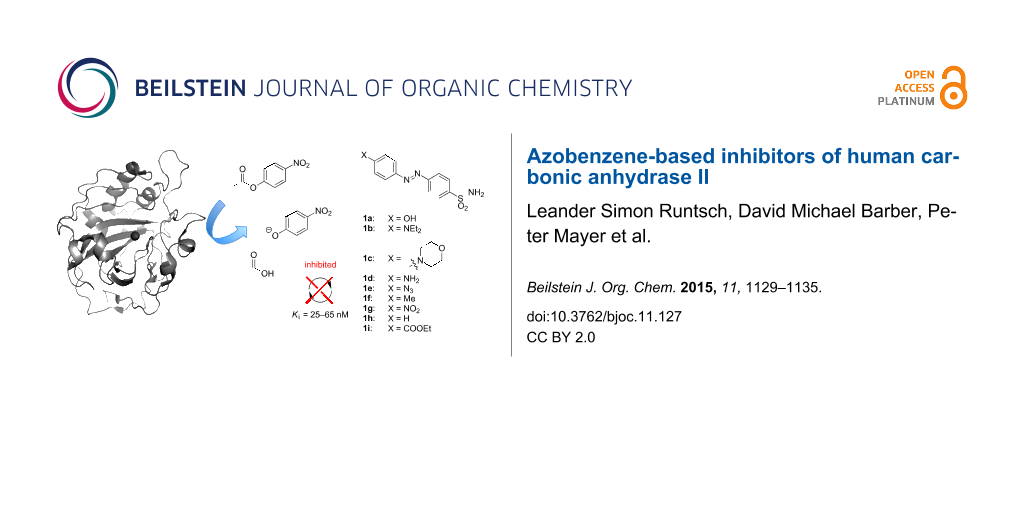
Abstract
Aryl sulfonamides are a widely used drug class for the inhibition of carbonic anhydrases. In the context of our program of photochromic pharmacophores we were interested in the exploration of azobenzene-containing sulfonamides to block the catalytic activity of human carbonic anhydrase II (hCAII). Herein, we report the synthesis and in vitro evaluation of a small library of nine photochromic sulfonamides towards hCAII. All molecules are azobenzene-4-sulfonamides, which are substituted by different functional groups in the 4´-position and were characterized by X-ray crystallography. We aimed to investigate the influence of electron-donating or electron-withdrawing substituents on the inhibitory constant Ki. With the aid of an hCAII crystal structure bound to one of the synthesized azobenzenes, we found that the electronic structure does not strongly affect inhibition. Taken together, all compounds are strong blockers of hCAII with Ki = 25–65 nM that are potentially photochromic and thus combine studies from chemical synthesis, crystallography and enzyme kinetics.

Graphical Abstract
Introduction
Carbonic anhydrase (CA) is an ubiquitously found zinc-containing metalloenzyme with many isoforms, which all catalyze the conversion of carbon dioxide and water to bicarbonate and a proton (Figure 1a, left) [1]. Despite its native purpose of pH and pressure regulation, its intrinsic esterase activity can be utilized to measure the catalytic activity by hydrolysis of p-nitrophenyl actetate (pNPA) to a phenolate, of which the product appearance can be observed colorimetrically (Figure 1a, right) [2]. In humans, isoform II (human carbonic anhydrase II; hCAII) is found in many tissues and is responsible for maintaining the inner eye pressure among other regulatory tasks [1]. Consequently, its failure is associated with glaucoma [1,3]. Treatment of this severe disease, that leads to blindness, is achieved with the application of aryl sulfonamides [3]. Being a transition-state analogue [4], this functional group exhibits excellent blocking characteristics of hCAII and culminates its power in many modern marketed drugs, such as acetazolamide (AAZ) or dorzolamide (Figure 1b) [5]. Furthermore, sulfonamide-containing azobenzenes exhibit affinity and blocking ability for hCAII (Figure 1c) [6]. With our knowledge in azobenzene chemistry and photopharmacology, we aimed to further understand how electronic substitution patterns on azobenzenes correlate to changes in enzyme affinity.
