. Dtsch. Chem. Ges. 1886, 19, 3218–3222. doi:10.1002/cber.188601902348
Return to citation in text: [1] [2] -
Anderson, G. W.; Zimmerman, J. E.; Callaha, F. M. J. Am. Chem. Soc. 1967, 89, 5012–5017. doi:10.1021/ja00995a032
Report about NMM preventing racemisation during peptide coupling due to its low basicity.
Return to citation in text: [1] -
Wissmann, H.; Kleiner, H.-J. Angew. Chem., Int. Ed. Engl. 1980, 19, 133–134. doi:10.1002/anie.198001331
Return to citation in text: [1] -
Wissmann, H. Phosphorus Sulfur Relat. Elem. 1987, 30, 645–648. doi:10.1080/03086648708079147
Return to citation in text: [1] -
Dunetz, J. R.; Xiang, Y.; Baldwin, A.; Ringling, J. Org. Lett. 2012, 13, 5048–5051. doi:10.1021/ol201875q
A recent study towards the amidation of an racemisation sensitive amino acid derivative.
Return to citation in text: [1] -
Smith, G. G.; Sivakua, T. J. Org. Chem. 1983, 48, 627–634. doi:10.1021/jo00153a001
Return to citation in text: [1] -
Cahiez, G.; Alexakis, A.; Normant, J. F. Tetrahedron Lett. 1978, 33, 3013–3014. doi:10.1016/S0040-4039(01)94926-3
Return to citation in text: [1] [2] -
Hanby, W. E.; Waley, S. G.; Watson, J. J. Chem. Soc. 1950, 3239–3249. doi:10.1039/JR9500003239
Selective monoesterification of glutamic acid.
Return to citation in text: [1] -
Gmeiner, P.; Feldman, P. F.; Chu-Moyer, M. Y.; Rapoport, H. J. Org. Chem. 1990, 55, 3068–3074. doi:10.1021/jo00297a023
Selective side chain monoesterifications of aspartic acid.
Return to citation in text: [1] -
Schwarz, H.; Bumpus, F. M.; Page, I. H. J. Am. Chem. Soc. 1957, 79, 5697–5703. doi:10.1021/ja01578a030
Return to citation in text: [1] -
Hannachi, J.-C.; Vidal, J.; Mulatier, J.-C.; Collet, A. J. Org. Chem. 2004, 69, 2367–2373. doi:10.1021/jo035700b
Reductive benzylation of amino acids with PhCHO and NaBH4 in MeOH.
Return to citation in text: [1] -
Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1972, 94, 7159–7161. doi:10.1021/ja00775a053
Initial discovery of L-Selectride.
Return to citation in text: [1] -
Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1973, 95, 1669–1671. doi:10.1021/ja00786a057
Initial development of Superhydride.
Return to citation in text: [1] -
Brown, H. C.; Kim, S. C.; Krishnamurthy, S. J. Org. Chem. 1980, 45, 1–12. doi:10.1021/jo01289a001
Initial development of Superhydride.
Return to citation in text: [1] -
Felpin, F.-X.; Fouquet, E. Chem.–Eur. J. 2010, 16, 12440–12445. doi:10.1002/chem.201001377
Return to citation in text: [1] [2] -
Khan, I. A.; Saxena, A. K. Tetrahedron 2012, 68, 1272–1279. doi:10.1016/j.tet.2011.11.047
Return to citation in text: [1] -
Kobrzycka, E.; Gryko, D.; Jurczak, J. Tetrahedron: Asymmetry 2002, 13, 2133–2139. doi:10.1016/S0957-4166(02)00545-1
Return to citation in text: [1] -
Griengl, H.; Hayden, W.; Schindler, E.; Wanek, E. Arch. Pharm. 1983, 316, 146–153. doi:10.1002/ardp.19833160211
Return to citation in text: [1] -
Appel, R.; Kleinstück, R. Chem. Ber. 1974, 107, 5–12. doi:10.1002/cber.19741070102
Return to citation in text: [1] -
Lange, G. L.; Gottardo, C. Synth. Commun. 1990, 20, 1473–1479. doi:10.1080/00397919008052864
Cyclodehydration with I2 and PPh3.
