1School of Environmental Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan
2Research Center for Material Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan
Corresponding author email
Associate Editor: R. Sarpong Beilstein J. Org. Chem.2015,11, 1241–1245.https://doi.org/10.3762/bjoc.11.138 Received 13 May 2015,
Accepted 16 Jul 2015,
Published 23 Jul 2015
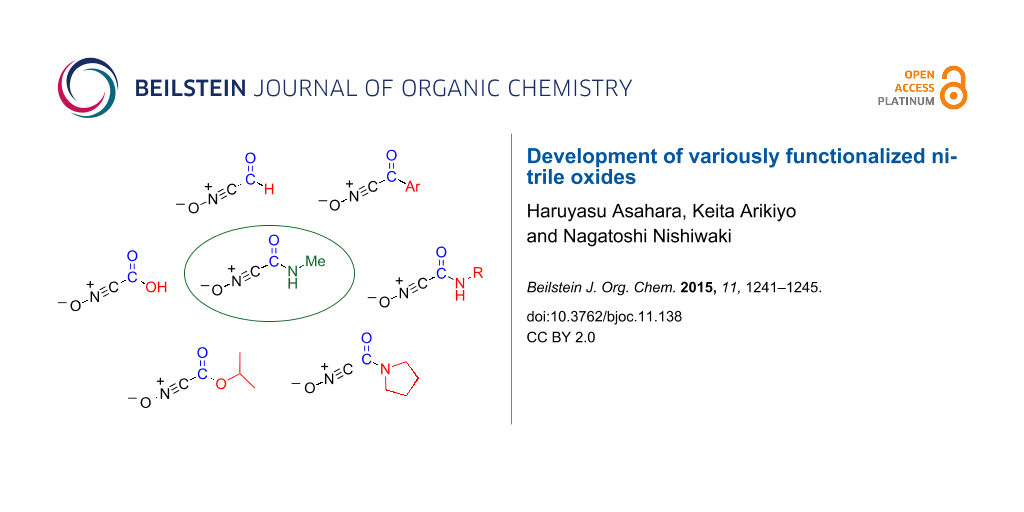
N-Methylated amides (N,4-dimethylbenzamide and N-methylcyclohexanecarboxamide) were systematically subjected to chemical transformations, namely, N-tosylation followed by nucleophilic substitution. The amide function was converted to the corresponding carboxylic acid, esters, amides, aldehyde, and ketone upon treatment with hydroxide, alkoxide, amine, diisobutylaluminium hydride and Grignard reagent, respectively. In these transformations, N-methyl-N-tosylcarboxamides behave like a Weinreb amide. Similarly, N-methyl-5-phenylisoxazole-3-carboxamide was converted into 3-functionalized isoxazole derivatives. Since the amide was prepared by the cycloaddition reaction of ethynylbenzene and N-methylcarbamoylnitrile oxide, the nitrile oxide served as the equivalent of the nitrile oxides bearing a variety of functional groups such as carboxy, alkoxycarbonyl, carbamoyl, acyl and formyl moieties.
Nitrile oxides are valuable synthetic synthons for the construction of heterocyclic compounds by cycloaddition reactions, which lead to the formation of two bonds in a single experimental step [1,2]. In addition, cycloadducts serve as precursors of polyfunctionalized compounds by ring opening reaction followed by N–O bond fission [2,3]. Hence, functionalized nitrile oxides are clearly useful for the preparation of more complex systems. However, because the precursors of the functionalized nitrile oxides are not always easily available, there are only a few reports that deal with these compounds compared to those on alkylated and arylated nitrile oxides [1]. Thus, it is highly desirable to develop methodologies for the facile generation of functionalized nitrile oxides.
In our previous work, we reported the preparation of nitrile oxide 2 bearing an N-methylcarbamoyl group by treatment of 2-methyl-4-nitroisoxazolin-5(2H)-one (1) only with water at room temperature. Nitrile oxide 2 undergoes cycloaddition reactions with alkenes [4,5], alkynes (Scheme 1) [4,5], nitriles [6], and 1,3-dicarbonyl compounds [7] to afford the corresponding polyfunctionalized heterocyclic compounds, which are not easily obtained by other approaches. Although this protocol is expected to be useful for synthesizing polyfunctionalized compounds, it is limited by the need of a N-methylcarbamoyl functional group. The synthetic utility of nitrile oxide 2 will be indeed improved by converting the N-methylcarbamoyl group into other functionalities, i.e., nitrile oxide 2 may serve as an equivalent of nitrile oxides 4 having versatile functional groups (Scheme 1).