![[1860-5397-11-127-1]](/bjoc/content/figures/1860-5397-11-127-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Function and inhibition of hCAII. a) hCAII (pdb: 2vva [7]) catalyzes the hydration of carbon dioxide to bicarbonate and a proton (left) as well as the hydrolysis of pNPA to acetate and a colored phenolate (λmax = 400 nm). b) Aryl sulfonamide-containing pharmacophores of hCAII. c) Aryl sulfonamide merged to azobenzenes.
Figure 1: Function and inhibition of hCAII. a) hCAII (pdb: 2vva [7]) catalyzes the hydration of carbon dioxide t...
Results and Discussion
Azobenzenes can be synthesized by a variety of known chemical transformations [8]. Among them the most widely used is the diazotization of aniline, followed by trapping of the diazonium salt with an electron-rich aromatic compound (such as anilines and phenols). Another commonly used method is the condensation between anilines and aryl nitroso compounds, known as the Mills reaction. According to these transformations, nine sulfonamide containing azobenzenes 1a–i with different moieties in the 4´-position were synthesized. The substitution in the 4´-position will have the biggest impact on the electronic properties of the sulfonamide group due to communication through the conjugated π-system of the aromatic units and the diazene unit. Commencing with the diazotization of sulfanilamide and subsequent reaction with phenol, N,N-diethylaniline or N-phenylmorpholine led to azobenzenes 1a [6], 1b [9] and 1c, in moderate to low yields (43%, 38% and 25%, respectively) (Scheme 1a). Employing methylene-protected aniline 2 (crystal structure depicted in Scheme 1e) according to the procedure from Supuran and co-workers [6], amino azobenzene 1d was isolated after a one-pot reaction over three steps in 25%. By diazotization of 1d and trapping the salt with TMS-azide, we obtained azido azobenzene 1e in 63% yield through a [3 + 2] and retro-[3 + 2] cycloaddition (Scheme 1b) according to the procedure of Barral et al [10]. For the Mills reaction, different nitroso compounds were generated that were all used without further purification for the following condensation reactions. For example, sulfanilamide reacted with Oxone® in a biphasic DCM/water mixture to its nitroso counterpart, which was condensed with p-toluidine to give methyl azobenzene 1f (Scheme 1c) in 45% yield over two steps. Additionally, several in situ generated nitroso compounds bearing a nitro and a carboxylic acid ester were reacted with sulfanilamide to obtain nitro azobenzene 1g and ethyl ester azobenzene 1i, respectively. Commercially available nitrosobenzene gave rise to the unsubstituted sulfonamide 1h (Scheme 1d). The yield was poor for nitro azobenzene 1g (9%). Furthermore, the reaction of nitrosobenzene to obtain 1h proceeded in low yield (38%), while 1i was isolated in quantitative yield (99%). Crystals suitable for X-ray diffraction were obtained for 2 (Scheme 1e) and all sulfonamide-containing azobenzenes 1a–i (Figure 2). The crystallization conditions can be found in Supporting Information File 1).
![[1860-5397-11-127-i1]](/bjoc/content/inline/1860-5397-11-127-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis and characterization of azobenzene-containing aryl sulfonamides by different strategies. a) Diazotization and trapping of the diazonium salt with an electron-rich aromatic compound yields azobenzenes 1a–c. b) Reaction of methylene sulfonate-protected aniline and one-pot deprotection yields 1d, which can be converted to 1e. c) Mills condensation to obtain 1f. d) Mills condensation to obtain 1g–i. e) Crystal structure of sulfonate 2.
Scheme 1: Synthesis and characterization of azobenzene-containing aryl sulfonamides by different strategies. ...
![[1860-5397-11-127-2]](/bjoc/content/figures/1860-5397-11-127-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structures for compounds 1a–i (co-solvents and/or multiple molecules in the asymmetric cell are omitted for clarity).
Figure 2: Crystal structures for compounds 1a–i (co-solvents and/or multiple molecules in the asymmetric cell...