Return to citation in text: [1] -
Stowell, J. K.; Widlanski, T. S. Tetrahedron Lett. 1995, 36, 1825–1826. doi:10.1016/0040-4039(95)00153-4
Return to citation in text: [1] -
Oza, V. B.; Corcoran, R. C. J. Org. Chem. 1995, 60, 3680–3684. doi:10.1021/jo00117a018
Return to citation in text: [1] -
Watanabe, Y.; Inada, E.; Jinno, M.; Ozaki, S. Tetrahedron Lett. 1993, 34, 497–500. doi:10.1016/0040-4039(93)85111-9
Return to citation in text: [1] -
Gemal, A. L.; Luche, J. L. J. Am. Chem. Soc. 1981, 103, 5454–5459. doi:10.1021/ja00408a029
Return to citation in text: [1] -
Huang, P.-Q.; Liu, L.-X.; Wei, B.-G.; Ruan, Y.-P. Org. Lett. 2003, 5, 1927–1929. doi:10.1021/ol034505g
Return to citation in text: [1]
| 49. | Kelly, B. D.; Lambert, T. H. Org. Lett. 2011, 13, 740–743. doi:10.1021/ol102980t |
| 50. | Flamme, E. M.; Roush, W. R. Beilstein J. Org. Chem. 2005, 1, No. 7. doi:10.1186/1860-5397-1-7 |
| 51. | Huy, P. H.; Koskinen, A. M. P. Org. Lett. 2013, 15, 5178–5181. doi:10.1021/ol4026588 |
| 52. |
Cherest, M.; Felkin, H.; Prudent, N. Tetrahedron Lett. 1968, 9, 2199–2204. doi:10.1016/S0040-4039(00)89719-1
Introduction of Felkin–Anh model. |
| 53. |
Stevens, C. L.; TerBeek, K. J.; Pillai, P. M. J. Org. Chem. 1974, 39, 3943–3946. doi:10.1021/jo00940a035
An early example of diastereodiscriminating reductions of amino ketones. |
| 64. |
Anderson, G. W.; Zimmerman, J. E.; Callaha, F. M. J. Am. Chem. Soc. 1967, 89, 5012–5017. doi:10.1021/ja00995a032
Report about NMM preventing racemisation during peptide coupling due to its low basicity. |
| 65. | Wissmann, H.; Kleiner, H.-J. Angew. Chem., Int. Ed. Engl. 1980, 19, 133–134. doi:10.1002/anie.198001331 |
| 66. | Wissmann, H. Phosphorus Sulfur Relat. Elem. 1987, 30, 645–648. doi:10.1080/03086648708079147 |
| 62. | Schotten, C. Ber. Dtsch. Chem. Ges. 1884, 17, 2544–2547. doi:10.1002/cber.188401702178 |
| 63. | Baumann, E. Ber. Dtsch. Chem. Ges. 1886, 19, 3218–3222. doi:10.1002/cber.188601902348 |
| 61. | Quitt, P.; Hellerbach, J.; Vogler, K. Helv. Chim. Acta 1963, 46, 327–333. doi:10.1002/hlca.19630460133 |
| 57. | Reyes, E.; Ruiz, N.; Vicario, J. L.; Badia, D.; Carrillo, L. Synthesis 2011, 443–450. doi:10.1055/s-0030-1258390 |
| 58. | Fraser, D. S.; Park, S. B.; Chong, J. M. Can. J. Chem. 2004, 82, 87–101. doi:10.1139/V03-165 |
| 59. | Chung, S.-K.; Lee, J.-M. Tetrahedron: Asymmetry 1999, 1441–1444. doi:10.1016/S0957-4166(99)00147-0 |
| 60. | Dondoni, A.; Perrone, A. Synthesis 1993, 1162–1176. doi:10.1055/s-1993-26021 |
| 61. | Quitt, P.; Hellerbach, J.; Vogler, K. Helv. Chim. Acta 1963, 46, 327–333. doi:10.1002/hlca.19630460133 |
| 54. |
Cram, D. J.; Wilson, D. R. J. Am. Chem. Soc. 1963, 85, 1245–1249. doi:10.1021/ja00892a008
Introduction of Cram chelate model. |
| 55. | Rittle, K. E.; Homnick, C. F.; Ponticello, G. S.; Evans, B. E. J. Org. Chem. 1982, 47, 3016–3018. doi:10.1021/jo00136a045 |
| 56. | Lubell, W. D.; Rapoport, H. J. Am. Chem. Soc. 1987, 109, 236–239. doi:10.1021/ja00235a035 |
| 67. |