Scheme 1:
A strategy for developing functionalized nitrile oxides.
Scheme 1:
A strategy for developing functionalized nitrile oxides.
Weinreb amide (N-methoxy-N-methylamide) is widely used for the conversion of less reactive amide moieties into other carbonyl functionalities [8,9]. The two oxygen atoms of the methoxy and carbonyl groups coordinate to the organometallic reagents, thereby suppressing further reaction with the obtained aldehyde or ketone. However, this protocol cannot be applied to the conversion of N-methylamides because methoxylation of N-methylamides is hitherto unknown. Thus, we speculated that the introduction of a sulfonyl group would be effective because of its high electron-withdrawing and coordinating abilities, serving as an equivalent of the Weinreb amide. Indeed, the use of N-methyl-3-phenyl-N-tosylbutanamide for this purpose was already reported by Itoh and coworkers [10]. However, a systematic study using simple amides has not yet been performed. In this context, we examined the sulfonylation and chemical conversion of N-methylated aromatic and aliphatic amides, and a similar conversion of N-methyl-5-phenylisoxazole-3-carboxamide (3), which is equal to the generation of variously functionalized nitrile oxides.
Results and Discussion
At the outset, methanesulfonylation (mesylation) of amides was studied. To a suspension of N,4-dimethylbenzamide (6A) and sodium hydride (2 equiv) in THF, mesyl chloride (7a, 1.3 equiv) was added, and the resultant mixture was stirred at room temperature for 9 h. After acidification, the reaction mixture was extracted with diethyl ether to afford the crude material, from which 32% of N-mesylated product 8Aa[11] was isolated and 61% of 6A was recovered. Thus, the conversion yield of 8Aa was 82% (Table 1, entry 1). On the other hand, the yield of 8Aa was considerably increased (up to 70%) when ethylmagnesium bromide was employed as a base instead of sodium hydride (Table 1, entry 2). However, the instability and gradual decomposition of mesylated product 8Aa by ambient moisture impedes its storage and use as a synthetic reagent. Hence, in the place of the mesyl group, the 4-methylbenzenesulfonyl (tosyl) moiety was employed as the activating group of the amide function. Indeed, tosylated product 8Ab[11] proved to be sufficiently stable under ambient conditions. Despite the considerable efforts for optimizing the reaction conditions, the highest yield obtained for 8Ab was 60%; however, 35% of starting material 6A was recovered, and the conversion yield was 92%. This tosylation method was applied to the aliphatic amide N-methylcyclohexanecarboxamide (6B) to afford 8Bb upon treatment with tosyl chloride (7b) using sodium hydride as a base (Table 1, entry 4). In this case, the use of an excess of 7b increased the yield of 8Bb up to 60% (85% of conversion yield, Table 1, entry 5).
Table 1:
Sulfonylation of N-methylamides.
entry
R1
R2
Base (equiv)
Temp./°C
Time/h
Product
Yield/%
1
4-MeC6H4
Me
NaH (2)
rt
9
8Aa
32 (82)a
2
4-MeC6H4
Me
EtMgBr (1.1)
0
1
8Aa
70
3
4-MeC6H4
4-MeC6H4
NaH (2)
0
24
8Ab
60 (92)a
4
c-Hexyl
4-MeC6H4
NaH (2)
0
12
8Bb
38
5b
c-Hexyl
4-MeC6H4
NaH (2)
0
12
8Bb
60 (85)a
aConversion yields were shown in parentheses; b5 equivalents of 7b were used.