The substitution patterns on the aromatic core together with their electronic characteristics determines the absorption spectra of the individual azobenzenes [11]. We assessed the π–π*-band wavelength of maximal absorption (λmax) by RP–LCMS equipped with a UV–vis diode array detector, when determining the purity of our library. Therefore, λmax is determined in a water/acetonitrile mixture, which mimics aqueous conditions that are also used for the biological assays. The results are given in Table 1. While the “naked” azobenzene 1h has the absorbance maximum in the bluest part of the spectrum (λmax (1h) = 322 nm), substitution on the azobenzene in the 4´-position leads to a bathochromic shift, which is smaller for electron-withdrawing groups (λmax (1g) = 328 nm; λmax (1i) = 342 nm) and more pronounced for electron-donating groups (λmax (1f) = 338 nm, λmax (1a) = 358 nm, λmax (1d) = 404 nm, λmax (1c) = 414 nm, and λmax (1b) = 460 nm). Interestingly, azide 1e with a Hammett constant of σ = 0.08 [12], exhibits a bathochromic shift (λmax (1e) = 356 nm) close to hydroxy azobenzene 1a although it can be considered neither electron-withdrawing, nor electron-donating.
Table 1: Maximal absorbance wavelength (λmax), Hammett constants (σ) and inhibitory characteristics (IC50 and Ki) of 1a–i.
| 4´-substitution pattern | λmax (nm) | Hammett constant σ [12] | IC50 (nM) | Ki (nM) | |
|---|---|---|---|---|---|
| 1a | OH | 358 | −0.37 | 165.6 | 29.7 |
| 1b | NEt2 | 460 | −0.83 | 139.6 | 25.0 |
| 1c | N-morpholine | 414 | −0.83a | 309.2 | 55.4 |
| 1d | NH2 | 404 | −0.66 | 171.4 | 30.7 |
| 1e | N3 | 356 | +0.08 | 257.1 | 46.1 |
| 1f | Me | 338 | −0.17 | 363.2 | 65.1 |
| 1g | NO2 | 328 | +0.78 | 159.4 | 28.6 |
| 1h | H | 322 | +0.00 | 249.7 | 44.8 |
| 1i | COOEt | 342 | +0.45 | 167.9 | 30.1 |
| AAZ | – | – | – | 55.5 | 10.0 |
aTo the best of our knowledge the Hammett constant for morpholine has not been previously determined, therefore we used the parameter for alkylated amines due to its similar electronic nature.
To gain a deeper understanding into the binding mode of the synthesized azobenzene-containing sulfonamides we set out to co-crystallize an inhibitor with the wild-type enzyme. Protein crystals bound to 1d (Figure 3a, pdb: 5byi) were obtained using a previously reported method [13]. Due to the rigidness of the azobenzene the sulfonamide nitrogen and the 4´-position are far apart (>12 Å). The moiety in this position is solvent exposed and should therefore not contribute to the binding affinities by direct interactions (Figure 3a and b). Apart from primary binding interactions between the sulfonamide to the zinc center and T199, both of which are well-described [1] (Figure 3b and c), we were looking for secondary interactions resulting from the azobenzene (Figure 3c). Indeed, we found that the methyl group of L198 interacts with the sulfonamide-bearing aromatic core with a carbon to centroid distance of 3.5 Å. Furthermore, P202 and F131 contribute to centroid interactions with the second aromatic ring with distances of 3.5 (NCH2 to centroid), 4.0 (NCH2CH2 to centroid) and 4.7 Å (CH to centroid), respectively. These interactions can be weaker or stronger depending on the functional group in the 4´-position, as they affect the electronic properties of the aromatic system, and this would also be reflected by the Hammett constant. Interestingly, the azobenzene does not adopt a completely planar shape but is distorted with dihedral angles of −143.3° and 23.8° at the N=N bond and at the second aromatic ring to the diazene unit, respectively. It should be noted that a water molecule is held by the peptide backbone in the gorge (2.6 Å to OH of T200 and 2.7 Å to CO of P202), which might have interactions with the diazene unit although the distance for classical hydrogen bonding is rather long (3.2 Å).