Dunetz, J. R.; Xiang, Y.; Baldwin, A.; Ringling, J. Org. Lett. 2012, 13, 5048–5051. doi:10.1021/ol201875q
A recent study towards the amidation of an racemisation sensitive amino acid derivative. |
| 68. | Smith, G. G.; Sivakua, T. J. Org. Chem. 1983, 48, 627–634. doi:10.1021/jo00153a001 |
| 69. | Cahiez, G.; Alexakis, A.; Normant, J. F. Tetrahedron Lett. 1978, 33, 3013–3014. doi:10.1016/S0040-4039(01)94926-3 |
| 77. | Felpin, F.-X.; Fouquet, E. Chem.–Eur. J. 2010, 16, 12440–12445. doi:10.1002/chem.201001377 |
| 78. | Khan, I. A.; Saxena, A. K. Tetrahedron 2012, 68, 1272–1279. doi:10.1016/j.tet.2011.11.047 |
| 79. | Kobrzycka, E.; Gryko, D.; Jurczak, J. Tetrahedron: Asymmetry 2002, 13, 2133–2139. doi:10.1016/S0957-4166(02)00545-1 |
| 80. | Griengl, H.; Hayden, W.; Schindler, E.; Wanek, E. Arch. Pharm. 1983, 316, 146–153. doi:10.1002/ardp.19833160211 |
| 74. |
Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1972, 94, 7159–7161. doi:10.1021/ja00775a053
Initial discovery of L-Selectride. |
| 75. |
Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1973, 95, 1669–1671. doi:10.1021/ja00786a057
Initial development of Superhydride. |
| 76. |
Brown, H. C.; Kim, S. C.; Krishnamurthy, S. J. Org. Chem. 1980, 45, 1–12. doi:10.1021/jo01289a001
Initial development of Superhydride. |
| 73. |
Hannachi, J.-C.; Vidal, J.; Mulatier, J.-C.; Collet, A. J. Org. Chem. 2004, 69, 2367–2373. doi:10.1021/jo035700b
Reductive benzylation of amino acids with PhCHO and NaBH4 in MeOH. |
| 62. | Schotten, C. Ber. Dtsch. Chem. Ges. 1884, 17, 2544–2547. doi:10.1002/cber.188401702178 |
| 63. | Baumann, E. Ber. Dtsch. Chem. Ges. 1886, 19, 3218–3222. doi:10.1002/cber.188601902348 |
| 69. | Cahiez, G.; Alexakis, A.; Normant, J. F. Tetrahedron Lett. 1978, 33, 3013–3014. doi:10.1016/S0040-4039(01)94926-3 |
| 70. |
Hanby, W. E.; Waley, S. G.; Watson, J. J. Chem. Soc. 1950, 3239–3249. doi:10.1039/JR9500003239
Selective monoesterification of glutamic acid. |
| 71. |
Gmeiner, P.; Feldman, P. F.; Chu-Moyer, M. Y.; Rapoport, H. J. Org. Chem. 1990, 55, 3068–3074. doi:10.1021/jo00297a023
Selective side chain monoesterifications of aspartic acid. |
| 72. | Schwarz, H.; Bumpus, F. M.; Page, I. H. J. Am. Chem. Soc. 1957, 79, 5697–5703. doi:10.1021/ja01578a030 |
| 83. | Stowell, J. K.; Widlanski, T. S. Tetrahedron Lett. 1995, 36, 1825–1826. doi:10.1016/0040-4039(95)00153-4 |
| 84. | Oza, V. B.; Corcoran, R. C. J. Org. Chem. 1995, 60, 3680–3684. doi:10.1021/jo00117a018 |
| 85. | Watanabe, Y.; Inada, E.; Jinno, M.; Ozaki, S. Tetrahedron Lett. 1993, 34, 497–500. doi:10.1016/0040-4039(93)85111-9 |
| 86. | Gemal, A. L.; Luche, J. L. J. Am. Chem. Soc. 1981, 103, 5454–5459. doi:10.1021/ja00408a029 |
| 81. | Appel, R.; Kleinstück, R. Chem. Ber. 1974, 107, 5–12. doi:10.1002/cber.19741070102 |
| 82. |
Lange, G. L.; Gottardo, C. Synth. Commun. 1990, 20, 1473–1479. doi:10.1080/00397919008052864
Cyclodehydration with I2 and PPh3. |
| 1. | Karjalainen, O. K.; Koskinen, A. M. P. Org. Biomol. Chem. 2012, 10, 4311–4326. doi:10.1039/c2ob25357g |