Next, nucleophilic substitutions of tosylated amides 8Ab and 8Bb were investigated (Table 2). Whereas water and methanol did not alter the amide moieties, hydroxide and methoxide converted amides 8Ab and 8Bb into the corresponding carboxylic acids (9a and 10a) and methyl esters (9b and 10b), respectively (Table 2, entries 1–4). In addition, the nucleophilic substitution also proceeded by employing neutral amines. The reaction of 8 with propylamine proceed smoothly, leading to N-propylamides 9c and 10c (Table 2, entries 5 and 6). The substitution reaction was influenced by the bulkiness of the amines. Heating at 120 °C was required for the substitution reaction by sec-butylamine. However, in the case of tert-butylamine, even when the reaction was conducted in a sealed tube at 120 °C, only a small amount of amide was observed (Table 2, entries 7–10). On the other hand, a cyclic secondary amine, pyrrolidine, afforded amides 9f and 10f quantitatively at room temperature (Table 2, entries 11 and 12).
Table 2:
Chemical conversions of N-methylamide to other functions.
aIntermediately produced 9 or 10 underwent the excess reactions to afford alcohols 11.
When more reactive butyllithium or isopropylmagnesium bromide was used as the nucleophile, the corresponding tertiary alcohols 11g and 11h were obtained in 51% and 58% yields, respectively, whereas ketones 9g and 9h were not detected (Table 2, entries 13 and 14). In the case of an aromatic Grignard reagent, the undesired reaction was suppressed. Although the reaction did not occur at −78 °C, the substitution proceeded successfully at −40 °C, affording ketones 9i and 10i (Table 2, entries 15 and 16). Sodium borohydride also proved to be highly reactive, causing a further addition reaction even at −78 °C to generate the corresponding alcohol 11j (Table 2, entry 17). This disadvantage was overcome by using bulkier diisobutylaluminium hydride (DIBAL) to furnish aldehydes 9j and 10j, although small amounts of alcohol 11j were also detected (Table 2, entries 18 and 19).
In contrast to tosylated amides 8Ab and 8Bb, tosylated N-methyl-5-phenylisoxazole-3-carboxamide 12 exhibited higher reactivity. Upon tosylation under similar conditions 3 did not afford product 12, and isoxazole-3-carboxylic acid 5a, a hydrolyzed product of 12, was observed. This problem was solved by quenching the reaction mixture with ice water instead of just water; yet, 12 was afforded in only 10% yield, and 5a was the main product in 38% yield. These findings imply that during the work-up procedure, further decomposition of 12 by water occurred even at low temperatures because of the higher electron deficiency of the isoxazole ring compared with carbocyclic rings. When, instead of water, less nucleophilic acetic acid was employed for the quench, 12 was isolated in 74% yield and the competitive hydrolysis of 12 was suppressed (6% of 5a).
Although tosylated amide 12 could be isolated, it gradually decomposed under ambient conditions. Hence, the chemical conversion was performed by adding a nucleophile to the mixture of the tosylation reaction, without the isolation of 12 (Table 3). As mentioned above, the use of a hydroxide anion for the hydrolysis was not required, and carboxylic acid 5a was obtained in 81% yield by the addition of only water (Table 3, entry 1). This protocol was applicable to a relatively bulky alcohol to afford isopropyl ester 5k in moderate yield (Table 3, entry 2). Similarly, primary and secondary amines underwent the substitution reaction, leading to 5c–f in good to moderate yields (Table 3, entries 3–6). It was noteworthy that, because of high reactivity of 12 caused by the isoxazole ring [20], the methylamino group could be replaced by a bulky tert-butylamino moiety. Conversion of the N-methylcarbamoyl group to an acyl or formyl group was achieved upon treatment with a Grignard reagent or with DIBAL (Table 3, entries 7 and 8).
Table 3:
Chemical conversion of N-methylisoxazole-3-carboxamide 3.
a2 Equiv of nucleophiles were used. bAt −40 °C. cAt −78°C.
The chemical conversion of N-methylamides to versatile carbonyl functions was systematically studied. In this protocol, the tosyl group was found to be effective for the activation of the carbamoyl group and for preventing over-addition by organometallic reagents. Isoxazole-3-carboxamide 3 was successfully converted in a similar fashion. Thus, as shown in Figure 1, nitrile oxide 2 serves as an equivalent of functionalized nitrile oxides 4, thereby improving the synthetic utility of carbamoylnitrile oxide 2.