![[1860-5397-11-127-3]](/bjoc/content/figures/1860-5397-11-127-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Crystal structure of hCAII bound to 1d (pdb: 5byi). a) The terminal amine of 1d is solvent-exposed, while the azobenzene is sticking in the cavity. b) Electron-density map of 1d bound to zinc with primary interactions. c) Interactions of 1d in the catalytic site in angstroms (Å). Primary interactions of the zinc-bound sulfonamide can be seen to T199 and L198 to the aromatic core. Secondary interactions can be observed from F131 and P202 towards the second aromatic ring of the azobenzene.
Figure 3: Crystal structure of hCAII bound to 1d (pdb: 5byi). a) The terminal amine of 1d is solvent-exposed,...
In order to determine the half-maximal inhibitory concentrations (IC50) and the inhibitory constants (Ki) towards hCAII for our library, we used a colorimetric endpoint measurement of the catalyzed pNPA hydrolysis (Figure 1a). Usually, a dansyl competition assay is employed for this purpose [1,14]. However, as this assay is fluorescence-based and azobenzenes can quench fluorescence [15], this might cause a distortion in the obtained data. Furthermore, irradiation with UV light (i.e., λ = 280 nm for tryptophan excitation) can result in azobenzene-cis-isomerization, which could lead to different binding characteristics. Therefore, we aimed at the endpoint absorbance system described herein. After expression and purification of wild-type hCAII we tested the benchmark blocker AAZ (Figure 1b) and obtained a Ki = 10.0 nM, which is in accordance with a previously reported inhibition constant (Ki (AAZ) = 12 nM [16]). Consequently, we were confident that our assay could assess the inhibitory characteristics of our library in a robust, reliable and reproducible manner.
By using the Cheng–Prusoff equation [17] with a Michaelis–Menten constant of Km = 1092.5 µM for pNPA (see Supporting Information File 1, Figure S2), we calculated the inhibitory constant Ki (Table 1) for each compound from the IC50 values obtained from sigmoidal fitting of the activity vs. concentration curve (see Figure 4a and Supporting Information File 1 for details). Azobenzenes 1a and 1d have been synthesized and tested previously (with a CO2 hydration assay), and the Ki values determined in the previous work are one order of magnitude higher than in our findings (Ki (1a) = 665 nM or 29.7; Ki (1d) = 106 nM or 30.7) towards hCAII [6]. Interestingly, in our studies the most efficient blocker turned out to be 1b with a Ki = 25.0 nM, which also shows the greatest red-shift in its maximal absorbance wavelength (π–π* band). Another electron-donating blocker bearing a methyl group substituent (1f), however, had the lowest affinity (Ki = 65.1 nM) of the library. Compound 1c offers the second-lowest affinity (Ki = 55.4 nM), which seems counter-intuitive, as the only difference with respect to 1b is the connection of the ethyl chains by an oxygen atom to a morpholine ring. This does not only affect the binding properties, but also the π–π* band, which is 46 nm blue-shifted relative to 1b. The inhibitors with the proton and azide substituents (1h and 1e, respectively) show very similar affinities towards hCAII with Ki = 44.8 nM and Ki = 46.1 nM. Sterics are also restrained, but should not affect binding, as the 4´-position is solvent-exposed (vide supra). Taking all of these findings into account, we conclude that the sulfonamide–zinc interaction dominates the binding affinity.