| 2. | Wijdeven, M. A.; Willemsen, J.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2010, 2831–2844. doi:10.1002/ejoc.200901494 |
| 3. | Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565 |
| 4. | Nemr, A. L. Tetrahedron 2000, 56, 8579–8629. doi:10.1016/S0040-4020(00)00790-0 |
| 5. | Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6 |
| 6. | Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. H.; Storer, R. Drug Discovery Today 2011, 16, 107–118. doi:10.1016/j.drudis.2010.08.017 |
| 7. | Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645–651. doi:10.1016/j.tetasy.2009.02.048 |
| 8. | Pruett, S. T.; Bushnev, A.; Hagedorn, K.; Adiga, M.; Haynes, C. A.; Sullards, M. C.; Liotta, D. C.; Merrill, A. H., Jr. J. Lipid Res. 2008, 49, 1621–1639. doi:10.1194/jlr.R800012-JLR200 |
| 9. | Brunner, M.; Koskinen, A. M. P. Curr. Org. Chem. 2004, 8, 1629–1645. doi:10.2174/1385272043369638 |
| 15. | Baker, R.; Harrison, T.; Swain, C. J.; Williams, B. J. J. Eur. Patent 0528495A1, 1993. |
| 16. | Harrison, T.; Williams, B. J.; Swain, C. J.; Ball, R. G. Bioorg. Med. Chem. Lett. 1994, 4, 2545–2550. doi:10.1016/S0960-894X(01)80280-8 |
| 37. | Cossy, J.; Dumas, C.; Pardo, D. G. Eur. J. Org. Chem. 1999, 1693–1699. doi:10.1002/(SICI)1099-0690(199907)1999:7<1693::AID-EJOC1693>3.0.CO;2-J |
| 38. | Cochi, A.; Burger, B.; Navarro, C.; Pardo, D. G.; Cossy, J.; Zhao, Y.; Cohen, T. Synlett 2009, 2157–2161. doi:10.1055/s-0029-1217568 |
| 1. | Karjalainen, O. K.; Koskinen, A. M. P. Org. Biomol. Chem. 2012, 10, 4311–4326. doi:10.1039/c2ob25357g |
| 2. | Wijdeven, M. A.; Willemsen, J.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2010, 2831–2844. doi:10.1002/ejoc.200901494 |
| 3. | Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565 |
| 4. | Nemr, A. L. Tetrahedron 2000, 56, 8579–8629. doi:10.1016/S0040-4020(00)00790-0 |
| 5. | Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6 |
| 6. | Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. H.; Storer, R. Drug Discovery Today 2011, 16, 107–118. doi:10.1016/j.drudis.2010.08.017 |
| 7. | Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645–651. doi:10.1016/j.tetasy.2009.02.048 |
| 37. | Cossy, J.; Dumas, C.; Pardo, D. G. Eur. J. Org. Chem. 1999, 1693–1699. doi:10.1002/(SICI)1099-0690(199907)1999:7<1693::AID-EJOC1693>3.0.CO;2-J |
| 12. | Singh, P. K.; Singh, V. K. Pure Appl. Chem. 2012, 84, 1651–1657. doi:10.1351/PAC-CON-11-10-16 |
| 13. | Nishiyama, H.; Ito, J.-i. Chem. Commun. 2010, 46, 203–212. doi:10.1039/b918923h |
| 14. | Desimoni, G.; Faita, G.; Jørgensen, K. A. Chem. Rev. 2006, 106, 3561–3651. doi:10.1021/cr0505324 |
| 2. | Wijdeven, M. A.; Willemsen, J.; Rutjes, F. P. J. T. Eur. J. Org. Chem. 2010, 2831–2844. doi:10.1002/ejoc.200901494 |
| 3. | Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565 |
| 4. | Nemr, A. L. Tetrahedron 2000, 56, 8579–8629. doi:10.1016/S0040-4020(00)00790-0 |
| 32. |
Cochi, A.; Pardo, D. G.; Cossy, J. Heterocycles 2012, 86, 89–116. doi:10.3987/REV-12-SR(N)2
Review on the synthesis of L-733,060 and L-733,061. |