Conversion of isoxazolecarboxamide 3 to 5c via tosyl derivative 12
To a solution of amide 3 (101 mg, 0.5 mmol) in THF (1 mL), a suspension of 60 wt % sodium hydride (100 mg, 2.5 mmol) in THF (3 mL) was added under argon. After the mixture was stirred vigorously for 10 min, the mixture was cooled to 0 °C and then a solution of tosyl chloride (191 mg, 1 mmol) in THF (1 mL) was slowly added. After the mixture was stirred for 3 h at 0 °C, propylamine (164 μL, 2 mmol) was added, and then the mixture was stirred at room temperature for 12 h. After evaporation of the solvent, the residue was dissolved into diethyl ether (5 mL), washed with water (5 mL), and the aqueous layer was extracted with diethyl ether (2 × 5 mL). The combined organic layer was dried over magnesium sulfate, and concentrated, and the residue was subjected to column chromatography on silica gel to afford N-propylcarboxamide 5c (85 mg, 0.37 mol, 74% yield), eluted with dichloromethane. 1H NMR (400 MHz, CDCl3, TMS) δ 1.01 (t, J = 7.2 Hz, 3H), 1.67 (tq, J = 7.2, 7.2 Hz, 2H), 3.43 (dt, J = 7.2, 7.2 Hz, 2H), 6.79–6.90 (br, 1H), 6.97 (s, 1H), 7.47–7.50 (m, 3H), 7.78–7.81 (m, 2H); 13C NMR (100 MHz, CDCl3, TMS) δ 11.4 (CH3), 22.7 (CH2), 41.2 (CH2), 99.2 (CH), 126.0 (CH), 126.9 (C), 129.1 (CH), 130.7 (CH), 158.9 (C), 159.3 (C), 171.5 (C).
References
Nishiwaki, N.; Asahara, H. Carbamoylnitrile Oxide and Inverse Electron-Demand 1,3-Dipolar Cycloaddition. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons: Hoboken, USA, 2014; pp 223–240. doi:10.1002/9781118778173.ch09
Return to citation in text:
[1]
[2]
Torssell, K. B. G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; VCH Publishers, Inc.: New York, USA, 1988; pp 55–74.
Return to citation in text:
[1]
[2]
Nishiwaki, N.; Maki, A.; Ariga, M. J. Oleo Sci.2009,58, 481–484. doi:10.5650/jos.58.481
Return to citation in text:
[1]
Nishiwaki, N.; Uehara, T.; Asaka, N.; Tohda, Y.; Ariga, M.; Kanemasa, S. Tetrahedron Lett.1998,39, 4851–4852. doi:10.1016/S0040-4039(98)00919-8
Return to citation in text:
[1]
[2]
Nishiwaki, N.; Kobiro, K.; Hirao, S.; Sawayama, J.; Saigo, K.; Ise, Y.; Okajima, Y.; Ariga, M. Org. Biomol. Chem.2011,9, 6750–6754. doi:10.1039/c1ob05682d
Return to citation in text:
[1]
Nishiwaki, N.; Kobiro, K.; Hirao, S.; Sawayama, J.; Saigo, K.; Ise, Y.; Nishizawa, M.; Ariga, M. Org. Biomol. Chem.2012,10, 1987–1991. doi:10.1039/c2ob06925c
Return to citation in text:
[1]
Balasubramaniam, S.; Aidhen, I. S. Synthesis2008, 3707–3738. doi:10.1055/s-0028-1083226
A review and references are cited in.
Return to citation in text:
[1]
Parhi, A. K.; Franck, R. W. Org. Lett.2004,6, 3063–3065. doi:10.1021/ol0489752
Nitrile oxide having a Weinreb amide can be generated from cinnamic acid within five reaction steps; however, this protocol does not involve a conversion of N-methylamide to the Weinreb amide.