The electronic differences of our azobenzene library is expressed by their absorption spectra (as an indicator for the electron richness of the azobenzene) or in their Hammett constants (as an indicator for electron-pushing or pulling effects). When plotted against each other a trend can be observed, which is reflected by a more bathochromic shift when the Hammett constant becomes more negative (Figure 4b). However, when plotting the Hammett constants or the maximal absorbance wavelength versus the IC50 (Figure 4c and d, respectively), no clear correlation can be found. In both cases morpholine 1c, azide 1e, methyl 1f and unsubstituted azobenzene sulfonamide 1h lie in the same region (50 µM), while all other inhibitors (hydroxy 1a, alkyl amine 1b, amine 1d, nitro 1h and ethyl carboxylate 1i) show higher affinities at around 30 µM. The latter also shows a distribution from UV to blue maximal absorbance. We therefore speculate that electronics do not have a primary effect on the binding properties towards hCAII, but rather the sole presence of an aryl sulfonamide is sufficient. It should be pointed out that “naked” azobenzene 1h does not follow the trend when correlating its Hammett constant of σ = 0.00 to its λmax = 322 nm. As the plots from Figure 4c and d can be considered mirror images of each other, compound 1h does not fit into this picture.
![[1860-5397-11-127-4]](/bjoc/content/figures/1860-5397-11-127-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Inhibition of hCAII by electronically different azobenzene sulfonamides and AAZ. a) Endpoint measurement for the determination of IC50 for compounds 1a–i. b) Hammett constants versus maximal absorbance wavelength shows decreasing trend. c) IC50 versus Hammett constants. d) IC50 versus λmax.
Figure 4: Inhibition of hCAII by electronically different azobenzene sulfonamides and AAZ. a) Endpoint measur...
Conclusion
In conclusion, we have synthesized a small library of nine azobenzene sulfonamides with differing substitution patterns in the 4´-position using either azo coupling reactions or the Mills reaction. We determined the π–π* band and the crystal structures of all nine compounds together with the protein crystal structure of hCAII bound to inhibitor 1d. The latter structure highlights the interactions of the sulfonamide and the azobenzene with the protein cavity. The inhibitory action on hCAII was tested for all compounds using an endpoint measurement of catalytic pNPA hydrolysis. The inhibitory constants were in close proximity to each other (Ki = 25–65 nM) and a correlation of electron density as characterized by the Hammett constant σ with the binding affinities was not observed. We have expanded the repertoire of sulfonamide blockers of hCAII and described synthetic routes to potentially photochromic representatives. Furthermore, the protein crystal structure of hCAII bound to 1d described herein can be used as a template for the rational design of novel hCAII blockers. The biological activity of these blockers is currently under investigation and the results will be published in due course.
Supporting Information
| Supporting Information File 1: Chemical procedures, spectral data and X-ray crystallographic tables. Protein purification, crystallization conditions and measurement of Michaelis–Menten constant. | ||
| Format: PDF | Size: 1.5 MB | Download |
Acknowledgements
D.M.B is grateful to the European Comission for a Marie Skłodowska-Curie Intra-European fellowship (PIEF-GA-2103-627990). J.B. is grateful to the Studienstiftung des deutschen Volkes for a Ph.D. fellowship. M.G. and D.T. thank the Munich Centre for Integrated Protein Science (CIPSM) as well as the Deutsche Forschungsgemeinschaft SFB749 for financial support. D.T. acknowledges support of the European Research Council for an Advanced Grant (268795). We are grateful to the staff of the beamline X06SA at the Paul Scherrer Institute, Swiss Light Source, Villigen (Switzerland) for assistance during data collection. Dr. David H. Woodmansee is acknowledged for helpful discussion and advice together with providing ethyl 4-nitrosobenzoate as well as Eric P. Trautman and Martin Maier for excellent synthetic assistance.
Author Contributions
D.T. supervised the research. J.B. and D.T. conceived and designed the study. J.B. and L.S.R. performed chemical, biological and small molecule crystallization experiments. J.B. performed protein purification, crystallization and binding data analysis. P.M. collected X-ray datasets of 1a–i and solved the structures. M.G. collected hCAII X-ray dataset and solved the structure. J.B., D.M.B and D.T. wrote the manuscript with input from all authors.