| 33. |
Cochi, A.; Pardo, D. G.; Cossy, J. Eur. J. Org. Chem. 2013, 809–829. doi:10.1002/ejoc.201201415
Reviews on the synthesis of (3-hydroxy)pipecolic acid derivatives. |
| 34. |
Cant, A. A.; Sutherland, A. Synthesis 2012, 44, 1935–1950. doi:10.1055/s-0031-1289767
Reviews on the synthesis of (3-hydroxy)pipecolic acid derivatives. |
| 35. |
Kadouri-Puchot, C.; Comesse, S. Amino Acids 2005, 29, 101–130. doi:10.1007/s00726-005-0193-x
Reviews on the synthesis of (3-hydroxy)pipecolic acid derivatives. |
| 38. | Cochi, A.; Burger, B.; Navarro, C.; Pardo, D. G.; Cossy, J.; Zhao, Y.; Cohen, T. Synlett 2009, 2157–2161. doi:10.1055/s-0029-1217568 |
| 40. | Pansare, S. A.; Paul, E. K. Org. Biomol. Chem. 2012, 10, 2119–2125. doi:10.1039/c2ob06644k |
| 10. | Zappia, G.; Cancelliere, G.; Gacs-Baitz, E.; Delle Monache, G.; Misiti, D.; Nevolam, L.; Botta, B. Curr. Org. Synth. 2007, 4, 238–307. doi:10.2174/157017907781369306 |
| 11. | Ager, D. J.; Prakash, I.; Schaad, D. R. Chem. Rev. 1996, 96, 835–875. doi:10.1021/cr9500038 |
| 36. | Lemire, A.; Grenon, M.; Pourashraf, M.; Charette, A. B. Org. Lett. 2004, 6, 3517–3520. doi:10.1021/ol048624n |
| 23. | Grauer, A.; König, B. Eur. J. Org. Chem. 2009, 5099–5111. doi:10.1002/ejoc.200900599 |
| 24. | Hanessian, S.; McNaughton-Smith, G.; Lombart, H.-G.; Lubell, W. D. Tetrahedron 1997, 53, 12789–12854. doi:10.1016/S0040-4020(97)00476-6 |
| 27. | Guengerich, F. P.; DiMari, S. J.; Broquist, H. P. J. Am. Chem. Soc. 1973, 95, 2055–2056. doi:10.1021/ja00787a080 |
| 28. | Elbein, A. D. FASEB J. 1991, 5, 3055–3063. |
| 38. | Cochi, A.; Burger, B.; Navarro, C.; Pardo, D. G.; Cossy, J.; Zhao, Y.; Cohen, T. Synlett 2009, 2157–2161. doi:10.1055/s-0029-1217568 |
| 39. | Bilke, J. L.; Moore, S. P.; O'Brien, P.; Gilday, J. Org. Lett. 2009, 11, 1935–1938. doi:10.1021/ol900366m |
| 40. | Pansare, S. A.; Paul, E. K. Org. Biomol. Chem. 2012, 10, 2119–2125. doi:10.1039/c2ob06644k |
| 22. | Linder, M. R.; Heckeroth, A. R.; Najdrowski, M.; Daugschies, A.; Schollmeyer, D.; Miculka, C. Bioorg. Med. Chem. Lett. 2007, 17, 4140–4143. doi:10.1016/j.bmcl.2007.05.053 |
| 29. | Teitelbaum, S. L.; Deselm, C. J. U.S. Pat. Appl. Publ. US 20110311519 A1, 2011. |
| 30. | Keller, T.; Mazitschek, R.; Whitman, M.; Lee, J. US Patent 2011/0263532 A1, 2011. |
| 31. | Keller, T.; Mazitschek, R.; Whitman, M. PCT Int. Appl. WO 2010019210 A2, 2010. |
| 87. | Huang, P.-Q.; Liu, L.-X.; Wei, B.-G.; Ruan, Y.-P. Org. Lett. 2003, 5, 1927–1929. doi:10.1021/ol034505g |
| 20. | Koepfli, J. B.; Mead, J. F.; Brockman, J. A., Jr. J. Am. Chem. Soc. 1947, 69, 1837. doi:10.1021/ja01199a513 |
| 21. | Koepfli, J. B.; Mead, J. F.; Brockman, J. A., Jr. J. Am. Chem. Soc. 1949, 71, 1048–1054. doi:10.1021/ja01171a080 |
| 59. | Chung, S.-K.; Lee, J.-M. Tetrahedron: Asymmetry 1999, 1441–1444. doi:10.1016/S0957-4166(99)00147-0 |
| 60. | Dondoni, A.; Perrone, A. Synthesis 1993, 1162–1176. doi:10.1055/s-1993-26021 |
| 17. | Desai, M. C.; Lefkowitz, S. L.; Thadeio, P. F.; Longo, K. P.; Snider, R. M. J. Med. Chem. 1992, 35, 4911–4913. doi:10.1021/jm00104a018 |