Return to citation in text:
[1]
Nagashima, H.; Ozaki, N.; Washiyama, M.; Itoh, K. Tetrahedron Lett.1985,26, 657–660. doi:10.1016/S0040-4039(00)89172-8
Return to citation in text:
[1]
Fang, C.; Qian, W.; Bao, W. Synlett2008, 2529–2531. doi:10.1055/s-2008-1078218
Return to citation in text:
[1]
Wu, X.; Larhed, M. Org. Lett.2005,7, 3327–3329. doi:10.1021/ol0512031
Return to citation in text:
[1]
Dokli, I.; Gredičak, M. Eur. J. Org. Chem.2015, 2727–2732. doi:10.1002/ejoc.201500051
Return to citation in text:
[1]
Zhou, F.; Ding, K.; Cai, Q. Chem. – Eur. J.2011,17, 12268–12271. doi:10.1002/chem.201102459
Return to citation in text:
[1]
Kovalenko, O. O.; Volkov, A.; Adolfsson, H. Org. Lett.2015,17, 446–449. doi:10.1021/ol503430t
Return to citation in text:
[1]
Kawano, M.; Kiuchi, T.; Matsuo, J.-i.; Ishibashi, H. Tetrahedron Lett.2012,53, 432–434. doi:10.1016/j.tetlet.2011.11.067
Return to citation in text:
[1]
Skillinghaug, B.; Sköld, C.; Rydfjord, J.; Svensson, F.; Behrends, M.; Sävmarker, J.; Sjöberg, P. J. R.; Larhed, M. J. Org. Chem.2014,79, 12018–12032. doi:10.1021/jo501875n
Return to citation in text:
[1]
Shivakumar, S. B.; Basavaraju, Y. B.; Umesha, B.; Krishna, M. H.; Mallesha, N. Eur. J. Chem.2014,5, 424–429. doi:10.5155/eurjchem.5.3.424-429.1020
Return to citation in text:
[1]
Tamura, M.; Nishimura, T.; Nishiwaki, N.; Ariga, M. Heterocycles2004,63, 1659–1665. doi:10.3987/COM-04-10085
Return to citation in text:
[1]
Desai, V. G.; Naik, S. R.; Dhumaskar, K. L. Synth. Commun.2014,44, 1453–1460. doi:10.1080/00397911.2013.854916
Return to citation in text:
[1]
Commercially available.
Return to citation in text:
[1]
[2]
Tait, B.; Cullen, M. Methods of modulating cftr activity. PCT Int. Appl. WO 2014210159, Dec 31, 2014.
Return to citation in text:
[1]
[2]
Maywald, V.; Freund, W.; Hamprecht, G.; Kuekenhoehner, T.; Plath, P.; Wuerzer, B.; Westphalen, K.-O. Carbonsäureamide. Eur. Pat. Appl. EP 418667, March 27, 1991.
Return to citation in text:
[1]
Nishiwaki, N.; Asahara, H. Carbamoylnitrile Oxide and Inverse Electron-Demand 1,3-Dipolar Cycloaddition. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons: Hoboken, USA, 2014; pp 223–240. doi:10.1002/9781118778173.ch09
2.
Torssell, K. B. G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; VCH Publishers, Inc.: New York, USA, 1988; pp 55–74.
Nishiwaki, N.; Asahara, H. Carbamoylnitrile Oxide and Inverse Electron-Demand 1,3-Dipolar Cycloaddition. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons: Hoboken, USA, 2014; pp 223–240. doi:10.1002/9781118778173.ch09
Balasubramaniam, S.; Aidhen, I. S. Synthesis2008, 3707–3738. doi:10.1055/s-0028-1083226
A review and references are cited in.
9.
Parhi, A. K.; Franck, R. W. Org. Lett.2004,6, 3063–3065. doi:10.1021/ol0489752
Nitrile oxide having a Weinreb amide can be generated from cinnamic acid within five reaction steps; however, this protocol does not involve a conversion of N-methylamide to the Weinreb amide.
Skillinghaug, B.; Sköld, C.; Rydfjord, J.; Svensson, F.; Behrends, M.; Sävmarker, J.; Sjöberg, P. J. R.; Larhed, M. J. Org. Chem.2014,79, 12018–12032. doi:10.1021/jo501875n


![[1860-5397-11-138-i1]](/bjoc/content/inline/1860-5397-11-138-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-138-i2.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-138-i3.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-138-i4.svg?max-width=637&scale=1.0)
![[1860-5397-11-138-1]](/bjoc/content/figures/1860-5397-11-138-1.svg?scale=2.0&max-width=1024&background=FFFFFF)