References
-
Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M. Chem. Rev. 2008, 108, 946–1051. doi:10.1021/cr050262p
Return to citation in text: [1] [2] [3] [4] [5] -
Pocker, Y.; Stone, J. T. Biochemistry 1968, 7, 2936–2945. doi:10.1021/bi00848a034
Return to citation in text: [1] -
Supuran, C. T. Nat. Rev. Drug Discovery 2008, 7, 168–181. doi:10.1038/nrd2467
Return to citation in text: [1] [2] -
Boriack-Sjodin, P. A.; Zeitlin, S.; Chen, H.-H.; Crenshaw, L.; Gross, S.; Dantanarayana, A.; Delgado, P.; May, J. A.; Dean, T.; Christianson, D. W. Protein Sci. 1998, 7, 2483–2489. doi:10.1002/pro.5560071201
Return to citation in text: [1] -
Masini, E.; Carta, F.; Scozzafava, A.; Supuran, C. T. Expert Opin. Ther. Pat. 2013, 23, 705–716. doi:10.1517/13543776.2013.794788
Return to citation in text: [1] -
Maresca, A.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C. T. Bioorg. Med. Chem. Lett. 2009, 19, 4929–4932. doi:10.1016/j.bmcl.2009.07.088
Return to citation in text: [1] [2] [3] [4] -
Sjöblom, B.; Polentarutti, M.; Djinović-Carugo, K. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10609–10613. doi:10.1073/pnas.0904184106
Return to citation in text: [1] -
Merino, E. Chem. Soc. Rev. 2011, 40, 3835–3853. doi:10.1039/c0cs00183j
Return to citation in text: [1] -
Broichhagen, J.; Schönberger, M.; Cork, S. C.; Frank, J. A.; Marchetti, P.; Bugliani, M.; Shapiro, A. M. J.; Trapp, S.; Rutter, G. A.; Hodson, D. J.; Trauner, D. Nat. Commun. 2014, 5, No. 5116. doi:10.1038/ncomms6116
Return to citation in text: [1] -
Barral, K.; Moorhouse, A. D.; Moses, J. E. Org. Lett. 2007, 9, 1809–1811. doi:10.1021/ol070527h
Return to citation in text: [1] -
Birnbaum, P. P.; Linford, J. H.; Style, D. W. G. Trans. Faraday Soc. 1953, 49, 735–744. doi:10.1039/tf9534900735
Return to citation in text: [1] -
Leffler, J. E.; Grunwald, E. Rates and equilibria of organic reactions as treated by statistical, thermodynamic, and extrathermodynamic methods; Wiley: New York, 1963; p 458.
Return to citation in text: [1] [2] -
Lesburg, C. A.; Huang, C.; Christianson, D. W.; Fierke, C. A. Biochemistry 1997, 36, 15780–15791. doi:10.1021/bi971296x
Return to citation in text: [1] -
Chen, R. F.; Kernohan, J. C. J. Biol. Chem. 1967, 242, 5813–5823.
Return to citation in text: [1] -
Vögtle, F.; Gorka, M.; Hesse, R.; Ceroni, P.; Maestri, M.; Balzani, V. Photochem. Photobiol. Sci. 2002, 1, 45–51. doi:10.1039/b106813j
Return to citation in text: [1] -
D'Ambrosio, K.; Smaine, F.-Z.; Carta, F.; De Simone, G.; Winum, J.-Y.; Supuran, C. T. J. Med. Chem. 2012, 55, 6776–6783. doi:10.1021/jm300818k
Return to citation in text: [1] -
Cheng, Y.-C.; Prusoff, W. H. Biochem. Pharmacol. 1973, 22, 3099–3108. doi:10.1016/0006-2952(73)90196-2
Return to citation in text: [1]
| 1. | Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M. Chem. Rev. 2008, 108, 946–1051. doi:10.1021/cr050262p |
| 12. | Leffler, J. E.; Grunwald, E. Rates and equilibria of organic reactions as treated by statistical, thermodynamic, and extrathermodynamic methods; Wiley: New York, 1963; p 458. |