| 18. | Rosen, T.; Seeger, T. F.; McLean, S.; Desai, M. C.; Guarino, K. J.; Bryce, D.; Pratt, K.; Heym, J. J. Med. Chem. 1993, 36, 3197–3201. doi:10.1021/jm00073a022 |
| 19. | Data, P.; Srivastava, S.; Coutinho, E.; Govil, G. Curr. Top. Med. Chem. 2004, 4, 75–103. doi:10.2174/1568026043451636 |
| 25. | Maillard, M. C.; Brookfield, F. A.; Courtney, S. M.; Eustache, F. M.; Gemkow, M. J.; Handel, R. K.; Johnson, L. C.; Johnson, P. D.; Kerry, M. A.; Krieger, F.; Meniconi, M.; Muñoz-Sanjuán, I.; Palfrey, J. J.; Park, H.; Schaertl, S.; Taylor, M. G.; Weddell, D.; Dominguez, C. Bioorg. Med. Chem. 2011, 19, 5833–5851. doi:10.1016/j.bmc.2011.08.020 |
| 26. | Quibell, M.; Benn, A.; Flinn, N.; Monk, T.; Ramjee, M.; Wang, Y.; Watts, J. Bioorg. Med. Chem. 2004, 12, 5689–5710. doi:10.1016/j.bmc.2004.07.054 |
| 77. | Felpin, F.-X.; Fouquet, E. Chem.–Eur. J. 2010, 16, 12440–12445. doi:10.1002/chem.201001377 |
| 38. | Cochi, A.; Burger, B.; Navarro, C.; Pardo, D. G.; Cossy, J.; Zhao, Y.; Cohen, T. Synlett 2009, 2157–2161. doi:10.1055/s-0029-1217568 |
| 39. | Bilke, J. L.; Moore, S. P.; O'Brien, P.; Gilday, J. Org. Lett. 2009, 11, 1935–1938. doi:10.1021/ol900366m |
| 40. | Pansare, S. A.; Paul, E. K. Org. Biomol. Chem. 2012, 10, 2119–2125. doi:10.1039/c2ob06644k |
| 46. | Véliz, E. A.; Beal, P. A. Tetrahedron Lett. 2006, 47, 3153–3156. doi:10.1016/j.tetlet.2006.02.138 |
| 47. | Chang, B. C.; Conrad, W. E.; Denney, D. B.; Denney, D. Z.; Edelman, R.; Powell, R. L.; White, D. W. J. Am. Chem. Soc. 1971, 93, 4004–4009. doi:10.1021/ja00745a031 |
| 48. | Denney, D. B.; Denney, D. Z.; Gigantino, J. J. J. Org. Chem. 1984, 49, 2831–2832. doi:10.1021/jo00189a044 |
| 43. | Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L., Jr.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c |
| 44. | Dandapani, S.; Curran, D. P. Chem.–Eur. J. 2004, 10, 3130–3138. doi:10.1002/chem.200400363 |
| 45. | Dembinski, R. Eur. J. Org. Chem. 2004, 2763–2772. doi:10.1002/ejoc.200400003 |
| 43. | Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L., Jr.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c |
| 44. | Dandapani, S.; Curran, D. P. Chem.–Eur. J. 2004, 10, 3130–3138. doi:10.1002/chem.200400363 |
| 45. | Dembinski, R. Eur. J. Org. Chem. 2004, 2763–2772. doi:10.1002/ejoc.200400003 |
| 39. | Bilke, J. L.; Moore, S. P.; O'Brien, P.; Gilday, J. Org. Lett. 2009, 11, 1935–1938. doi:10.1021/ol900366m |
| 41. | Shioiri, T.; Izawa, K.; Konoike, T.; Karpf, M. Pharmaceutical Process Chemistry; Wiley-VCH: Weinheim, Germany, 2010; pp 1–37. |
| 42. | Roberge, D. M. Org. Process Res. Dev. 2004, 8, 1049–1053. doi:10.1021/op0400160 |
| 40. | Pansare, S. A.; Paul, E. K. Org. Biomol. Chem. 2012, 10, 2119–2125. doi:10.1039/c2ob06644k |
| 35. |
Kadouri-Puchot, C.; Comesse, S. Amino Acids 2005, 29, 101–130. doi:10.1007/s00726-005-0193-x
Reviews on the synthesis of (3-hydroxy)pipecolic acid derivatives. |
© 2014 Huy et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)