| 13. | Lesburg, C. A.; Huang, C.; Christianson, D. W.; Fierke, C. A. Biochemistry 1997, 36, 15780–15791. doi:10.1021/bi971296x |
| 1. | Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M. Chem. Rev. 2008, 108, 946–1051. doi:10.1021/cr050262p |
| 11. | Birnbaum, P. P.; Linford, J. H.; Style, D. W. G. Trans. Faraday Soc. 1953, 49, 735–744. doi:10.1039/tf9534900735 |
| 1. | Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M. Chem. Rev. 2008, 108, 946–1051. doi:10.1021/cr050262p |
| 3. | Supuran, C. T. Nat. Rev. Drug Discovery 2008, 7, 168–181. doi:10.1038/nrd2467 |
| 12. | Leffler, J. E.; Grunwald, E. Rates and equilibria of organic reactions as treated by statistical, thermodynamic, and extrathermodynamic methods; Wiley: New York, 1963; p 458. |
| 1. | Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M. Chem. Rev. 2008, 108, 946–1051. doi:10.1021/cr050262p |
| 6. | Maresca, A.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C. T. Bioorg. Med. Chem. Lett. 2009, 19, 4929–4932. doi:10.1016/j.bmcl.2009.07.088 |
| 6. | Maresca, A.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C. T. Bioorg. Med. Chem. Lett. 2009, 19, 4929–4932. doi:10.1016/j.bmcl.2009.07.088 |
| 2. | Pocker, Y.; Stone, J. T. Biochemistry 1968, 7, 2936–2945. doi:10.1021/bi00848a034 |
| 10. | Barral, K.; Moorhouse, A. D.; Moses, J. E. Org. Lett. 2007, 9, 1809–1811. doi:10.1021/ol070527h |
| 7. | Sjöblom, B.; Polentarutti, M.; Djinović-Carugo, K. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10609–10613. doi:10.1073/pnas.0904184106 |
| 6. | Maresca, A.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C. T. Bioorg. Med. Chem. Lett. 2009, 19, 4929–4932. doi:10.1016/j.bmcl.2009.07.088 |
| 16. | D'Ambrosio, K.; Smaine, F.-Z.; Carta, F.; De Simone, G.; Winum, J.-Y.; Supuran, C. T. J. Med. Chem. 2012, 55, 6776–6783. doi:10.1021/jm300818k |
| 6. | Maresca, A.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C. T. Bioorg. Med. Chem. Lett. 2009, 19, 4929–4932. doi:10.1016/j.bmcl.2009.07.088 |
| 9. | Broichhagen, J.; Schönberger, M.; Cork, S. C.; Frank, J. A.; Marchetti, P.; Bugliani, M.; Shapiro, A. M. J.; Trapp, S.; Rutter, G. A.; Hodson, D. J.; Trauner, D. Nat. Commun. 2014, 5, No. 5116. doi:10.1038/ncomms6116 |
| 17. | Cheng, Y.-C.; Prusoff, W. H. Biochem. Pharmacol. 1973, 22, 3099–3108. doi:10.1016/0006-2952(73)90196-2 |
| 5. | Masini, E.; Carta, F.; Scozzafava, A.; Supuran, C. T. Expert Opin. Ther. Pat. 2013, 23, 705–716. doi:10.1517/13543776.2013.794788 |
| 1. | Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M. Chem. Rev. 2008, 108, 946–1051. doi:10.1021/cr050262p |
| 14. | Chen, R. F.; Kernohan, J. C. J. Biol. Chem. 1967, 242, 5813–5823. |
| 4. | Boriack-Sjodin, P. A.; Zeitlin, S.; Chen, H.-H.; Crenshaw, L.; Gross, S.; Dantanarayana, A.; Delgado, P.; May, J. A.; Dean, T.; Christianson, D. W. Protein Sci. 1998, 7, 2483–2489. doi:10.1002/pro.5560071201 |
| 15. | Vögtle, F.; Gorka, M.; Hesse, R.; Ceroni, P.; Maestri, M.; Balzani, V. Photochem. Photobiol. Sci. 2002, 1, 45–51. doi:10.1039/b106813j |
© 2015 Runtsch et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)